
Abstract
These toxicity studies aimed to assess the safety and tolerability of a novel intravenous diclofenac sodium (37.5 mg/mL) formulation containing povidone K12 (80 mg/mL) as the key excipient in Wistar rats. This formulation was tested at doses of 3, 7, and 15 mg/kg/day and was administered daily for 28 days by intravenous route. Toxicokinetic estimation revealed a dose-proportional increase in plasma exposure to diclofenac. The formulation was well tolerated in males; however, mortality was observed in females (2/15) at the highest dose (15 mg/kg/day). Adverse gastrointestinal events related to NSAIDS and a few other treatment-related effects on clinical and anatomic pathology were noted at the 15 mg/kg/day dose, which normalized at the end of the 2-week recovery period. In addition, the excipient povidone K12 was present in a higher amount than the approved Inactive Ingredient Database (IID) limit in the proposed novel formulation. It was qualified through a separate 28-day repeated dose toxicity study by intravenous route in Wistar rats. Povidone K12 was found to be well tolerated and safe up to a dose of 165 mg/kg/day. No treatment-related adverse effects were observed in this study. In conclusion, repeated administration of a novel intravenous formulation containing diclofenac sodium was found to be safe up to the dose of 7 mg/kg/day in female rats and 15 mg/kg/day in male rats.
Introduction
Diclofenac is a highly effective and well-tolerated nonselective non-steroidal anti-inflammatory drug (NSAID) recommended for use in the treatment of acute and chronic pain and inflammatory conditions. 1 The mechanism of action of diclofenac involves the relatively equipotent inhibition of cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) enzymes through the inhibition of the synthesis of prostaglandin-E2 (PGE2), prostacyclins, and thromboxanes, which are essential components of the inflammatory response of the body.2,3
Composition of the Novel Diclofenac Sodium Injection.
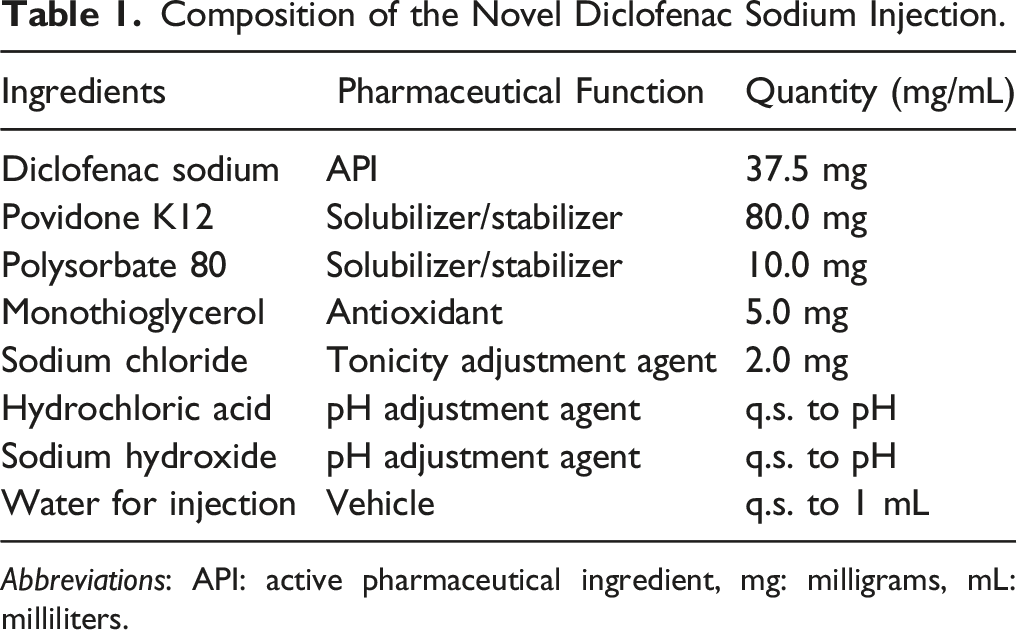
Abbreviations: API: active pharmaceutical ingredient, mg: milligrams, mL: milliliters.
Materials and Methods
Animals, Husbandry, Ethics, and Accreditations
A total of 130 male and 130 female Wistar rats (aged 6–8 weeks, weighing approximately 100–300 grams) were obtained from the Animal Research Facility of Zydus Research Centre, Ahmedabad, India. The animals were acclimated for a minimum period of 5 days before the experimental procedures were initiated. Rats were housed in clean, sterilized solid floor individually-ventilated cages covered with a stainless-steel grill top with a provision for keeping pellet feed and a water bottle. Autoclaved corncob was used as the bedding material. The following environmental conditions were maintained: temperature: 18 to 29°C; relative humidity: 30 to 70%; 12 hours of light–dark photoperiod; and up to 15 room air changes per hour. Certified rodent pellet feed (Teklad 2014C) and drinking water (filtered by reverse osmosis followed by UV treatment) were provided ad libitum.
Experiments involving rats were reviewed and approved by the Instructional Animal Ethics Committee (approval numbers: ZRC/TOX/NP/244/05-2K19, ZRC/TOX/NP/265/05-2K20). These studies were conducted in a GLP-compliant and AAALAC International-accredited test facility. The animal facility is registered with The Committee for Control and Supervision of Experiments on Animals (CCSEA), a statutory committee under the Government of India (GoI) (Facility Registration Number: 77/PO/RcBi/SL/99/CPCSEA). The Clinical Pathology Laboratory (CPL-II) at the Zydus Research Centre is accredited by the National Accreditation Board for Testing and Calibration Laboratories (NABL), GoI, and the National GLP Compliance Monitoring Authority (NGCMA), GoI. The laboratory has an established in-house quality control program and an external quality assessment program with the College of American Pathologists (CAP).
Test Formulation Details
Diclofenac Sodium Injection (37.5 mg/mL)
Each mL contained 37.5 mg of diclofenac sodium in an aqueous solution containing povidone K12, polysorbate 80, monothioglycerol, sodium chloride, pH-adjusting agents (sodium hydroxide and hydrochloric acid), and water for injection. The physicochemical attributes of the drug product during manufacturing and storage were controlled according to the recommended specifications. Diclofenac Sodium USP (IUPAC name: Sodium [2-[(2,6-Dichlorophenyl)amino]phenyl]acetate; CAS number: 15307-79-6; molecular weight: 318.13 g/mole) was found to be not less than 33.75 mg/mL and not more than 41.25 mg/mL, and the concentration of monothioglycerol NF was found to be not less than 1.25 mg/mL and not more than 5.50 mg/mL (25.0% and 110.0% of the labeled amount) in an HPLC assay of the final formulation.
Povidone K-12 Solution, 80 mg/mL
Each ml was found to contain 72.0 mg/mL of povidone K-12 (CAS number: 9003-39-8; molecular weight: between 2000 to 3000 Dalton) (90.1% of the label amount), as determined in an assay carried out in the final formulation. The formulations were provided under ready-to-use conditions for the animal studies. Both formulations met the predefined acceptance criteria. The formulations were stored at 20°C to 25°C (68° to 77°F); excursions were permitted at 15° to 30°C (59° to 86°F) (USP controlled room temperature) and protected from light.
The qualitative and quantitative compositions of the proposed diclofenac sodium injection mixture are listed in Table 1.
Study Designs
Animals were randomly assigned to different groups using SAS® software (Statistical Analysis System, version 9.4). Animals whose body weight was within ±20% of the mean body weight were selected for the studies. The animals not selected for the study were humanely euthanized. Two rat studies were conducted to evaluate the toxic effects of povidone K-12 and diclofenac injection.
