
Abstract
Methamphetamine (METH) is a highly addictive psychostimulant and one of the most widely abused drugs worldwide. The continuous use of METH eventually leads to neurotoxicity and drug addiction. Studies have shown that neurotoxicity is strongly associated with METH-induced neuroinflammation, and microglia are the key drivers of neuroinflammation. Triggering receptor expressed on myeloid cells 2 (TREM2) is reported to play a key role in activation of microglia and neuroinflammation. Yet, the molecular mechanisms by which METH causes neuroinflammation and neurotoxicity remain elusive. In the current study, we investigated the role of TREM2 in neuroinflammation induced by METH in BV2 cells and the wild-type (WT) C57BL/6J mice, CX3CR1GFP/+ transgenic mice, and TREM2 knockout (KO) mice. Postmortem samples from the frontal cortex of humans with a history of METH use were also analyzed to determine the levels of TREM2, TLR4, IBA1, and IL-1β. The expression levels of TREM2, TLR4, IBA1, IL-1β, iNOS, and Arg-1 were then assessed in the BV2 cells and frontal cortex of mice and human METH users. Results revealed that the expression levels of TREM2, TLR4, IBA1, and IL-1β were significantly elevated in METH-using individuals and BV2 cells. Microglia were clearly activated in the frontal cortex of WT C57BL/6 mice and CX3CR1GFP/+ transgenic mice, and the protein levels of IBA1, TREM2, TLR4, and IL-1β were elevated in the METH-induced mouse models. Moreover, TREM2-KO mice showed further increased microglial activation, neuroinflammation, and excitotoxicity induced by METH. Thus, these findings suggest that TREM2 may be a target for regulating METH-induced neuroinflammation.
Keywords
Introduction
Methamphetamine (METH) is a highly addictive psychostimulant drug and one of the most widely abused drugs worldwide. As a highly lipid-soluble substance, METH can easily cross the blood-brain barrier (BBB).1,2 Long-term METH exposure can lead to neurotoxic effects through oxidative stress, mitochondrial impairment, endoplasmic reticulum stress, astrocyte and microglial activation, axonal transport barriers, autophagy, and apoptosis. The continuous use of METH eventually leads to drug addiction and serious health complications, including memory loss, attention deficit, and cognitive impairment. 1 These neurological complications are strongly associated with METH-induced neurotoxicity and neuroinflammation. 3 Furthermore, METH can disrupt the BBB, leading to neuroinflammation and ultimately neuronal degeneration. METH-induced neuroinflammation makes the brain more susceptible to neuropathology both directly and indirectly.4-6
Neuroinflammation is a process of complex innate immune responses to brain injury. As an essential component of the innate immune system of the central nervous system (CNS), neuroinflammation plays a critical role in neural tissue repair and restoration. 7 METH is regarded as a contributor to neuronal inflammation through the excessive release of dopamine (DA) and glutamate (Glu). 6
Recent research has demonstrated that abnormal activation of microglia, a consequence of METH-induced neuroinflammation, can regulate the release of numerous pro- and anti-inflammatory cytokines and chemokines and play a crucial role in METH-induced neuroinflammation. 8 Microglia are regarded as prototypic tissue-resident macrophage-like innate immune cells of the CNS, which interact with various types of cells, including neurons and astrocytes. 8 Microglial activation results in the elevation of potentially neurotoxic molecules, such as pro-inflammatory cytokines, proteinases, and reactive oxygen species (ROS), which causes the neuroinflammation. 9 Activated microglia are also responsible for the secretion of high amounts of excitotoxic glutamate, which mediates excitotoxicity and neuroinflammation, leading to neurodegeneration. 10 Once activated, microglia promote neurological inflammation by releasing various pro-inflammatory factors, such as tumor necrosis factor-α (TNF-α), interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and monocyte chemotactic protein-1 (MCP-1).6,11 Microglia are categorized as either the M1 pro-inflammatory phenotype or the M2 anti-inflammatory phenotype. 12 Classically activated microglia (M1) express polarization markers, such as inducible nitric oxide synthase (iNOS) and TNF-α, which exacerbate neuronal damage and lead to tissue injury, whereas alternatively activated microglia (M2) are characterized by the expression of polarization markers, such as triggering receptor expressed on myeloid cells 2 (TREM2), arginase-1 (Arg-1), and interleukin-10 (IL-10), which promote angiogenesis and prevent neuronal death.13,14 TREM2, a single transmembrane receptor member of the immunoglobulin superfamily, is mainly expressed in microglial cells within the brain 15 and is implicated in several neurodegenerative disorders, including Alzheimer’s disease (AD) and Parkinson’s disease. 16 TREM2 is also involved in the regulatory process of neurodegenerative diseases and neuroinflammation 17 and mediates crucial functions in the microglia, including suppression of pro-inflammatory responses and stimulation of engulfment of apoptotic neurons and Aβ deposits.18-20
Studies have shown that Toll-like receptor 4 (TLR4)/TREM2 imbalance may be a potential link between AD and systemic inflammation. TREM2 can serve as a potential therapeutic target for treating systemic inflammation in AD progression. 17 It is currently unclear whether TREM2 is involved in the process of METH induced neuroinflammation, and whether TREM2 can regulate TLR4 to participate in the regulation of METH induced neuroinflammation is still unclear.
Numerous studies have shown that TLR4 is involved in METH-induced neuroinflammation. Indeed, indirect and direct activation of microglia by METH associated with TLR4 21 is not clear yet. TLR4, an immunological pattern recognition receptor that can be activated both in myeloid differentiation primary response protein 88 (Myd88)-dependent and Myd88-independent pathways. 22 Activation of microglia due to METH increases inflammatory mediators, such as IL-1β, TNF-α, and IL-6.23,24 However, the molecular and cellular mechanisms underlying microglial activation and neuroinflammation induced by METH remain unclear.
In the current study, we hypothesized that TREM2 and TLR4 may be two key regulators of inflammation that play essential roles in METH-induced neuroinflammation. Thus, we investigated the roles of TREM2 and TLR4 in METH-activated microglia and METH-induced neuroinflammation.
Materials and Methods
Reagents
The METH (items No.171212-200603) used in the study was purchased from the National Institutes for Food and Drug Control. The purity of METH is 98%.
Antibodies
The following antibodies were used in the study: TREM2 (Cell Signaling Technology, 15101), TLR4 (Cell Signaling Technology, 67824), IBA1 (Abcam, ab178846), and IL-1β (Abcam, ab254360).
Human Brain Tissue
The prefrontal cortex of humans with a history of METH use and healthy controls were obtained from the Forensic Appraisal Center of Kunming Medical University. METH users were identified based on forensic toxicology analysis and police intelligence investigations. Controls consisted of non-drug-addicted and healthy individuals with no CNS disease. Immunohistochemical (IHC) analysis was used to detect the expression levels of TREM2, TLR4, IBA1, and IL-1β, while western blot analysis was used to detect the expression levels of TREM2, TLR4, and IL-1β. This work was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving human samples. All protocols were approved by the Medical Ethics Committee of Kunming Medical University and carried out in accordance with established Guiding Principles for Postmortem Brain Research.
