
Abstract
DNA damage repair has been the key mechanism of cisplatin resistance in hepatocellular carcinoma (HCC). The present study elucidated the molecular mechanism by which nucleolar and spindle-associated protein 1 (NUSAP1) influenced cisplatin tolerance in HCC by regulating DNA damage. First, high mRNA expression of E2F8 and NUSAP1 in HCC was detected by real-time quantitative PCR in cells and tumor tissue. The interaction between E2F8 and NUSAP1 was confirmed by chromatin immunoprecipitation (ChIP) and dual-luciferase reporter assays that E2F8 bound to the promoter region of NUSAP1 and regulated its transcriptional activity. The effects of the E2F8/NUSAP1 axis on cell viability, cell cycle, DNA damage protein γ-H2AX, and cisplatin resistance were investigated by CCK-8, flow cytometry, comet detection, and western blot. The results showed that NUSAP1 knockdown blocked the cell cycle in G0/G1 phase, promoted cisplatin-induced DNA damage, and enhanced cisplatin sensitivity in HCC. Overexpressed E2F8 promoted cell cycle arrest by silencing NUSAP1 in HCC, and promoting DNA damage as well as cisplatin sensitivity. In conclusion, our results suggested that E2F8 enhanced the chemoresistance of HCC cells to cisplatin by activating NUSAP1 to inhibit DNA damage, which provides a basis for describing new therapeutic targets that effectively exacerbate DNA damage and improve the chemical sensitivity of HCC to cisplatin.
Introduction
Liver cancer emerges as the sixth most common cancer as well as the fourth major cause of cancer-related deaths worldwide. 1 In terms of the global cancer statistics in 2018, there were about 841,000 new cases and 782,000 deaths per year. 1 Hepatocellular carcinoma (HCC) remains the primary histological subtype of liver cancer and accounts for approximately 80% of primary liver cancer cases. Currently, there are diverse forms of therapeutic regimens for liver cancer such as hepatectomy, chemotherapy, immunotherapy, radiofrequency ablation, or alcohol ablation, 2 as well as liver transplantation.3–6 Though the aforementioned treatment options can improve patient’s survival, chemotherapy remains the optimal method of clinical care. At present, platinum-based combined chemotherapy is one of the important means of treatment for patients with advanced HCC, among which cisplatin is an effective and widely used first-line drug. In recent years, tumor resistance has been the prominent challenge for the failure of cancer treatment, limiting the selection and use of cancer drugs. A prior study demonstrated that DNA damage repair is one of the most crucial mechanisms of cisplatin resistance in HCC. For instance, the knockdown of the THOC1 gene gives rise to DNA damage in HCC cells and enhances tumor sensitivity to cisplatin. 7 Zhang et al. 8 observed that the knockdown of XRCC1 brings about an increase in DNA damage as well as a decrease in DNA repair capacity, therefore sensitizing HepG2 cells to cisplatin. In conclusion, with the deepening understanding of the drug resistance mechanism of liver cancer, how to utilize these research outcomes to transform them into treatment regimens is now an urgent problem to be solved. The current study probes molecular targets that can improve cisplatin resistance, hoping to find out approaches that can reduce tumor resistance and develop corresponding drugs.
Nucleolar and spindle-associated protein 1 (NUSAP1) is a microtubule-associated protein that exerts essential roles in various biological functions, including spindle assembly, chromosome segregation, cell division, microtubule cross-linking, bundling, and attachment to chromosomes.9–12 Previous research showed that NUSAP1 serves as an oncogene in gastric cancer (GC), 13 glioblastoma 14 as well as HCC, 15 and its high expression is closely coupled to malignant tumor progression. Clinical research illuminated that highly expressed NUSAP1 has a positive correlation with the poor prognoses of non-small cell lung cancer, 16 HCC, 17 and cervical cancer. 18 Mechanistic studies have confirmed that down-regulated NUSAP1 can repress the proliferation, migration, and invasion of inflammatory breast cancer cells via regulating CDK1 and DLGAP5 expression, and boost the drug sensitivity to epirubicin. 19 Guo et al. 13 disclosed that NUSAP1 promotes the migration and invasion of GC cells by YAP1. Although the biological effects of NUSAP1 have been reported in numerous cancers, the underlying mechanism of NUSAP1 in HCC has not been well elucidated. Bioinformatics revealed the presence of the upstream regulatory molecule E2F8 of NUSAP1. As a result, the present study further probed the impact of the E2F8/NUSAP1 axis on DNA damage repair with cisplatin resistance in HCC through cell experiments.