Povidone K12 Qualification Study: 28-Day Repeat Dose Toxicity of Povidone K-12 Solution
Three treatment groups (20 animals/group) comprising 10 male and 10 female Wistar Han rats were intravenously administered the excipient povidone K-12 solution at dosages of 33.0, 82.5, and 165.0 mg/kg/day for a minimum of 28 consecutive days. Concurrent control group animals were treated with 0.9% sodium chloride as the vehicle. Additionally, 10 male and 10 female rats were maintained in the control and high-dose groups for a 2-week treatment-free recovery period to determine the persistence, recovery, or delayed toxic effects, if any.
Diclofenac Sodium Toxicity Study: 28-Day Repeat Dose Toxicity of Diclofenac Sodium Injection
Three treatment groups (20 animals/group comprising 10 male and 10 female Wistar rats) received daily intravenous injections of diclofenac sodium at dosages of 3, 7, and 15 mg/kg/day for a minimum period of 28 consecutive days. The control group received a placebo alone. Additionally, 5 male and 5 female rats in the control and high-dose groups were maintained for a treatment-free recovery period of 2 weeks to determine the persistence, recovery, or delayed toxic effects. Independent groups of 5 male and 5 female rats per dose were maintained for the toxicokinetic assessment on day 1 and day 28 of the treatment period.
Dose Levels and Justification
Povidone K12 Qualification Study
As per the proposed clinical formulation, the maximum daily intravenous intake of povidone K-12 is 320 mg. Based on the body surface area factor, 320 mg is equivalent to 33 mg/kg/day in rats. The selected doses of the povidone K-12 solution used in the present study were 33, 82.5, and 165 mg/kg/day. The dose volume administered to each animal was less than 5 mL/kg body weight. Additionally, the vehicle control group animals were used in this study and treated with vehicle alone, 0.9% w/v sodium chloride solution.
Diclofenac Sodium Toxicity Study
Diclofenac sodium was administered at dose levels of 0 (vehicle), 3, 7, and 15 mg/kg/day in rats based on the reported toxicity findings of diclofenac sodium injection. 6 The dose volume administered to each animal was 0.4 mL/kg body weight.
The dosing volume was calculated based on the most recent body weight of each animal. The test formulations were administered by intravenous route (bolus) into the tail vein of rats using a graduated syringe fitted with a 26½ gauge needle.
Clinical Observations
Cage-side observations were carried out twice a day to assess morbidity and mortality. All animals were observed once a day for visible treatment-related clinical signs, including reactions at the injection site until the end of treatment and recovery periods.
Ophthalmic Examination
Ophthalmological examination was performed with an ophthalmoscope on all the animals before the start of treatment and thereafter on the vehicle control and high-dose groups near the end of the treatment and recovery periods.
Body Weight Measurements and Food Consumption
Body weights were recorded for all animals upon receipt and before randomization. Body weight was recorded on day 1 (before the start of the treatment) and thereafter at weekly intervals until the end of the treatment and recovery periods. Food consumption of the main and recovery groups was measured weekly throughout the study period.
Neurobehavioral Observations
Neurobehavioral observations following the Irwin test and grip strength tests were carried out for control and high-dose group animals near the end of the treatment and recovery periods. Neurobehavioral observations included (a) home cage observations for any spontaneous activity; (b) handling observations: ease of removing, handling reactivity, palpebral closure, lacrimation, eye and skin examination, salivation, and nasal discharge; (c) open field observations: posture, locomotion, gait, arousal, vocalization, respiration, tremors, clonic and/or tonic convulsions, urination, defecation, stereotype, and bizarre behaviors; and (d) sensory responses: approach, touch, startle pupil constriction, pinna reflex, tail pinch, surface righting reflex, visual placing, and air righting responses.
Clinical Pathology Investigations
Detailed clinical pathology investigations were carried out for all study groups at the end of the treatment and recovery period. These investigations were conducted at the Clinical Pathology Laboratory at Zydus Research Centre, Ahmedabad, India. All animals from the respective dose groups were fasted overnight (with water allowed). Blood samples were drawn from the retro-orbital plexus under mild isoflurane anesthesia. At the time of blood collection, an aliquot of blood was collected in tubes containing 2% K2EDTA for hematology parameters and in a vacutainer (gel + clot activator) for clinical chemistry analytes. For clinical chemistry analysis, the serum was separated via centrifugation at 4000 rpm for 10 minutes.
Hematology parameters were analyzed using an Advia 2120i hematology analyser (Siemens Healthineers, USA). Blood smears were prepared from each animal at the end of the treatment and recovery periods. Clinical chemistry parameters were analyzed using a Cobas c311 analyzer (Roche Diagnostics, Switzerland).
Urine analysis included physical, chemical, and microscopic examination of the urine samples collected from overnight fasted animals housed in metabolic cages before the scheduled necropsy examination of the main and recovery groups. The physical examination included an estimation of volume, color, and clarity. A chemical examination was performed for different analytes. The microscopic examination included evaluation for the presence of crystals, epithelial cells, casts, erythrocytes, and leukocytes.
Terminal Procedures
Necropsy was performed on all animals at the end of the treatment and recovery periods. All the animals were fasted overnight before euthanasia. Animals were euthanized by carbon dioxide (CO2) asphyxiation followed by exsanguination. At necropsy, the animals were examined visually for external abnormalities, including at the application site. The abdominal, thoracic, and cranial cavities and their contents were examined for abnormalities, and the organs were removed, examined, and weighed. Tissue samples, except eyes and testes, were collected and placed in a 10% neutral buffer until processing. Histopathological examination was carried out on control and high-dose group animals in povidone K12 qualification study and from control (0 mg/kg/day), mid dose (7 mg/kg/day) and high dose (15 mg/kg/day) groups in female rats and control (0 mg/kg/day) and high dose (15 mg/kg/day) groups in male rats from diclofenac sodium toxicity study. All gross lesions were processed for histopathological examination. A peer review of histopathological findings was performed. The absolute and relative weights (organ-to-fasting body weight ratios) of the following organs were measured: adrenal glands, brain, epididymides, heart, kidneys, liver, lungs, ovaries with oviducts, spleen, testes, thymus, and uterus with cervix.
Statistical Analysis
Statistical analysis was performed using SAS® (Statistical Analysis System), version 9.4 software. Normality within groups was analyzed by the Kolmogorov‒Smirnov test, where if the results of the Kolmogorov‒Smirnov test were found to be not statistically significant, the parametric test was applied; otherwise, the nonparametric test was applied. The homogeneity of variances across groups was analyzed by Levene’s test. If the results of Levene’s test were not statistically significant, one-way ANOVA was applied, and if the results were significant, the Kruskal‒Wallis test was used. For more than two groups, one-way ANOVA (parametric test) or the Kruskal‒Wallis test (nonparametric test) was applied to analyze the equality of means across groups. If the one-way ANOVA results were statistically significant, then Dunnett’s test was applied for pairwise comparisons across groups. If the results of the Kruskal‒Wallis test were statistically significant, then Dunn’s test was applied to analyze pairwise comparisons across groups. For two groups, the Student’s t-test (parametric test) or the Mann‒Whitney U test (nonparametric test) were applied to analyze the equality of means across groups. All the statistical analyses and comparisons were evaluated at the 5% (*/$) and 1% (**/$$) levels of significance.
Types of comparisons:
1. Vehicle control (I) vs. Treatment groups (II, III, IV) marked by * and/or ** 2. Vehicle control recovery (I-R) vs. High dose recovery (IV-R) marked by $ and/or $$
Results
Povidone K12 Qualification Study
All animals tolerated the intravenous dose of povidone K-12 solution without any reactions at the injection site or signs of toxicity up to a high dose of 165 mg/kg/day in both sexes. No abnormal observations were noted during detailed clinical, ophthalmic, and neurobehavioral observations. Body weights and food consumption remained unaffected and were comparable to those of the vehicle control group in both sexes. No treatment-related adverse effects were observed in the clinical or anatomic pathology parameters up to the high dose of 165 mg/kg/day at the end of the treatment or recovery periods in either sex.