Animals
Male TREM2 knockout (KO) mice (18-20 g) and age-matched wild-type (WT) C57BL/6J mice and CX3CR1GFP/+ transgenic mice were used. TREM2 KO mice and CX3CR1GFP/+ transgenic mice were gift from prof. Fan Li's lab of Yunnan University. The wild-type (WT) C57BL/6J mice were purchased from Experimental Animal Center of Kunming Medical University. CX3CR1 is only expressed in microglia in the brain and is often used as a specific biomarker of microglia residing in the brain.25,26 To investigate the effect of METH on microglia, this study constructed a METH toxicity model using CX3CR1GFP/+ transgenic mice. The animals were housed in clean polypropylene cages under constant temperature and humidity conditions, with a 12 h-12-h day-night cycle and free access to food and water. All animal experiments were carried out in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals. All protocols were approved by the Institutional Animal Committee of Kunming Medical University.
The TREM2-KO mice, WT C57BL/6J mice, and CX3CR1GFP/+ transgenic mice were randomly divided into two groups (Saline and METH groups), respectively. METH (5 mg/kg/d) was administered to mice via intraperitoneal injection for 5 consecutive days. The methamphetamine doses were selected based on the literature.27,28
Open Field Test
An open field test, which consisted of a 50 cm × 50 cm × 40 cm sized box, was used to observe changes in animal behavior. SuperMaze animal behavior analysis software and VisuTrack software were used to automatically record activity distance. At the start of the experiment, the mice were quickly placed in the central area of the box, and the software automatically recorded their activities in the box for a period of 15 min.
Cell Culture
BV-2 cell line is an immortal cell line derived from mouse microglia. BV2 cells obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) were cultured at 37°C in a 5% CO2 atmosphere with high-sugar Dulbecco’s Modified Eagle Medium (DMEM) (Biological Industries; Cat# 2036135) containing 10% fetal bovine serum (Gibco; Cat# 42A0378 K) and 50 μg/mL penicillin and streptomycin (Gibco; Cat# 2076673). The BV2 cells were treated with different concentrations of METH (0-.8 mM) for 24 h or with .1 mM METH for different times (0-24 h).
Western Blot Analysis
Total cellular lysates were prepared using cell lysate buffer supplemented with complete protease inhibitor cocktail and phosphatase inhibitor cocktail (Beyotime, P0013 B). Protein concentrations were determined using the BCA assay. Western blotting was performed to detect the relative expression levels of TREM2, TLR4, IL-1β, TNF-α, Arg-1, and iNOS.