To sum up, this research demonstrated highly expressed NUSAP1 in HCC, and explored the cisplatin resistance effect and possible molecular mechanism of NUSAP1 in HCC by regulating DNA damage. We observed that E2F8 could regulate NUSAP1 to suppress DNA damage and then induce cisplatin resistance in HCC cells. The current study lays the vital groundwork for seeking a new therapeutic target for HCC and improving the chemosensitivity of HCC.
Materials and Methods
Bioinformatics Analysis
mRNA expression data of HCC were derived from The Cancer Genome Atlas (TCGA) (Normal: 50, Tumor: 374), and the mRNA expression differences between the normal group and tumor group were analyzed using “edgeR” package (|logFC|>2, padj<.05) to identify differential mRNAs. Then the target gene mRNAs of the research were determined by literature citation. Functional enrichment analysis of high and low NUSAP1 expression groups was performed by utilizing Gene Set Enrichment Analysis (GSEA), based on TCGA data. Potential transcription factors upstream of mRNA were predicted utilizing the hTFtarget database. The correlation and binding site of mRNA and transcription factors were predicted by Pearson's correlation and JASPAR to determine the final transcription factor. Target genes were grouped by median, and high and low expression groups were selected for GSEA analysis.
Cell Culture
Human hepatocyte cells THLE-3 (CRL-11233) and human HCC lines HepG2 (HB-8065), SK-HEP-1 (HTB-52), as well as SNU-182 (CRL-2235), were obtained from American Type Culture Collection (USA). The above-mentioned cells were placed into the Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, USA) with 10% fetal bovine serum (FBS) (Invitrogen, USA), and cultured in 5% CO2 at 37°C.
Cell Transfection
Si-NUSAP1, oe-E2F8, and corresponding negative controls were all derived from Ribobio (China). Si-NUSAP1, oe-E2F8, and corresponding negative controls were transfected into HCC cells by using Lipofectamine 2000 kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
Cell Counting Kit-8 (CCK-8) Detects Cell Viability and IC50
To evaluate the cell viability, CCK-8 kits were used for CCK-8 detection in the light of the manufacturer’s instructions (Beyotime, China). After transfecting for 24 h, HCC cells at a density of 5 × 103/well were seeded in a 96-well plate. After 1 h of culture in the presence of 10 μL CCK-8 solution optical density (OD) was determined at 450 nm with a microplate reader (SpectraMax M5e, USA) at different time points (1, 2, 3, 4, and 5 days).
Cisplatin was purchased from Merck Life Sciences (USA). IC50 value was determined in HCC cells treated with cisplatin (0, 1, 2, 4, 8, 16, 32 μg/mL) for 48 h. 20
Cell Cycle Assay
HCC cells transfected with different plasmids were obtained by trypsin digestion and fixed with 70% ethanol. Cells were stained with 50 μg/mL propidium iodide (PI) and 100 μg/mL ribonuclease I (RNase I) at 37°C for 30 min. FACScan flow cytometer (BD Bioscience, USA) and BD CellQuest software (BD Bioscience, USA) were accessible for sample analysis. 21
Comet Assay
Alkaline comet assay was conducted with Comet Assay Kit (Bio-Techne, USA). Experimental procedures were carried out as previously described. 22 The slides were soaked in .75% agarose solution and coated with agarose. Cells (1 × 106/mL) were resuspended in phosphate-buffered saline, and 10 μL cell suspension was supplemented with 90 μL of low-melting agarose. With gentle stirring, the mixture was laid on the slide. Upon solidification, the slide was dissolved in a 4°C dissolving solution for 1 h. The slide was rinsed with electrophoresis buffer and electrophoresed at 50 V for 5 min. Cells were maintained with ethanol after rinsing, and the slide was dried at 37°C for 10 min. The slide was stained with gel staining in a dark room for 10 min. Upon rinsing the slide, the DNA fluorescence was assessed by a fluorescence microscope (Nikon Eclipse E800, Japan) and a digital camera (Nikon DXM1200F, Japan). The degree of DNA damage was determined based on the proportion of DNA content in the comet tail. The extent of DNA damage was calculated based on the proportion of the fluorescence intensity of the comet tail to that of the entire comet. At least 5 DNA comets were chosen at random to calculate their degree of DNA damage. All experiments were repeated three times.