Based on the live-phase observations and lack of histopathological findings of this study, the NOAEL of povidone K-12 solution is considered to be 165 mg/kg/day in Wistar rats via the intravenous route.
Diclofenac Sodium Toxicity Study
No mortality occurred at doses of 3 and 7 mg/kg/day in females or up to 15 mg/kg/day in males upon repeated intravenous dosing of diclofenac sodium for 28 consecutive days. However, one female rat was found dead on study day 16 and one moribund female was euthanized on study day 26; both animals belonged to the 15 mg/kg/day dose, and had exhibited lethargy, a hunchback posture, decreased motor activity, and piloerection during the treatment period. The cause of death was considered to be treatment related based on poor health condition and gross pathological observations attributed to persistent debilitation of general health due to ulcers in duodenum and jejunum. No signs of toxicity were observed in male rats up to 15 mg/kg/day. In females, discoloration of feces (dark black) was observed in the majority of animals at 15 mg/kg/day throughout the treatment period; however, it recovered within 24 h after cessation of the treatment during the recovery period. At the injection site, no apparent treatment-related changes were observed in the diclofenac sodium injection-treated rats compared to those in the control groups in either sex.
Effect of Diclofenac Sodium on Body Weight of Wistar Rats.
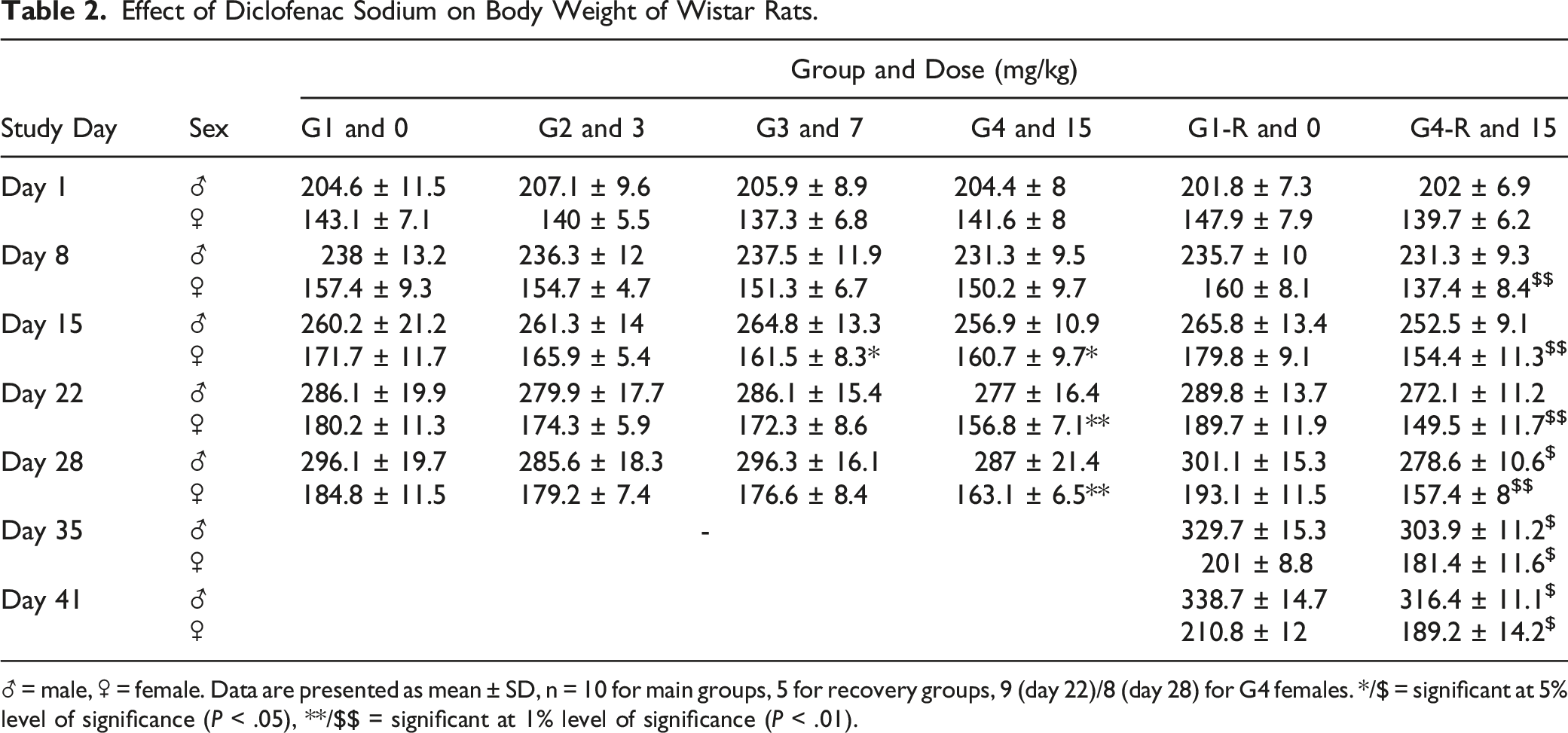
♂ = male, ♀ = female. Data are presented as mean ± SD, n = 10 for main groups, 5 for recovery groups, 9 (day 22)/8 (day 28) for G4 females. */$ = significant at 5% level of significance (P < .05), **/$$ = significant at 1% level of significance (P < .01).
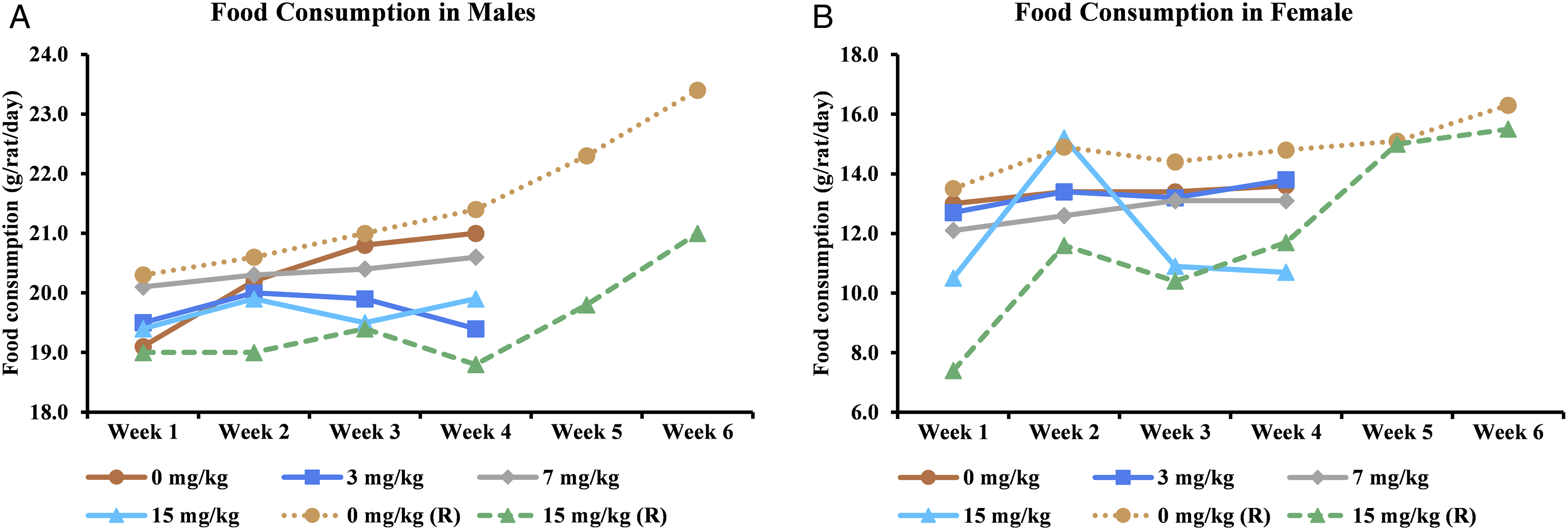
Mean weekly food consumption in (A) male and (B) female animals.