Equal amounts of soluble protein (25 μg) were subjected to sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to a polyvinylidene fluoride (PVDF) membrane. The blotted membranes were then blocked with 5% non-fat dry milk in 1 × Tris-buffered saline (TBS) with Tween 20 (TBST) buffer for 1 h at room temperature, followed by an overnight incubation with the indicated primary antibodies at 4°C. The membranes were then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature, followed by image exposure using the Super Chemiluminescent ECL detection system. Protein band density was quantified by ImageJ software (National Institutes of Health) and normalized to β-actin.
Immunofluorescence (IF) Staining
Paraffin brain sections were deparaffinized in xylene and dehydrated in absolute ethanol, then treated with .2% Triton X-100 and blocked with 10% goat serum (Solarbio, SL038) before incubation with primary antibodies (anti-TREM2, anti-TLR4, anti-IBA1) overnight at 4°C. The samples were washed with phosphate-buffered saline (PBS) and incubated with fluorescent secondary antibodies for 1 h at 37°C, followed by 4’,6-diamidino-2-phenylindole (DAPI) staining for nuclear visualization. Samples were then imaged by fluorescence microscopy.
Immunohistochemical (IHC) Staining
Paraffin brain sections were deparaffinized in xylene and dehydrated in absolute ethanol. The sections were then incubated overnight at 4°C with primary antibodies against TREM2, TLR4, IBA1, and IL-1β. After washing in PBS for 15 min, the sections were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. 3,3’-Diaminobenzidine (DAB) was used to visualize TREM2, TLR4, IBA1, and IL-1β.
Statistical Analysis
All experimental data were expressed as mean ± standard deviation (SD). Each experiment was repeated at least three times. The western blotting and open-field test results were assessed using one-sample t test, student’s t test, and one-way analysis of variance (ANOVA). The paired t-tests were used to determine differences before and after self-administration, while one-way ANOVA was used to compare groups. Statistical analyses were performed using GraphPad Prism v6.0 and Adobe Illustrator CC. Here, P ≤ .05 was used to indicate statistical significance.
Results
Expression Levels of TREM2, TLR4, IBA1, and IL-1β Were Significantly Elevated in Individuals with a History of METH Uses
We assessed the expression levels of TREM2, TLR4, IBA1, and IL-1β in the prefrontal cortex of humans using immunocytochemistry and western blot analysis. As shown in Figure 1, the expression levels of TREM2, TLR4, and IL-1β were significantly elevated and the number of microglia was significantly increased in the METH group compared to the control group. These results suggest that METH use can activate TREM2, TLR4, and microglial levels and induce neuroinflammation. Effects of METH on TREM2, TLR4, IBA1, and IL-1β expression in METH-using individuals. (A) Expression levels of TREM2 (a), TLR4 (b), IBA1 (c), and IL-1β (d) were observed in human prefrontal cortex by IHC. (B) Western blot and quantitative analyses were performed to determine the expression levels of TREM2, TLR4, and IL-1β in the prefrontal cortex of humans. β-Actin was used as the loading control. Data are expressed as TREM2/β-actin, TLR4/β-actin, and IL-1β/β-actin and presented as mean ± SD of three independent experiments (**P < .01, ***P < .001 vs control).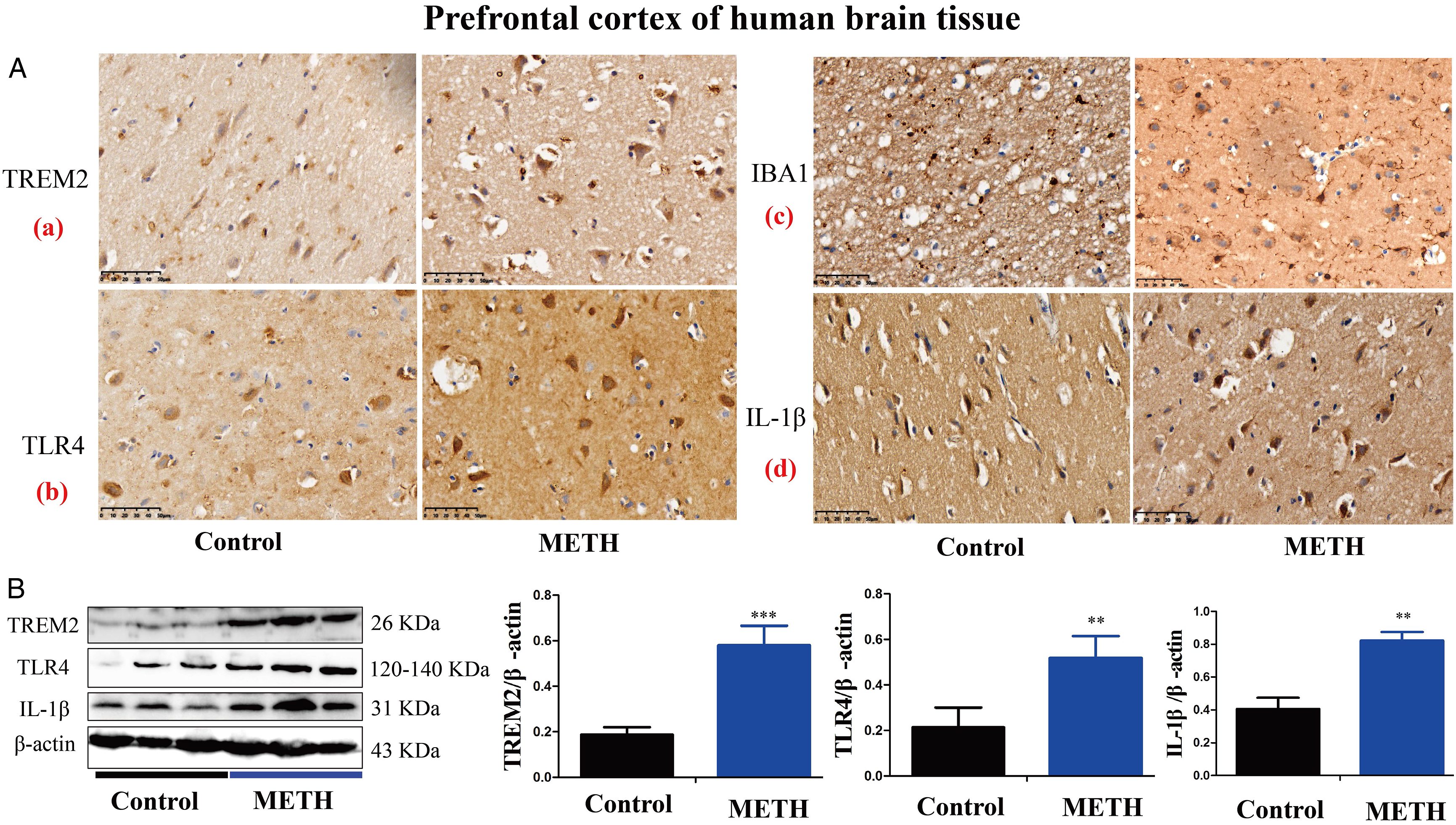