Western Blot (WB)
WB was performed in this study in terms of the method described by Fang et al. 23 Primary rabbit anti-human γ-H2AX, and β-actin antibodies, and the secondary goat anti-rabbit IgG antibody were obtained from Thermo Fisher Scientific (Invitrogen, USA).
Real-Time Quantitative Polymerase Chain Reaction (qRT-PCR)
qRT-PCR Primer Sequence.
Chromatin Immunoprecipitation (ChIP)
Dual-Luciferase Assay
First, we constructed NUSAP1-WT and NUSAP1-MUT luciferase reporter vectors (Promega, USA). Then oe-NC/oe-E2F8 was co-transfected with the above two plasmids into HCC cells for 48 h. Luciferase activity in each transfected group was determined using the luciferase activity assay kit (Promega, USA) according to the manufacturer’s instructions, and the experiment was repeated three times.
Data Analysis
All quantification assays were done in triplicate. Data were expressed as mean ± SD and statistically evaluated by GraphPad Prism 6 software (GraphPad Software, USA). The distinction was compared through t-test or one-way analysis of variance, with *, # representing P < .05, indicating statistical significance.
Results
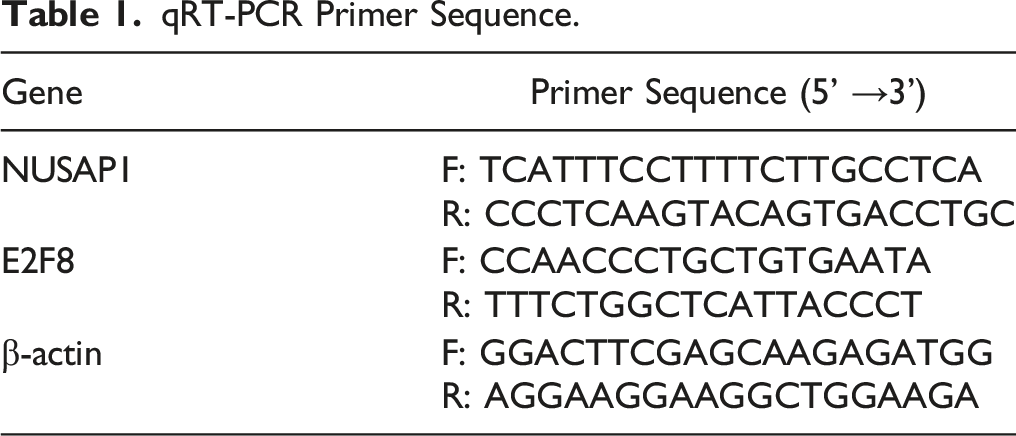
NUSAP1 Is Highly Expressed in HCC
The current study showed a prominently high expression of NUSAP1 in HCC tissue via bioinformatics analysis (Figure 1A). The outcomes of Gene Set Enrichment Analysis (GSEA) analysis revealed that ectopic expression of NUSAP1 may influence the cellular functions such as cell cycle, DNA replication as well as mismatch repair (Figure 1B-D). Subsequently, we tested the mRNA expression of NUSAP1 at the cellular level to validate the predicted NUSAP1 expression in HCC. qRT-PCR results demonstrated that NUSAP1 was significantly highly expressed in HCC cell lines (HepG2, SK-HEP-1, and SNU-182) compared to human normal hepatocytes THLE-3 (Figure 1E). In conclusion, we concluded that NUSAP1 acted as a cancer-promoter in HCC progression. NUSAP1 shows high expression in HCC: (A) NUSAP1 expression analyzed by bioinformatics analysis; (B-D) GSEA analysis was conducted in high and low NUSAP1 expression groups; (E) NUSAP1 expression in HCC cell lines (HepG2, SK-HEP-1, and SNU-182) and human normal hepatocytes (THLE-3); *P < .05 relative to THLE-3 group, mean ± SD, n = 3. SD, standard deviation.