Effect of Diclofenac Sodium on Hematological Parameters of Wistar Rats.
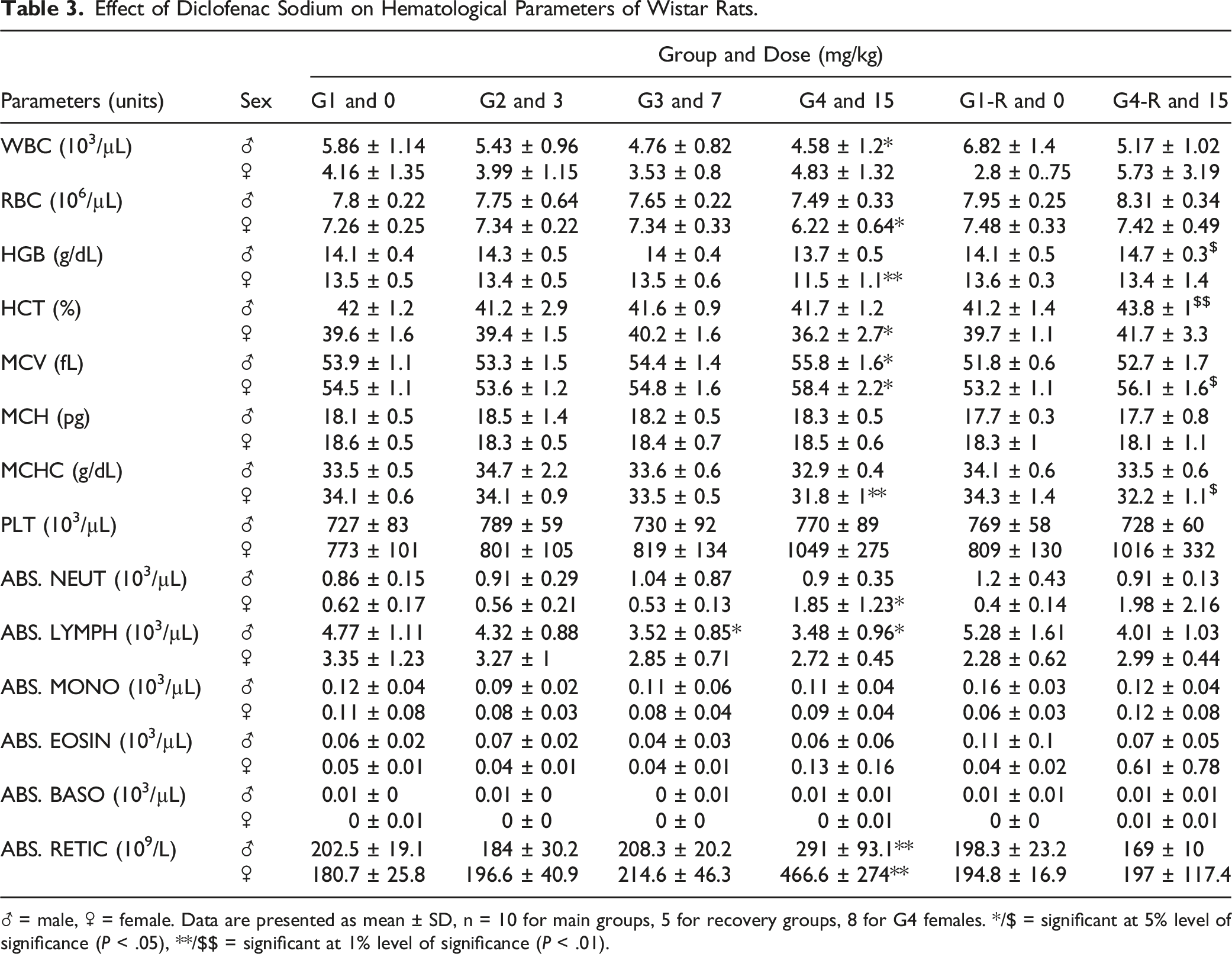
♂ = male, ♀ = female. Data are presented as mean ± SD, n = 10 for main groups, 5 for recovery groups, 8 for G4 females. */$ = significant at 5% level of significance (P < .05), **/$$ = significant at 1% level of significance (P < .01).
Effect of Diclofenac Sodium on Biochemical Parameters of Wistar Rats.
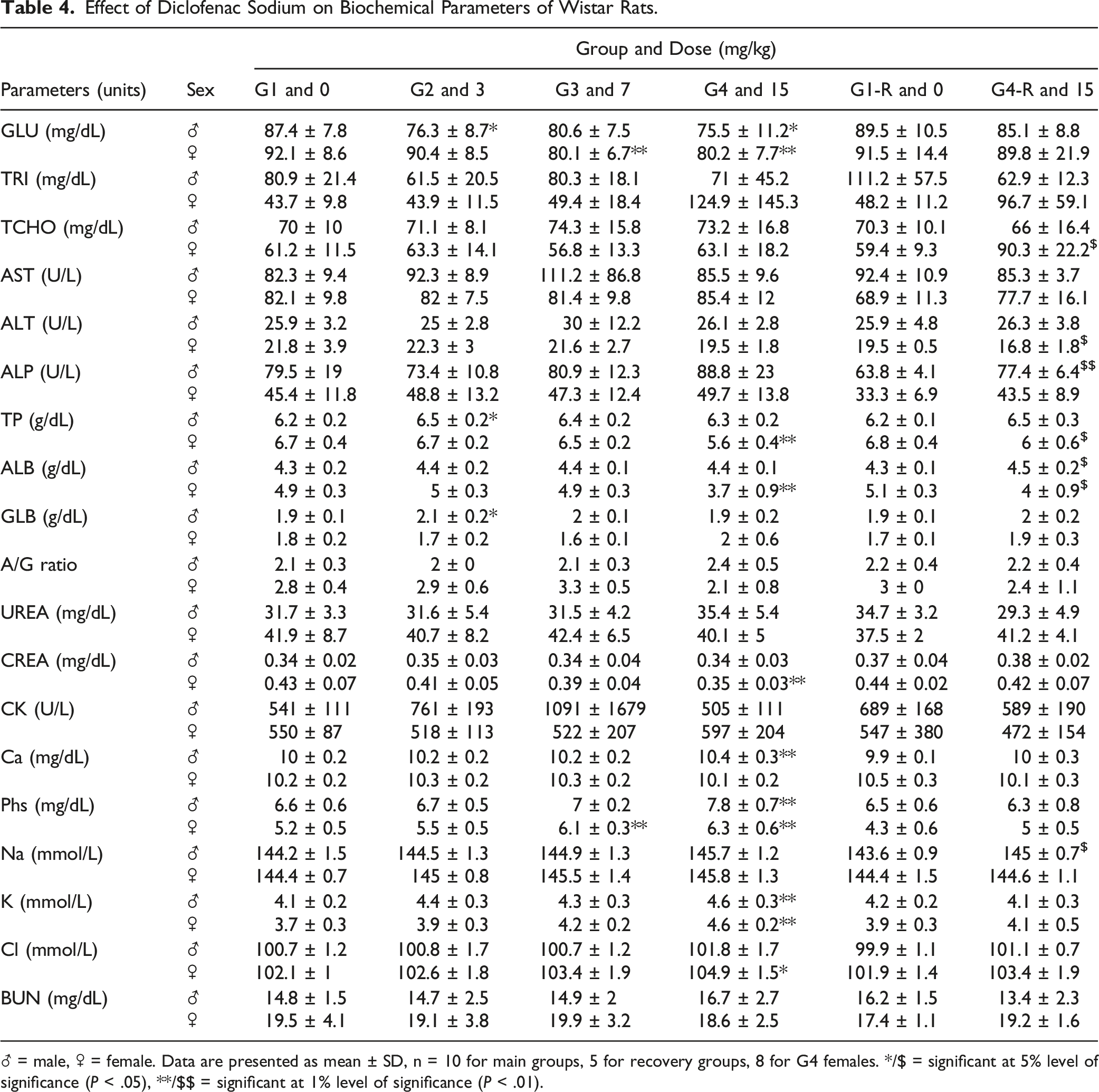
♂ = male, ♀ = female. Data are presented as mean ± SD, n = 10 for main groups, 5 for recovery groups, 8 for G4 females. */$ = significant at 5% level of significance (P < .05), **/$$ = significant at 1% level of significance (P < .01).