Expression Levels of TREM2, TLR4, IBA1, and IL-1β in Prefrontal Cortex of METH-Induced Mice
We established a protocol for treating WT-C57BL/6J and TREM2-KO mice with METH to investigate behavioral changes and expression levels of TREM2, TLR4, IBA1, and IL-1β in the prefrontal cortex. METH (5 mg/kg/d) was administered to the mice daily via intraperitoneal injection for 5 consecutive days, followed by behavioral assessments using the open-field test, as shown in Figure 2A. Results demonstrated a significant increase in total movement distance in both WT-C57BL/6J and TREM2-KO mice following 5-day METH administration, compared to pre-administration levels (Figure 2B). Moreover, total movement distance was significantly higher in the TREM2-KO METH group than in the WT METH group. We also performed immunocytochemical analysis to detect the expression levels of TREM2, TLR4, and IL-1β in the prefrontal cortex of WT-C57BL/6J mice. Results showed that the expression levels of TREM2, TLR4, and IL-1β were significantly increased in the METH group compared to the Saline group, as shown in Figure 2C. In addition, IF was conducted to detect the expression of the microglial marker IBA1. Results showed that the microglia were markedly activated in the METH group compared to the Saline group, as shown in Figure 2C and D. These findings suggest that TREM2 and TLR4 may be involved in the process of METH-induced neuroinflammation. Effects of METH on movement distance in mice and TREM2, TLR4, IBA1, and IL-1β expression in the prefrontal cortex of mice. (A) Protocol for establishing METH-treated WT-C57BL/6J and TREM2-KO mice. METH (5 mg/kg/d) was administered to the mice daily via intraperitoneal injection for 5 consecutive days. Open-field test was performed to analyze mouse movement distance before, on the day of, and after administration of METH. (B) Behavioral analysis of mouse movement distance. (C) Expression levels of TREM2 (a), TLR4 (b), and IL-1β (c) were detected by IHC, and expression level of IBA1 (d) was detected by IF.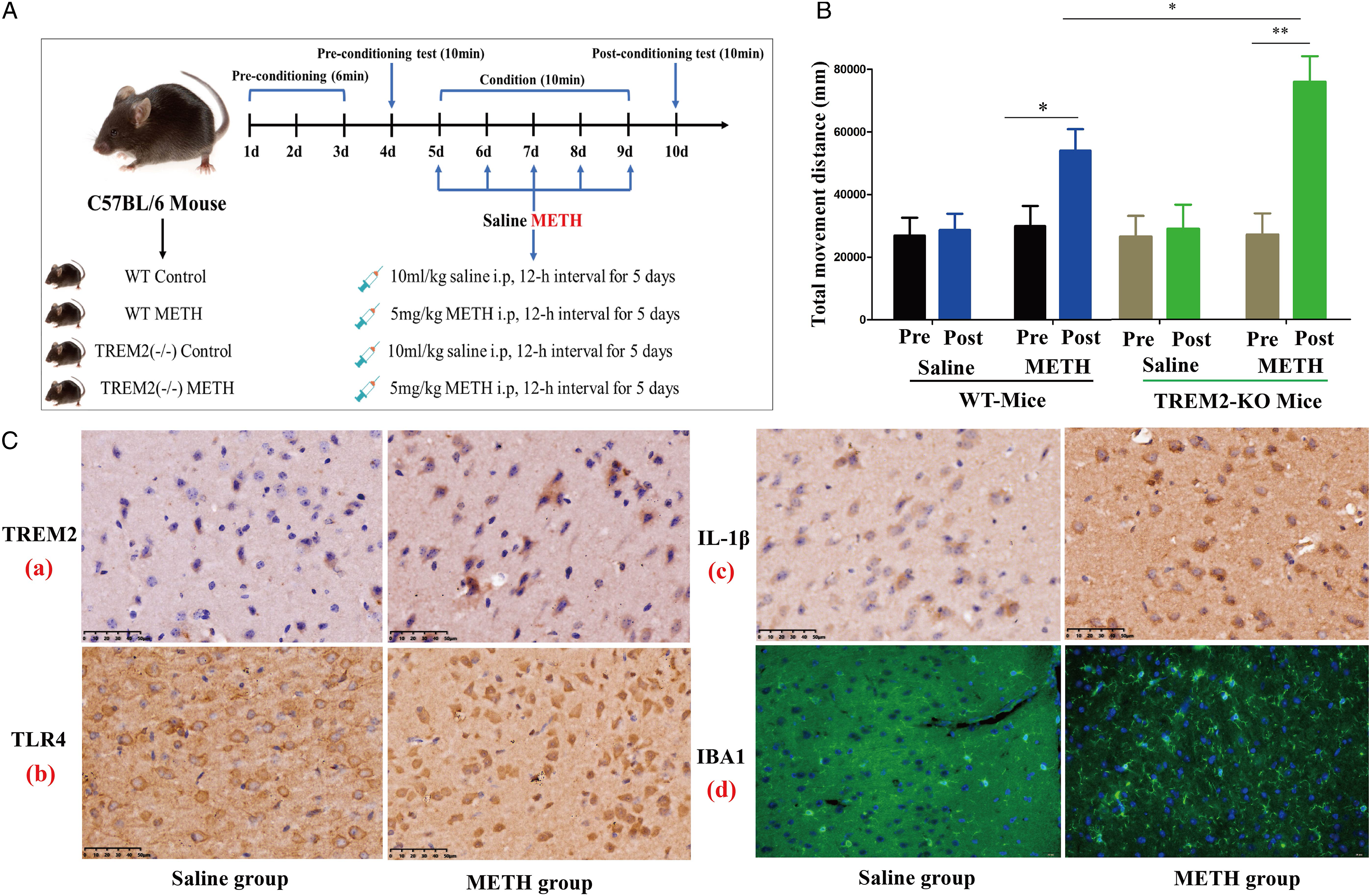
METH Promoted Microglial Activation and Enhanced Expression of TREM2 and TLR4 in Prefrontal Cortex of CX3CR1GFP/+ Transgenic Mice
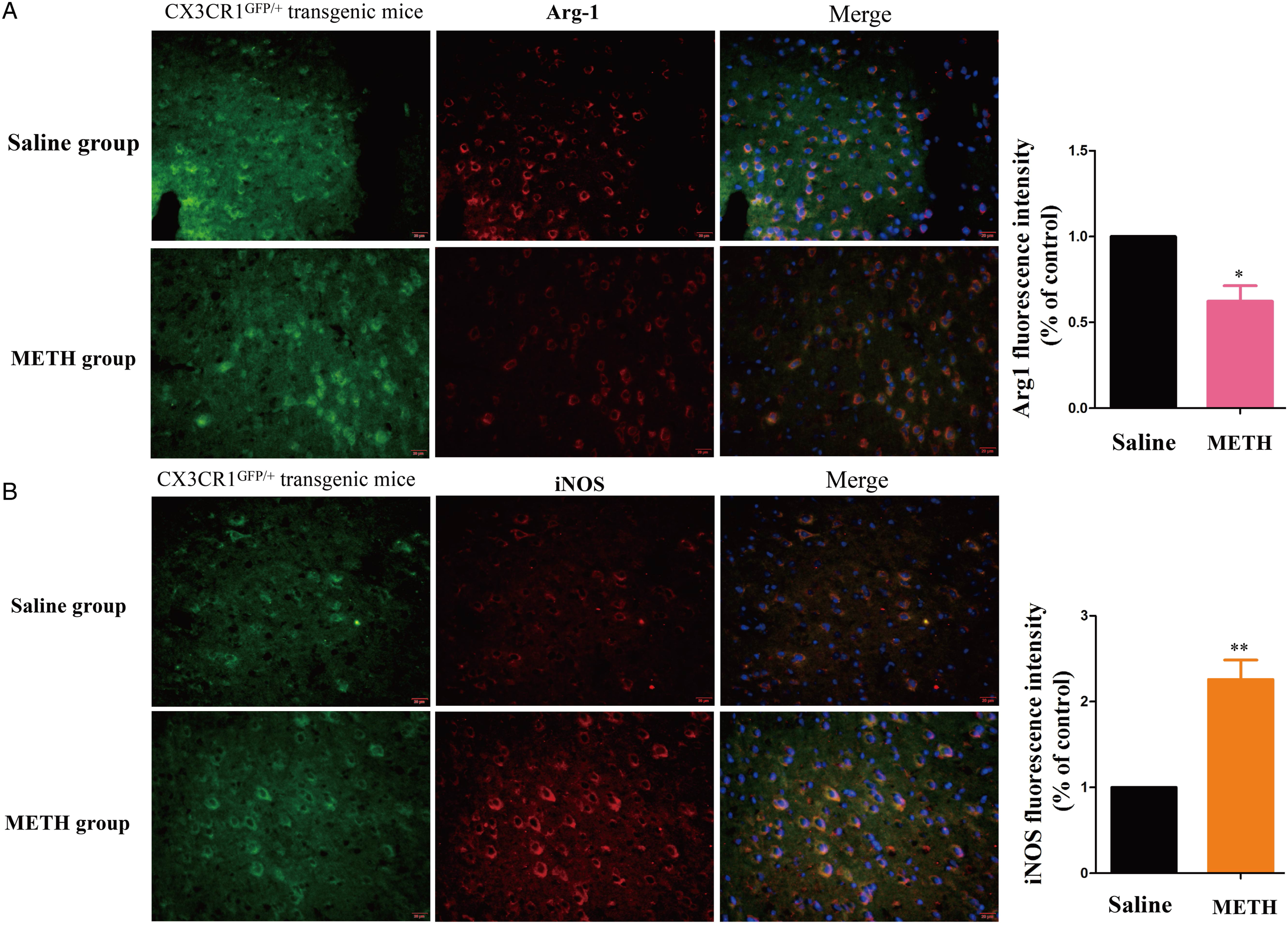