The Impact of NUSAP1 on HCC Progression and Cisplatin
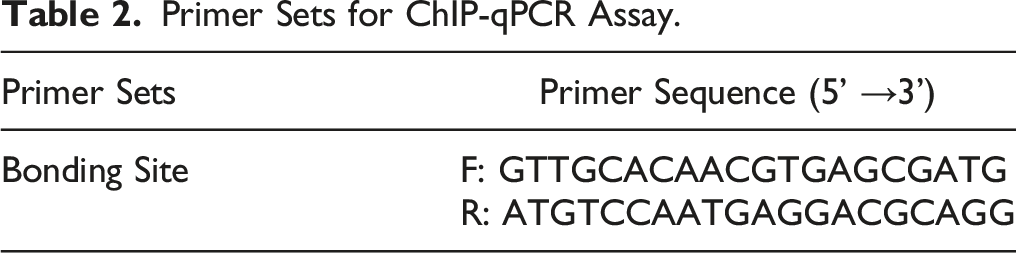
To verify the functions of NUSAP1 in HCC development, we selected two cell lines (HepG2 and SK-HEP-1) with relatively higher NUSAP1 expression for in vitro cell experiments. First, we established two HCC cell lines with knockdown NUSAP1 expression. Then the transfection effectiveness was verified via qRT-PCR (Figure 2A). NUSAP1 expression in the si-NUSAP1 group was significantly decreased in comparison with the control group. The results of the CCK-8 assay showed a notable decrease in cell viability in the si-NUSAP1 group compared with the control group (Figure 2B). Cell cycle outcomes indicated a significant increase in the number of cells in the G0/G1 phase in si-NUSAP1 group while an evident decrease in the number of cells in S phase (Figure 2C). Prior studies have identified that abnormal DNA damage repair remains one of the mechanisms by which cisplatin chemotherapy resistance occurs in tumors.
25
Then we further explored the impact of NUSAP1 on cisplatin resistance in HCC. CCK-8 detected cell activity under different concentrations of cisplatin ((0, 1, 2, 4, 8, 16, 32 μg/mL) treatment and calculated the IC50 value of cells to cisplatin. The results showed that compared with the si-NC group, the si-NUSAP1 group cells showed a more sensitive response to cisplatin, indicating that knockdown of NUSAP1 could enhance the sensitivity of HCC to cisplatin (Figure 2D). According to the IC50 value, 5.0 μg/mL was selected as cisplatin treatment concentration for subsequent studies. To probe related mechanisms, we performed comet assay and detected the protein expression of γ-H2AX, a marker of the degree of DNA damage, so as to assess the impact of NUSAP1 on cisplatin-induced DNA damage. As the results showed, HCC cells treated with cisplatin in the si-NUSAP1 group presented more significant DNA damage (Figure 2E), while the protein expression of γ-H2AX was also evidently increased in this group of cells (Figure 2F). The aforementioned findings exhibited that knockdown NUSAP1 could repress the viability of HCC cells and upgrade the DNA damage in HCC cells induced by cisplatin. Impact of NUSAP1 on HCC progression and cisplatin resistance: (A) qRT-PCR was used to detect the expression of NUSAP1 in each group of cells; (B) CCK-8 assay was used to test the cell viability of each group; (C) flow cytometry was used to measure the cell cycle progression of each group; (D) CCK-8 was used to determine the IC50 value of each group of cells; (E) comet assay was used to examine the degree of DNA damage; (F) western blot was used to detect the protein expression of γ-H2AX; *P < .05 relative to si-NC group, mean ± SD, n = 3. SD, standard deviation.