Effect of Diclofenac Sodium on Absolute Organ Weights (grams).
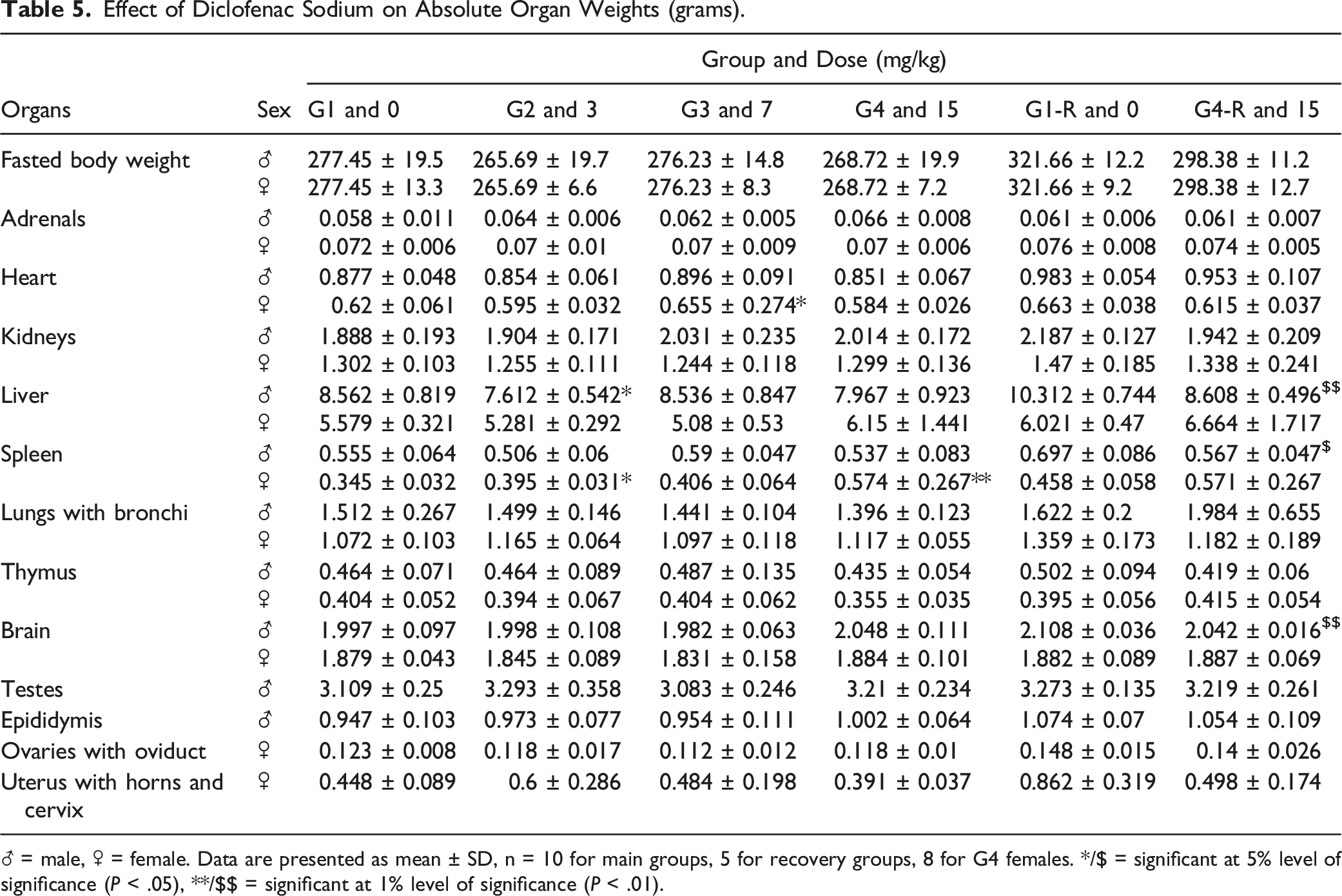
♂ = male, ♀ = female. Data are presented as mean ± SD, n = 10 for main groups, 5 for recovery groups, 8 for G4 females. */$ = significant at 5% level of significance (P < .05), **/$$ = significant at 1% level of significance (P < .01).