CX3CR1GFP/+ transgenic mice were used to examine the activation of microglia and the expression levels of TREM2 and TLR4 following intraperitoneal administration of METH (5 mg/kg) for 5 days. The expression levels of TREM2, TLR4, Arg-1, and iNOS were detected in the prefrontal cortex of the transgenic mice using IF, as shown in Figure 3 (A, B) and Figure 4A and B. Upon METH administration, the expression levels of TREM2, TLR4, and iNOS in microglia showed a significant increase, while that of Arg-1 showed a significant decrease, compared to that in the Saline group. These results suggest that METH can activate and induce microglia to express TREM2 and TLR4. Effects of METH on TREM2, and TLR4 expression in the prefrontal cortex of CX3CR1GFP/+ transgenic mice. Expression levels of TREM2, and TLR4 were detected by IF. (A) Merged images of prefrontal cortex stained with DAPI, CX3CR1GFP/+ (green), and anti-TREM2 antibodies (red). (B) Merged images of prefrontal cortex stained with DAPI, CX3CR1GFP/+ (green), and anti-TLR4 antibodies (red) (**P < .01, ***P < .001 vs Saline group). Effects of METH on Arg-1, and iNOS expression in the prefrontal cortex of CX3CR1GFP/+ transgenic mice. Expression levels of Arg-1,and iNOS were detected by IF. (A) Merged images of prefrontal cortex stained with DAPI, CX3CR1GFP/+ (green), and anti-Arg-1 antibodies (red). (B) Merged images of prefrontal cortex stained with DAPI, CX3CR1GFP/+ (green), and anti-iNOS antibodies (red) (*P < .05, **P < .01 vs Saline group).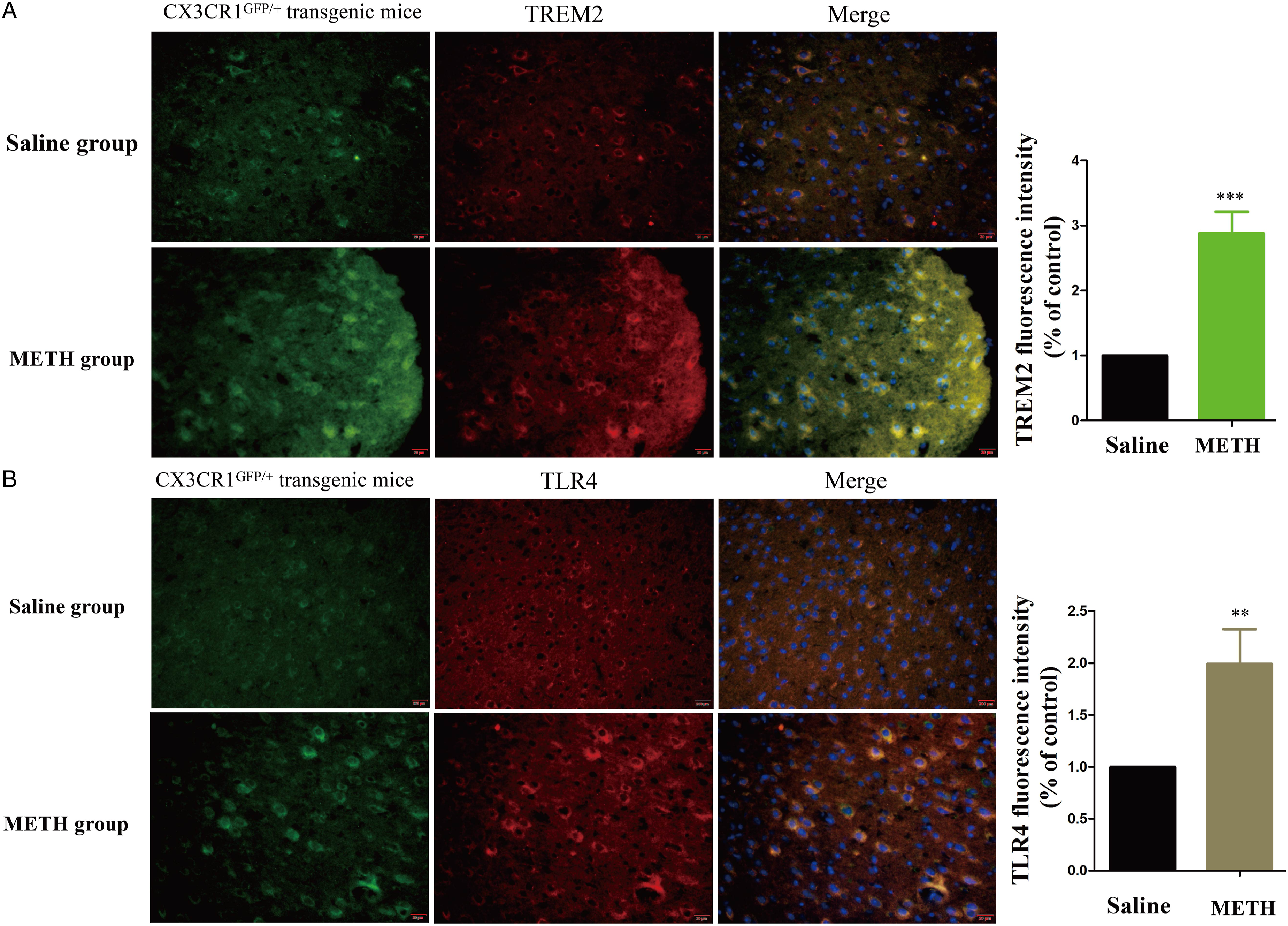
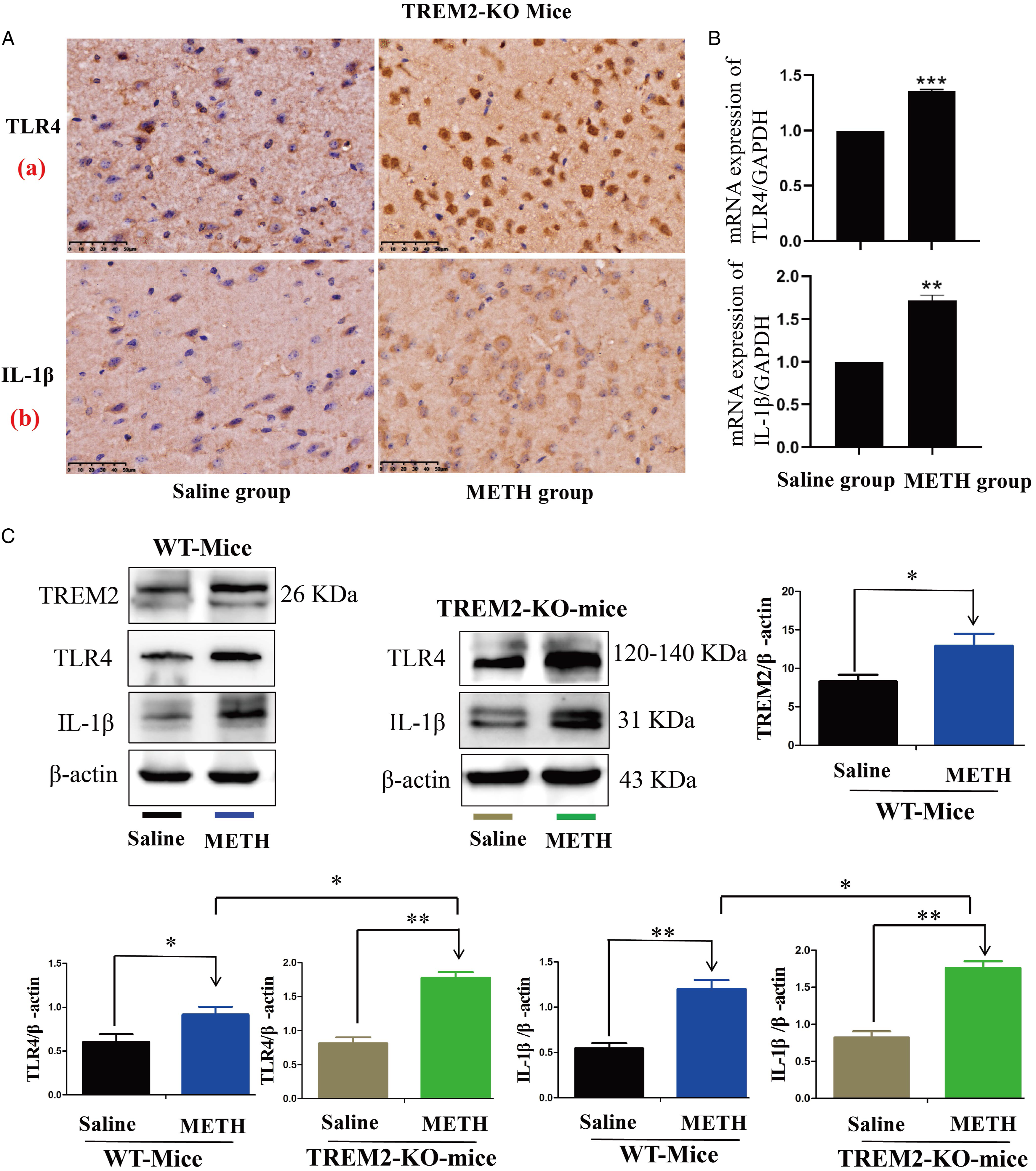
TREM2 Knockout Aggravated Microglial Activation and Neuroinflammation Induced by METH
TREM2 KO mice were used to explore the role of TREM2 in the activation of microglial and induction of neuroinflammation following intraperitoneal administration of METH (5 mg/kg) for 5 days. The expression levels of Arg-1 and iNOS were detected by IF in the prefrontal cortex of TREM2-KO mice. The expression of Arg-1 in microglia was reduced, while the expression of iNOS was increased in the TREM2-KO METH group compared with the Saline group (Figure 5A and B). The expression levels of TLR4 and IL-1β were detected by IHC, quantitative polymerase chain reaction (q-PCR), and western blot. The IHC and q-PCR results revealed a significant increase in the expression levels of TLR4 and IL-1β in the prefrontal cortex of TREM2-KO mice following 5 days of METH administration compared to the Saline group (Figure 6A and B). Moreover, after 5 days of METH administration, the expression levels of TREM2, TLR4, and IL-1β were significantly increased in both WT and TREM2-KO mice. Notably, the expression levels of TLR4 and IL-1β were significantly higher in the TREM2-KO METH group than in the WT-METH group (Figure 6C). These findings suggest that TREM2 knockout exacerbates microglial activation and neuroinflammation induced by METH exposure. Effects of METH on Arg-1, and iNOS expression in the prefrontal cortex of TREM2-KO