E2F8 Is the Upstream Regulatory Molecule of NUSAP1
To further clarify the molecular mechanism by which NUSAP1 affects cisplatin resistance in HCC, the hTFtarget database was accessed to predict the possible upstream transcription factors of NUSAP1, and the predicted results were compared with differentially up-regulated mRNAs in HCC. Finally, 15 candidate transcription factors were obtained (Figure 3A). Pearson's correlation analysis of NUSAP1 expression and the expression of 15 transcription factors suggested that NUSAP1 showed a significant positive correlation with E2F8 (Figure 3B), and E2F8 was significantly up-regulated in HCC tumor tissue (Figure 3C). Meanwhile, the JASPAR database predicted a motif of E2F8 at 2000 bp upstream of the NUSAP1 promoter region (Figure 3D), so E2F8 was a possible upstream transcription factor of NUSAP1. To verify the above hypothesis, E2F8 expression in human HCC cell lines was examined via qRT-PCR. The findings showed that E2F8 expression was significantly up-regulated in HCC cell lines (Figure 3E). Subsequently, we verified the binding relationship of E2F8 to the NUSAP1 gene promoter through ChIP assay, and the results indicated that E2F8 bound to the NUSAP1 gene promoter (Figure 3F). Furthermore, the targeted relationship between the two was confirmed by dual-luciferase assay, and it could be seen that overexpression of E2F8 increased wild-type NUSAP1 luciferase activity, but had no effect on mutant-type NUSAP1 luciferase activity (Figure 3G). The above results clarified that E2F8 was an upstream transcription factor of NUSAP1. NUSAP1 has an upstream regulatory molecule E2F8: (A) The UpSet plot of Human Transcription Factor Targets (hTFtarget) database and up-regulated mRNAs; (B) Pearson's correlation plot of NUSAP1 and E2F8; (C) bioinformatics of E2F8 expression in HCC; (D) bioinformatics predicted the binding site of NUSAP1 to E2F8; (E) detection of E2F8 expression in HCC cell line and human normal hepatocytes; *P < .05 relative to THLE-3 group, mean ± SD, n = 3; (F) ChIP verified the binding and targeted relationship between NUSAP1 and E2F8; *P < .05 relative to IgG group, mean ± SD, n = 3; (G) dual-luciferase assay verified the binding and targeted relationship between NUSAP1 and E2F8; *P < .05 relative to oe-NC group, mean ± SD, n = 3. ChIP, Chromatin Immunoprecipitation; WT, wild type; SD, standard deviation.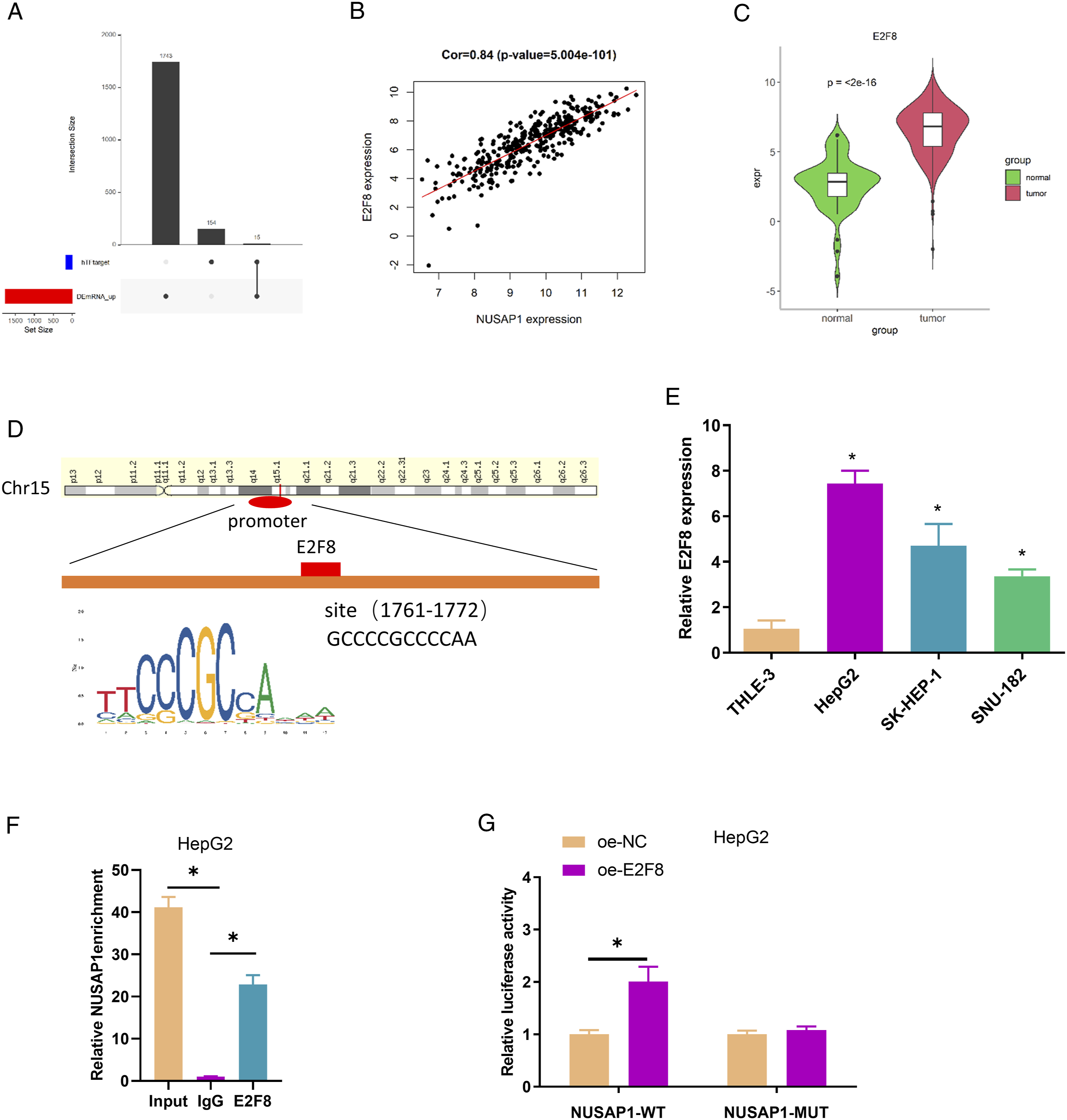