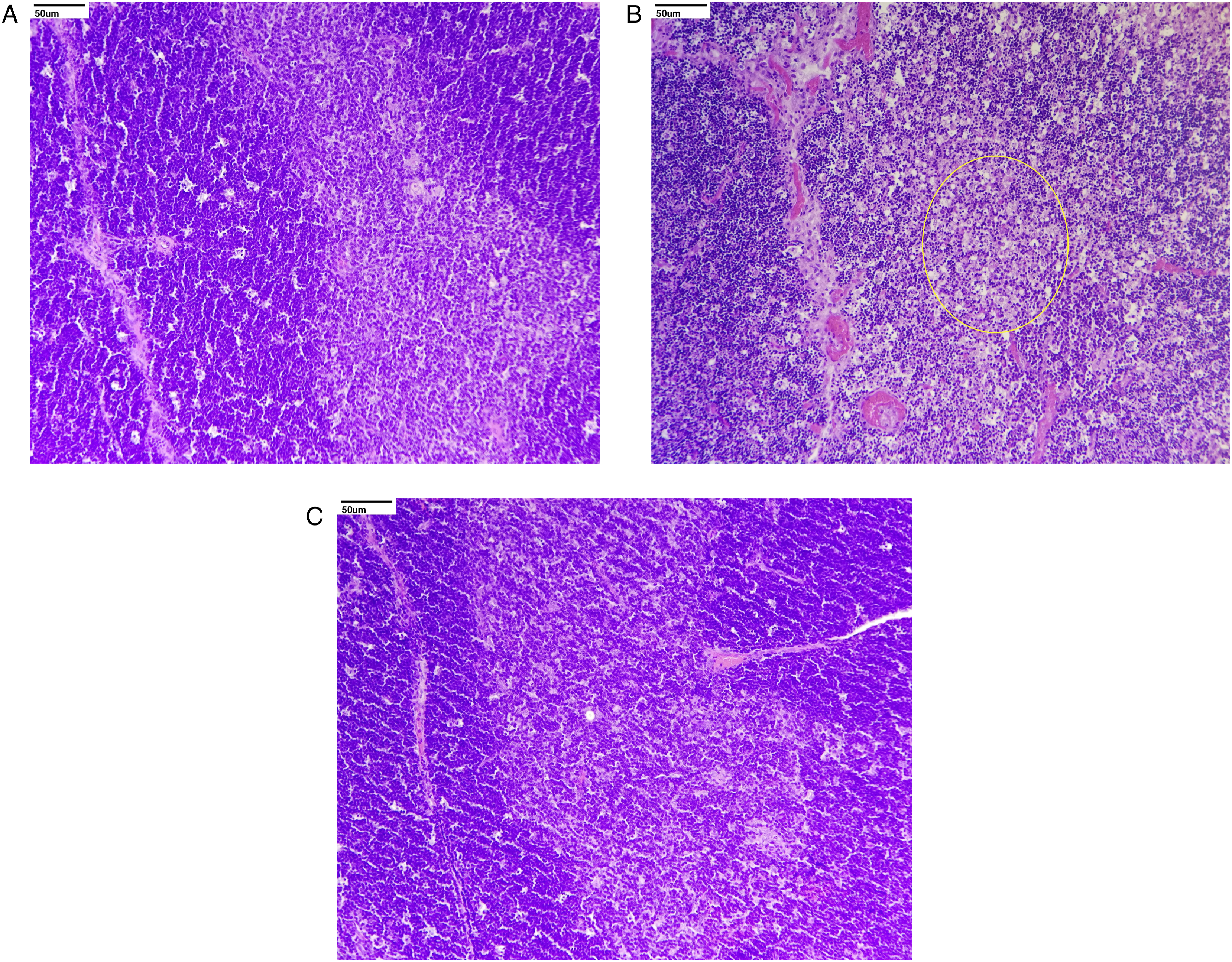
Histopathological evaluation revealed no treatment-related adverse changes in animals at 3 and 7 mg/kg/day and in male rats at 15 mg/kg/day. In females at 15 mg/kg/day, treatment-related adverse effects included moderate to severe ulcers in the duodenum and jejunum (Figures 2 and 3). Other treatment-related changes observed at 15 mg/kg/day were focal minimal erosion in the glandular mucosa and serosal inflammation in the stomach in females; minimal infiltration of inflammatory cells in the mucosa and/or submucosa of the duodenum and jejunum in both sexes and the ileum, cecum, and stomach in females; mildly to moderately increased extramedullary hematopoiesis in the spleen; mildly to moderately increased myeloid cellularity in the sternum and femur; mild infiltration of polymorphonuclear cells; minimally to mildly increased cellularity of histiocytes; and mild sinus ectasia in mesenteric lymph nodes (Figures 2, 4 and 5). Photomicrograph shows moderate to severe ulcers in the duodenum and minimal infiltration of inflammatory cells in mucosa and/or submucosa of the duodenum marked in the circle: (A) control and (B) high dose (H&E, 20X). Photomicrograph of the injection site shows minimal to mild inflammation characterized by perivascular infiltration of inflammatory cells, occasional hemorrhage, and degenerative changes of the blood vessel with surrounding connective tissue marked in different circles. These effects were found to be reversible at the end of the recovery period: (A) control, (B) high dose, and (C) high-dose recovery groups (H&E, 20X). Photomicrograph of the thymus shows decreased lymphoid cellularity, found to be reversible at the end of the recovery period marked in the circle: (A) control, (B) high dose, and (C) high-dose recovery groups (H&E, 20X). Photomicrograph of the spleen shows mild to moderate increased extramedullary hematopoiesis marked in different circles: (A) control and (B) high dose (H&E, 20X).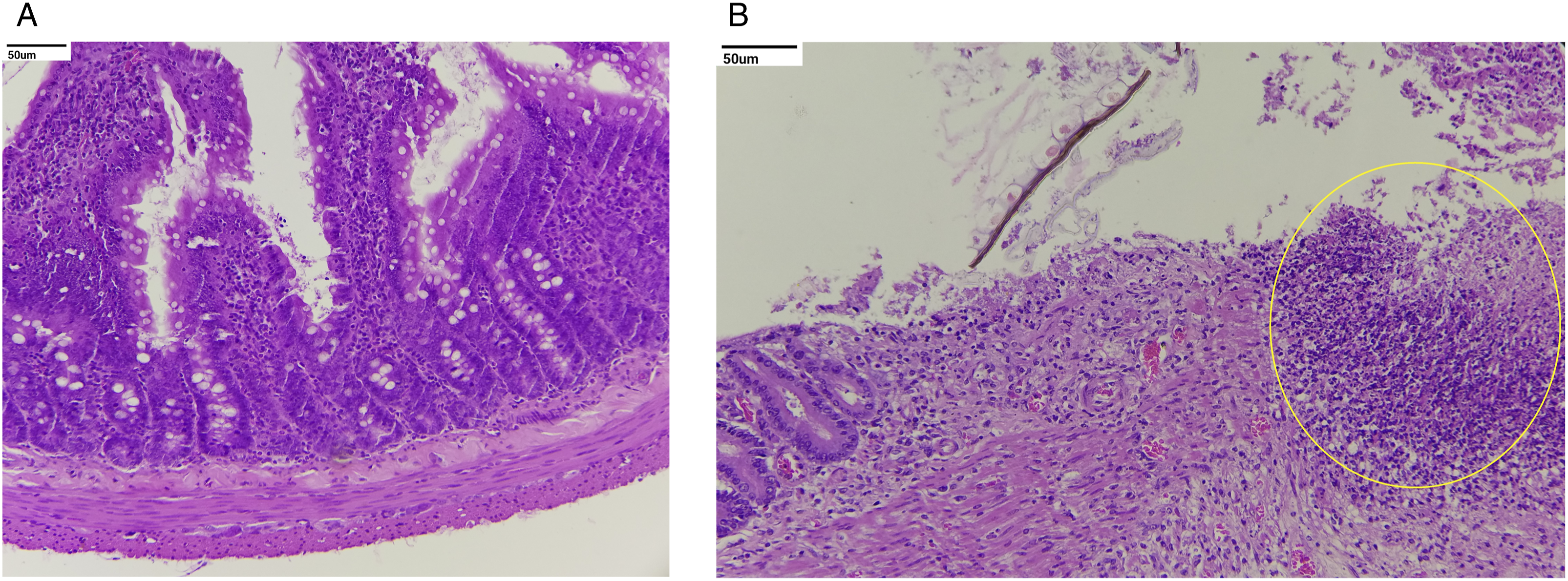