mice. (A) Expression levels of IBA1 and Arg-1 were detected by IF. Merged images of prefrontal cortex stained with DAPI, anti-IBA1 antibodies (green), and anti-Arg-1 antibodies (red). (B) Expression levels of IBA1 and iNOS were detected by IF. Merged images of prefrontal cortex stained with DAPI, anti-IBA1 antibodies (green), and anti-iNOS antibodies (red) (*P < .05, **P < .01 vs Saline group). Effects of METH on TREM2, TLR4, and IL-1β expression in the prefrontal cortex of TREM2-KO mice. (A) Expression levels of TLR4 (A) and IL-1β (B) were detected by IHC. (B) Expression levels of TLR4 and IL-1β were detected by q-PCR. (C) Western blot and quantitative analyses were performed to determine expression levels of TREM2, TLR4, and IL-1β in prefrontal cortex of WT and TREM2-KO mice. β-actin was used as a loading control. Data are expressed as TREM2/β-actin, TLR4/β-actin, and IL-1β/β-actin and presented as mean ± SD of three independent experiments (*P < .05, **P < .01).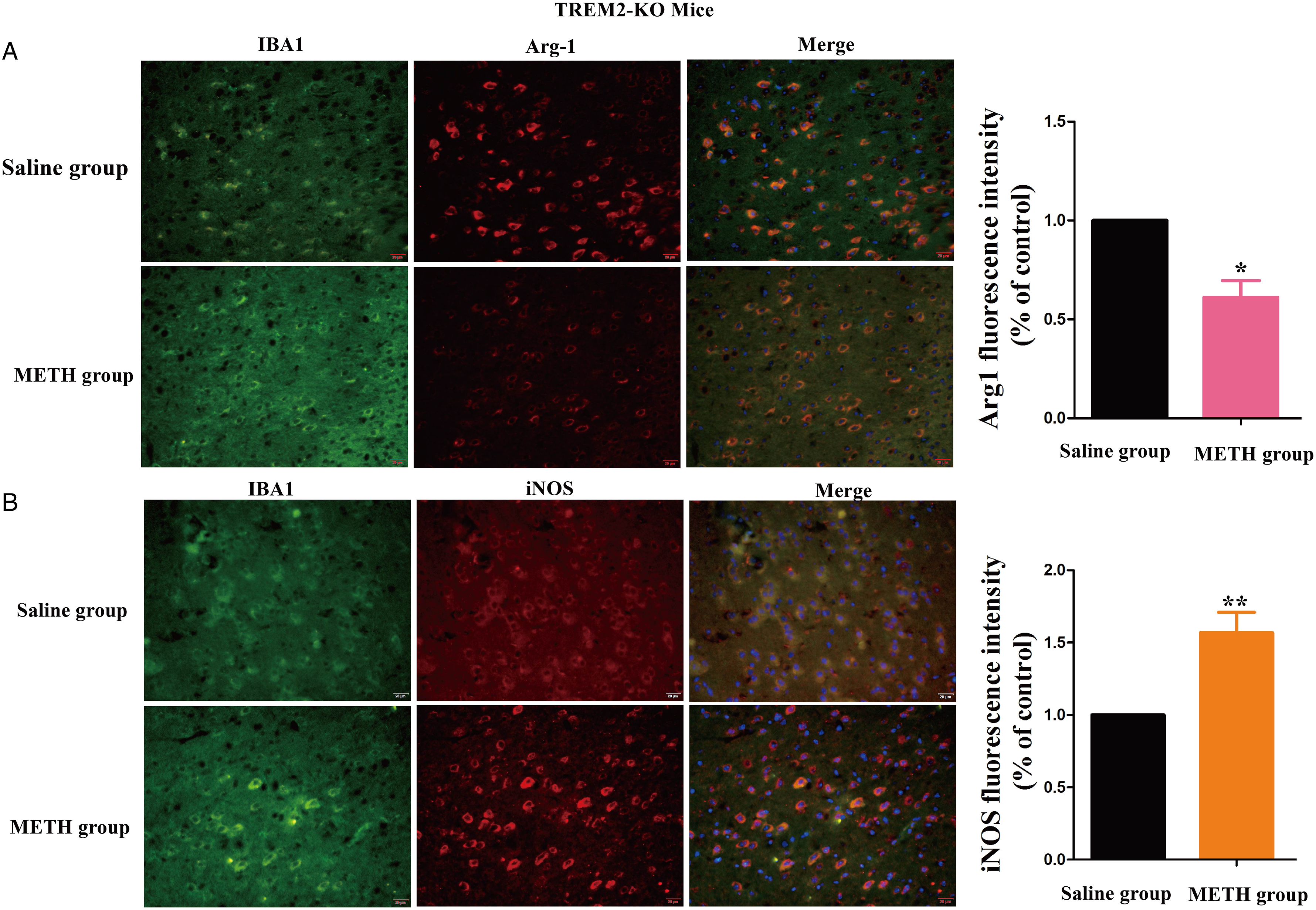
METH Activated TREM2 and Induced Neuroinflammation in BV2 Cells
BV2 cells were subjected to different concentrations of METH (0-.8 mM) for 24 h or treated with .1 mM METH for varying time intervals (0-24 h). The expression levels of TREM2, IL-1β, TNF-α, iNOS, and Arg-1 in the BV2 cells were then detected by western blot analysis, as shown in Figure 7. With increasing concentration and duration of METH treatment, the expression levels of TREM2 and IL-1β in the BV2 cells displayed varying degrees of elevation compared to the control group. Furthermore, METH administration resulted in a significant increase in the expression levels of TNF-α and iNOS in the BV2 cells, while Arg-1 expression was significantly reduced. These findings indicate that METH can activate microglia and TREM2 expression and induce neuroinflammation. METH administration increased TREM2, IL-1β, TNF-α, and iNOS expression and decreased Arg-1 expression in BV2 cells. BV2 cells were treated with different concentrations of METH for 24 h (A) or with .1 mM METH for different times (B). Western blot and quantitative analyses were performed to determine TREM2, IL-1β, TNF-α, iNOS, and Arg-1 expression levels in BV2 cells. β-Actin was used as a loading control. Data are expressed as TREM2/β-actin, IL-1β/β-actin, TNF-α/β-actin, iNOS/β-actin, and Arg-1/β-actin and presented as mean ± SD of three independent experiments (*P < .05, **P < .01, ***P < .001 vs control).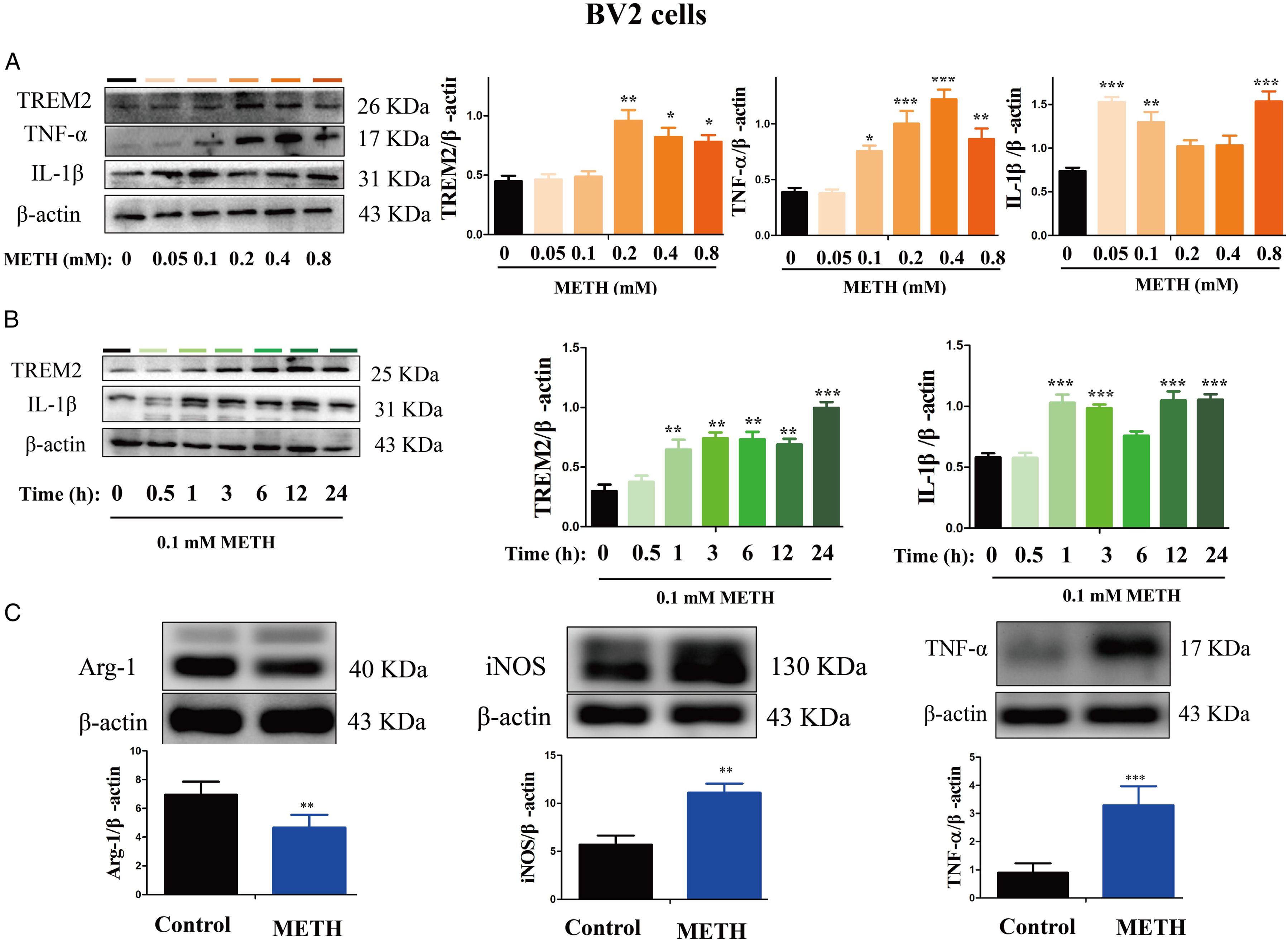
Discussion
METH, an amphetamine-type psychostimulant, has become a serious public health concern in many countries. Long-term METH exposure can induce neurotoxic effects and addiction. However, the molecular and cellular mechanisms underlying METH-induced neurotoxicity and addiction have yet to be fully elucidated.