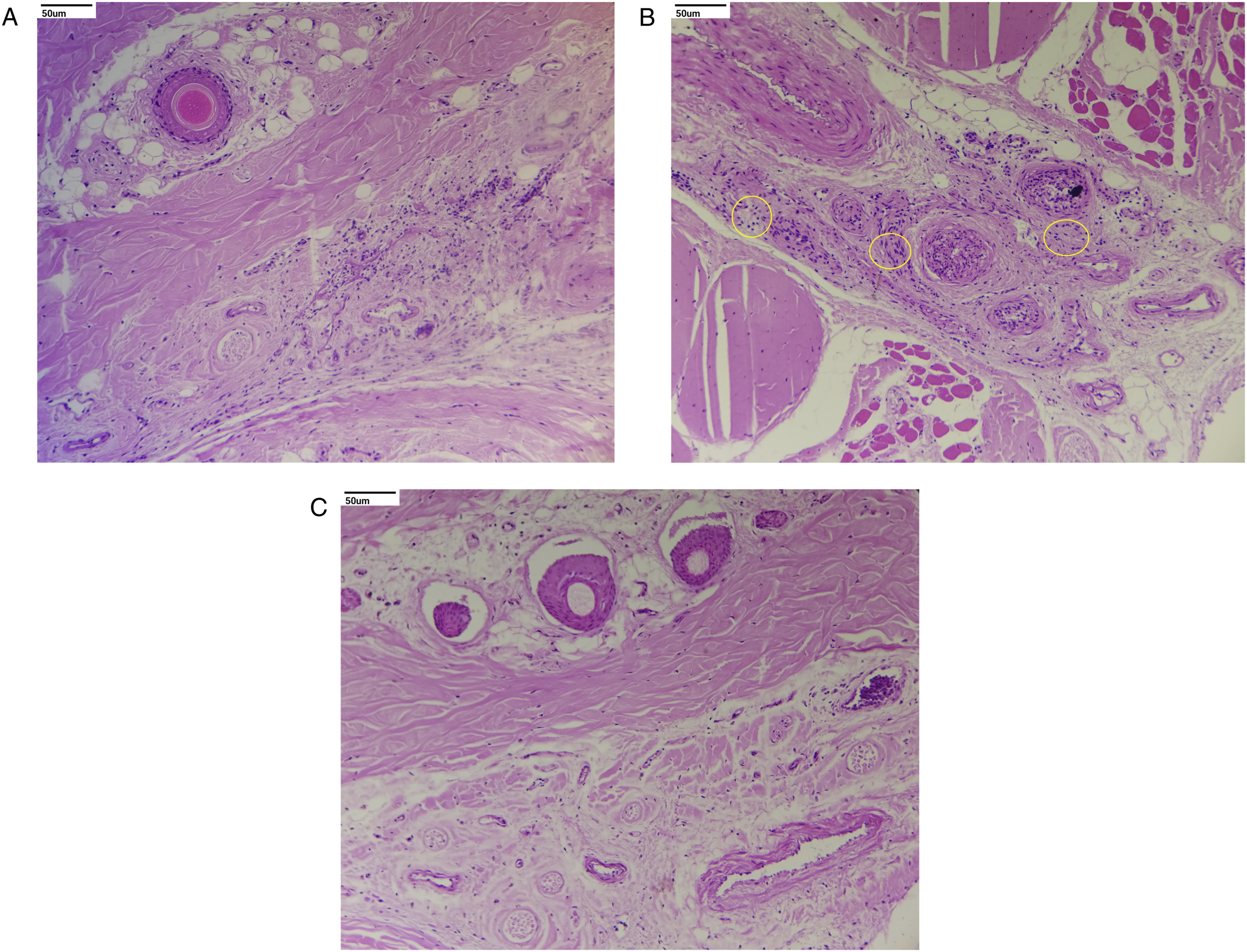
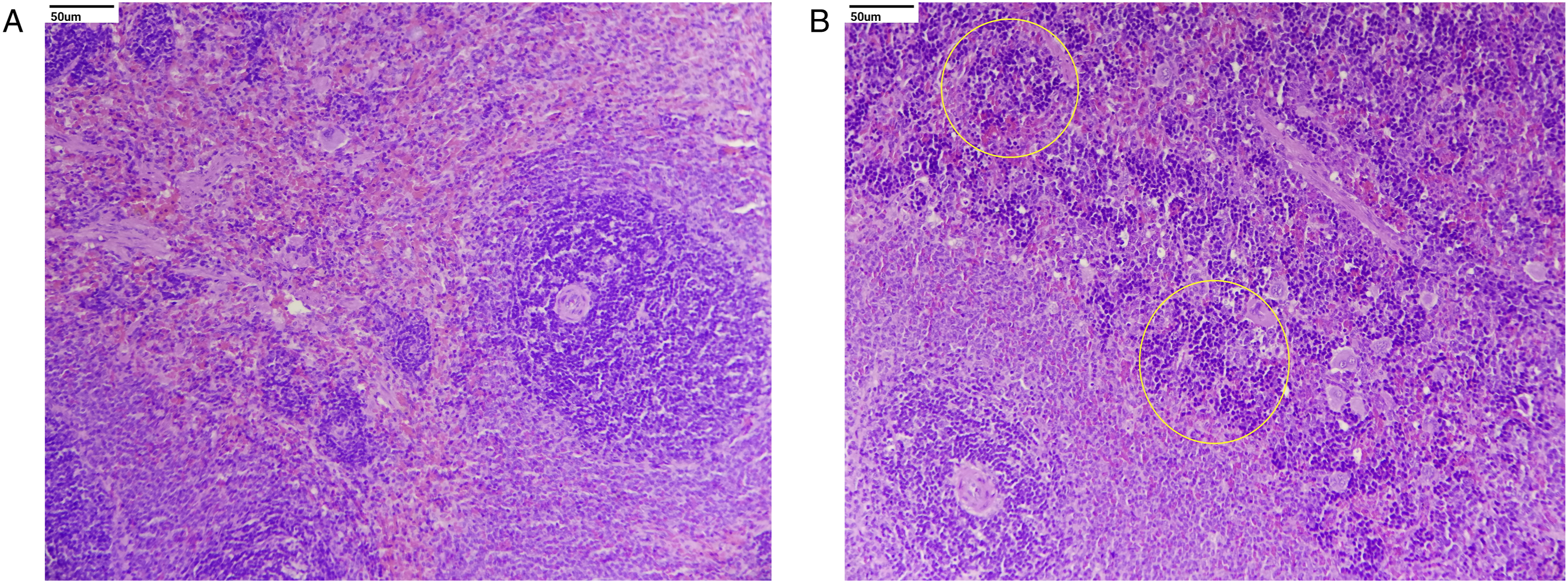
At the injection site (tail), minimal to mild inflammation was observed at 15 mg/kg/day in both sexes. Inflammation is characterized by perivascular infiltration of inflammatory cells, occasional hemorrhage, and degenerative changes in blood vessels with surrounding connective tissue. These changes appeared to be test article-related local effects. However, in control animals of both sexes, inflammatory changes observed at the injection site were of minimal severity, comparable to those in the low- and mid-dose groups and were characterized by infiltration of inflammatory cells alone with occasional hemorrhages, which could be attributed to mechanical injury upon repetitive injections (Figure 3).
At the end of the 2-week treatment-free recovery period, the treatment-related changes were found to be completely reversible in the small intestine (duodenum and ileum), stomach, and injection site (tail), whereas the changes observed in the jejunum, mesenteric lymph nodes, spleen, and myeloid cellularity in bone (sternum and femur) showed a trend of reversibility in terms of severity and incidence of findings.
Mean Plasma Exposure for Diclofenac Sodium Injection.
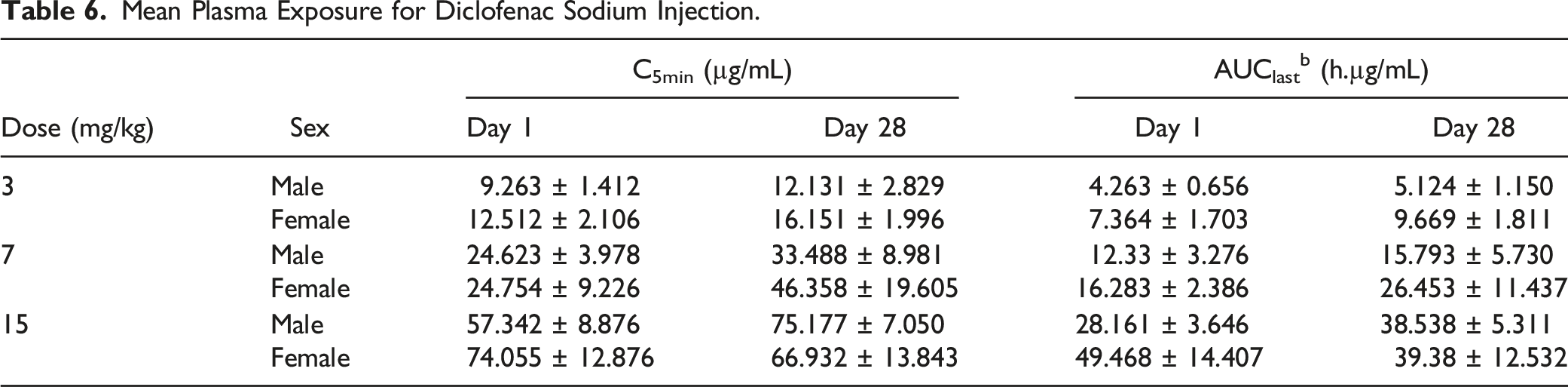
In conclusion, repeated intravenous administration of diclofenac sodium was well tolerated in males at 15 mg/kg/day and at 7 mg/kg/day in females. Compared with male rats, female rats appeared more sensitive to gastrointestinal toxicity at the 15 mg/kg/day dose. The NOAEL for diclofenac sodium injection is considered to be 7 mg/kg/day in females and 15 mg/kg/day in males.
Discussion
The novel intravenous diclofenac sodium-K12 formulation developed by ZWD was a sterile, clear, and colorless liquid with an 8.5 pH and osmolality of 323 mOsmol/kg and fulfilled the sterility, particulate matter, and other test requirements listed under the United States Pharmacopoeia.
The amount of povidone K12 per unit dose in the novel intravenous diclofenac-K12 formulation (80 mg) is greater than its inactive ingredient database limit published by the US FDA (20 mg). In the excipient qualification study, povidone K-12 was administered at doses of 33, 82.5, and 165 mg/kg/day. This study did not report any toxicologically relevant adverse effects up to the highest dose of 165 mg/kg/day, therefore, 165 mg/kg/day is considered the NOAEL for povidone K12. Considering the effective human dose based on the DylojectTM label, the dosages of povidone K-12 tested in the present study were approximately 1X, 2.5X, and 5X of the intended human dose. Thus, it can be inferred that the povidone K-12 intravenous formulation would not raise any safety concerns when administered to humans.
In the diclofenac sodium toxicity study, the novel diclofenac sodium formulation was administered to Wistar rats at doses of 3, 7, and 15 mg/kg/day for 28-days, and the GI tract was shown to be the primary target organ of toxicity. Diclofenac, through the inhibition of COX-1, decreases the synthesis of prostaglandins such as PGE2, which serve gastroprotective purposes. The inhibition of PGE2 leads to a decrease in mucin production, bicarbonate secretion, and epithelial cell turnover from the gastric epithelium, resulting in GI injuries such as gastric erosion and ulcers.2,7 In the present study, diclofenac doses of 3 and 7 mg/kg/day were well tolerated. Toxicologically significant gross and histopathological adverse changes were observed in the duodenum and jejunum at the highest dose tested, 15 mg/kg/day. Adverse effects such as moderate to severe ulcers; infiltration of inflammatory cells in the mucosa and/or submucosa of the duodenum and jejunum in both sexes and the ileum, cecum, and stomach; focal minimal erosion in the glandular mucosa; and serosal inflammation in the stomach were observed. These histopathological changes can be correlated with the clinical signs observed in the present study, such as discolored feces (black), lethargy, hunchback posture, and decreased motor activity. The results from hematological evaluations, such as lower RBC count, hemoglobin, hematocrit, and MCHC values, higher MCV and reticulocyte count, and the occurrence of polychromasia in blood smear examinations, could be due to hemolysis. 8 The observed GI adverse effects, including GI hemorrhage, perforation, and anemia, among others, are consistent with various findings reported for diclofenac. 7 However, these adverse effects are observed over an extended period of exposure to diclofenac and are reversible upon cessation of treatment. 2 The reversal of diclofenac-induced toxicity in the present study was observed through the normalization of weekly body weight gain, food consumption, clinical pathology parameters, cessation of clinical signs, and reversal of histopathological changes in target organs.
In this study, sex-based differences in the mean exposure levels of diclofenac sodium were observed. Compared with male animals, systemic exposure to diclofenac in female animals was higher in all dose groups. As such, the NOAEL dose for this study was different for both sexes, owing to the severity of clinical signs and pathological findings. Although some research suggests that women are more susceptible to diclofenac-related liver injury than males and that men are at greater risk of gastric bleeding than women are, available evidence on the effect and pharmacokinetics of diclofenac does not support any differences in treatment regimens for males and females.4,9
Upon oral administration, diclofenac is rapidly absorbed. Only approximately 60% of intact diclofenac reaches systemic circulation following first-pass metabolism in the liver. Diclofenac absorption is strongly influenced by gastric pH, drug precipitation in the stomach, gastric emptying time, and enterohepatic circulation. Diclofenac has a shorter biological half-life (∼2 hours) and faster elimination rate (∼1.2–1.8 hours), requiring frequent administration of the drug to maintain a therapeutic plasma concentration, which, in turn, can increase the risk of drug-related adverse events.4,10,11 Diclofenac is known to cause dose-dependent gastrointestinal and cardiovascular adverse events, such as gastro-duodenal lesions, ulcers, thrombosis, and hypertension. 12 The need for frequent dosing due to a faster elimination rate compromises the tolerability of diclofenac. Intravenous formulations are advantageous over oral formulations; they offer quicker onset of action, provide 100% bioavailability, and bypass first-pass metabolism, thereby effectively reducing the administered dose and dose-dependent adverse events. Previously conducted studies have reported that diclofenac-related GI toxicity is a result of local as well as systemic effects and due to enterohepatic recirculation of the drug. It has been suggested that modifications in the formulation or route of administration might reduce GI toxicity.13,14 This tested novel intravenous diclofenac-povidone K12 formulation did not cause any renal toxicity, which can be attributed to the absence of HPβCD.
Conclusion
A novel intravenous diclofenac-povidone K-12 formulation was developed to address the unmet medical need for a fast-acting IV formulation of diclofenac sodium. The data here support a NOAEL for diclofenac sodium injection of 15 mg/kg/day in male rats and 7 mg/kg/day in female rats based on the observed GI toxicity in females at the high dose. Evaluation of the povidone K12 excipient demonstrated that it was well tolerated in rats with a NOAEL dose of 165 mg/kg/day (the highest dose administered), which translates to a human equivalent dose (HED) of approximately 1604 mg/day. With 80 mg of povidone K12 present in the novel diclofenac drug product, this study provides a safety margin of at least 20-fold above the HED at the NOAEL dose in rats. Thus, these data support the safety of the novel diclofenac-povidone K-12 intravenous formulation in humans.
Footnotes
Acknowledgments
The authors are grateful to the management and scientific team at Zydus Research Centre, Ahmedabad, for their assistance in preparing this manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were funded by Zydus Lifesciences Limited, Ahmedabad, India.
Author Contribution
Bhatt, L., contributed to conception and design, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Shah, C., and Patel, J., contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Rajwadi, V., contributed to design, contributed to acquisition, drafted manuscript, and critically revised manuscript; Patel, R., contributed to conception, contributed to analysis, drafted manuscript, and critically revised manuscript; Ranvir, R., contributed to conception, contributed to analysis and interpretation, drafted manuscript, and critically revised manuscript; Patel, H., contributed to conception and design, contributed to analysis, drafted manuscript, and critically revised manuscript; and Sundar, R., and Jain, M., contributed to conception and design and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Animal Welfare and Ethics Statement
All experimental procedures involving wistar rats were conducted according to the guidelines provided by the Committee for Control and Supervision of Experiments on Animals, Government of India and approved by the Institutional Animal Ethics Committee (Facility Registration Number: 77/PO/RcBi/SL/99/CPCSEA).
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.