Many studies have provided evidence implicating neuroinflammation in the mechanisms underlying METH-induced neurotoxicity. Microglia, resident immune cells of the CNS, play a crucial role in maintaining CNS homeostasis and orchestrating immune responses during METH-induced neuroinflammation. Recent research has shed light on the association between microglial activation mediated by TREM2 and the pathogenesis of neurodegenerative diseases and neuroinflammatory processes. 29
In the current study, we examined the levels of TREM2, TLR4, IBA1, and IL-1β in postmortem brain samples from individuals with a history of METH use. We observed a significant elevation in the expression levels of TREM2, TLR4, IBA1, and IL-1β in METH-using individuals, suggesting the involvement of TREM2, TLR4, microglial activation, and neuroinflammation in the context of METH addiction and neurotoxicity. In addition, based on in vitro experiments, we demonstrated that exposure of BV2 cells to METH increased the expression levels of TREM2, IL-1β, TNF-α, and iNOS, but led to a reduction in Arg-1 expression. These results suggest that METH exposure activates microglia and TREM2 may be involved in the process of METH-induced neuroinflammation. Moreover, to further investigate the behavioral changes associated with METH exposure, we established mouse models using both WT and TREM2-KO mice through intraperitoneal injection of METH and subsequent behavioral assessment using the open field test. Following administration of METH, we observed a significant increase in the total movement distance of both WT and TREM2-KO mice. Interestingly, the effect was more pronounced in the TREM2-KO mice, indicating that the absence of TREM2 heightened the excitatory neurotoxicity induced by METH. Additionally, METH administration led to marked activation of microglia in the frontal cortex, accompanied by increased expression of TREM2, TLR4, and IL-1β. We used CX3CR1GFP/+ transgenic mice subjected to intraperitoneal administration of METH to investigate microglial activation and TREM2 and TLR4 expression. Our results demonstrated elevated levels of TREM2, TLR4, and iNOS expression in microglia, along with reduced Arg-1 expression. These findings indicate that METH can activate microglia and induce neuroinflammation, with the involvement of TREM2 and TLR4. To clarify the regulatory effects of TREM2 on microglia and neuroinflammation, we found that TREM2-KO resulted in a significant increase in TLR4 and IL-1β expression, decrease in Arg-1 expression, and increase in iNOS expression in microglia. Furthermore, the expression levels of TLR4 and IL-1β were significantly higher in the TREM2-KO METH group than in the WT-METH group. These findings highlight the crucial role of TREM2 in the activation of microglia and the induction of neuroinflammation in the context of METH exposure.
Within the CNS, TREM2 is predominantly expressed in microglia and regulates inflammatory cytokine production, apoptotic neuron phagocytosis, and cell survival. 30 Recent evidence has suggested that TREM2 modulates neuroinflammation based on its effects on microglia and macrophages.31,32 Furthermore, studies have found that TREM2 deficiency in microglia and macrophages may cause inflammation. 33 Silencing of TREM2 signaling has been shown to increase the expression levels of macrophage inhibitory protein-2 (MIP-2), TNF-α, IL-6, and IL-1β, while overexpression of TREM2 reduces the transcription of TNF-α, IL-1β, and nitric oxide synthase 2 (NOS2). 34 Activation of TREM2 modulates microglia/macrophages by stimulating phagocytosis, promoting cell survival, and suppressing inflammation. 32 Studies have also suggested that TREM2 down-regulates inflammation by inhibiting activation of the TLR/IL-1R, mitogen-activated protein kinase (MAPK), and nuclear factor kappa-B (NF-κB) pathways.35,36 Furthermore, the influence of TREM2 on neuroinflammation and its neuroprotective effects appear to be partially mediated by increased expression of interleukin-1 receptor-associated kinase 3 (IRAK3). Notably, IRAK3 knockdown reverses the beneficial effects observed upon TREM2 overexpression. 37 Furthermore, the influence of TREM2 on neuroinflammation and its neuroprotective effects appear to be partially mediated by increased expression of IRAK3. Notably, IRAK3 knockdown reverses the beneficial effects observed upon TREM2 overexpression. 38 These findings highlight the crucial roles of both TREM2 and TLR4 in the inflammatory process. Dysregulation of microglial TLR4/TREM2 balance may serve as a potential link between AD and systemic inflammation. 17 Furthermore, TLR4 gene silencing has been shown to alleviate METH-induced hepatotoxicity by inhibiting LPS-mediated inflammation in mice. 39 Consistent with these studies, our results demonstrated the involvement of TREM2 and TLR4 in the activation of microglia and induction of neuroinflammation under METH exposure.
In conclusion, our study revealed elevated protein levels of TREM2, TLR4, and IL-1β in METH-induced BV2 cells, mouse models and humans with a history of METH use. Notably, TREM2 knockout aggravated microglial activation, neuroinflammation, and excitotoxicity induced by METH (Figure 8). These findings suggest that TREM2 may serve as a potential target for regulating METH-induced neuroinflammation. However, further research is required to elucidate the specific mechanisms underlying the role of TREM2 in regulating METH-induced neuroinflammation. Schematic depicting the regulatory role of TREM2 in METH-induced neuroinflammation in microglia.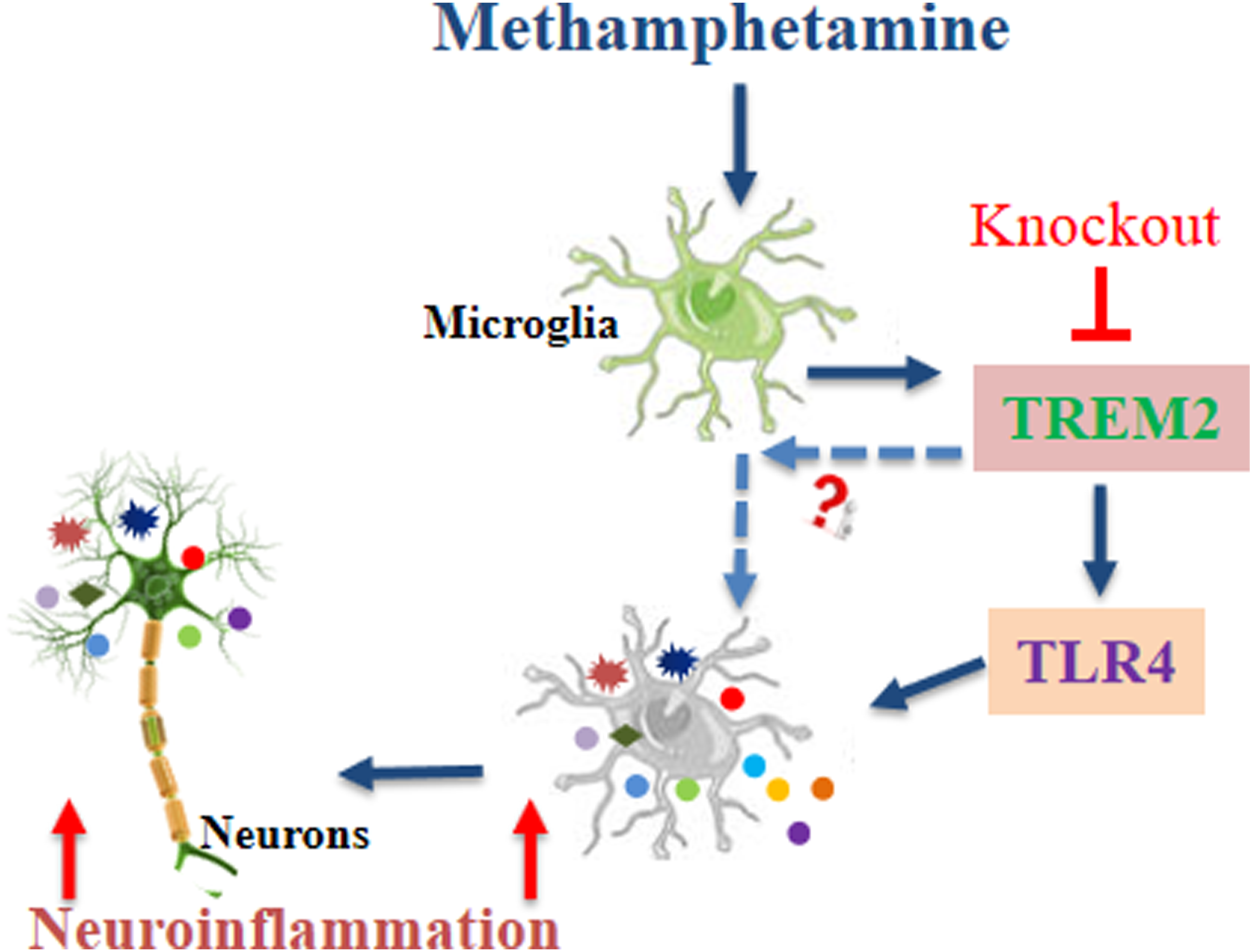
Footnotes
Author Contributions
Peng, Y.X., contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Yang, G.M., contributed to conception, contributed to acquisition, analysis, and interpretation, and drafted manuscript; Wang, S.W., contributed to conception, contributed to analysis and interpretation, and drafted manuscript; Lin, W.R., contributed to acquisition and analysis and drafted manuscript; Zhu, L.H., contributed to acquisition and drafted manuscript; Dong, W.J., contributed to acquisition and drafted manuscript; Shen, B.Y., contributed to analysis and drafted manuscript; Nie, Q.Y., contributed to acquisition and drafted manuscript; Hong, S.J., contributed to conception and design, contributed to interpretation, and critically revised manuscript; Li, L.H., contributed to conception and design, contributed to interpretation, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (82202081 and 82260336), Yunnan Applied Basic Research Projects Joint Special Project (202301AY070001-026, 202301AY070001-267, and 202301AY070001-234), Basic Research Program of Yunnan Province (202301AT070269), and Yunnan Education Department Research Project (2022J0207).