E2F8 Promotes Cisplatin Resistance in HCC by Regulating DNA Damage Repair via Activation of NUSAP1
To confirm the role of the E2F8/NUSAP1 regulatory axis during HCC progression, we selected HepG2 cells with more significant phenotypes in the cell function assays in the previous section for rescue experiments. First, the transfection efficiency was confirmed using qRT-PCR (Figure 4A). The findings showed that the mRNA expression of NUSAP1 was dramatically decreased in cells from the si-NUSAP1 group and notably increased from the oe-E2F8 group in comparison with the control group, while NUSAP1 expression of the oe-E2F8 + si-NUSAP1 group returned to the control level. Next, the CCK-8 assay results revealed that cell viability was increased in cells from the oe-E2F8 group compared to the control group, while further silencing NUSAP1 reversed the promoting function of oe-E2F8 in cell viability (Figure 4B). Cell cycle results ascertained that cell number in G0/G1 phase was reduced and cell number in S phase was significantly increased in the oe-E2F8 group, while further silencing NUSAP1 restored cell cycle progression to the control level (Figure 4C). These results suggested that E2F8 might promote the proliferation of HCC cells by accelerating cell cycle through NUSAP1. Then we further explored the impact of the E2F8/NUSAP1 axis on cisplatin resistance in HCC. CCK-8 results revealed that overexpression of E2F8 resulted in decreased cisplatin sensitivity in HCC compared with controls, and further silencing of NUSAP1 reversed the inhibitory function of overexpression of E2F8 on cisplatin sensitivity in HCC (Figure 4D). Finally, the effect of the E2F8/NUSAP1 regulatory network on cisplatin-induced DNA damage was evaluated by comet assay and protein expression of γ-H2AX. The outcomes showed that the degree of DNA damage in cisplatin-treated HCC cells was significantly decreased in the oe-E2F8 group (Figure 4E). Meanwhile, the protein expression of γ-H2AX, a marker of the degree of DNA damage, was also dramatically decreased in this group of cells (Figure 4F). However, overexpression of E2F8 and silencing of NUSAP1 restored the degree of cisplatin-induced DNA damage as well as the protein expression of the marker γ-H2AX to control levels in HCC cells (Figure 4E-F). The aforementioned outcomes displayed that E2F8 repressed cisplatin sensitivity in HCC through regulating DNA damage repair via activation of NUSAP1. E2F8 suppresses cisplatin sensitivity in HCC by activating NUSAP1 to regulate DNA damage repair: (A) detection of NUSAP1 expression in HCC; (B) CCK-8 tested cell viability; (C) flow cytometry measured cell cycle progression; (D) CCK-8 examined the IC50 value of HCC cells to cisplatin; (E) comet assay detected the degree of DNA damage; (F) expression of γ-H2AX by western blot; The above experimental cells were grouped as (oe-NC + si-NC; oe-E2F8 + si-NC; oe-NC + si-NUSAP1; oe-E2F8 + si-NUSAP1); *P < .05 relative to oe-NC + si-NC group, #P < .05 relative to oe-NC + si-NUSAP1 group, mean ± SD, n = 3; SD, standard deviation.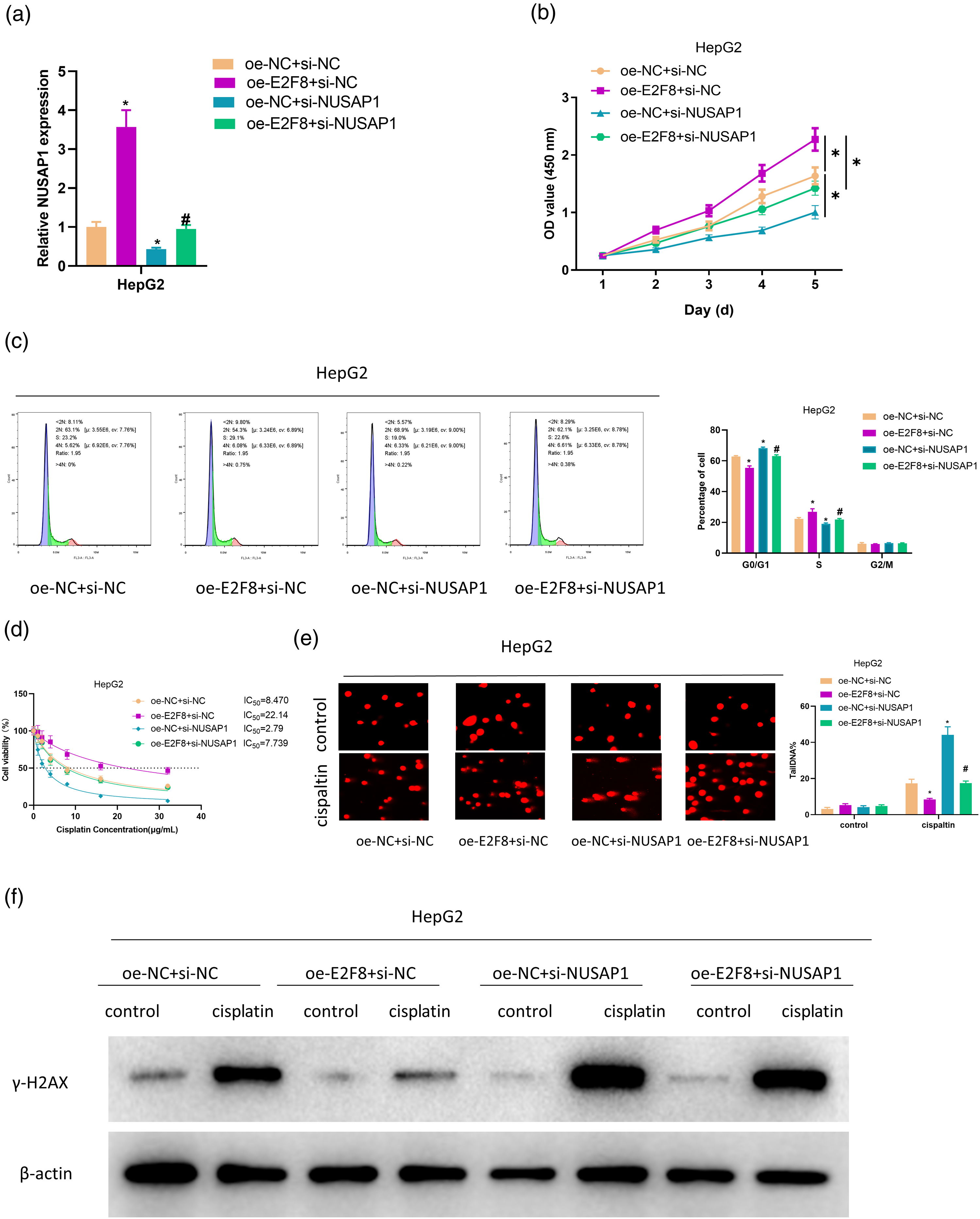
Discussion
The mechanism of tumor drug resistance is a complicated process that has become a research hotspot in recent years. Its influencing factors mainly include DNA damage repair, individual genetic discrepancies, tumor microenvironment, cancer stem cells, drug inactivation, reduced drug absorption, and changes in anti-tumor drug metabolism.26–28 It has been shown that abnormalities in DNA damage repair function can affect cisplatin sensitivity in tumor cells. For example, silencing MARK2 can prevent cisplatin resistance in osteosarcoma stem cells by regulating DNA damage and repair. 29 Let-7e suppresses DNA damage repair and sensitizes ovarian cancer to cisplatin by targeting PARP1. 30 In this work, we also observed that silencing NUSAP1 could advance DNA damage and thereby improve cisplatin sensitivity in HCC. Prior studies have reported that NUSAP1 acts as an important regulator of mitotic progression, spindle assembly, and chromosome attachment. Yang et al. 31 confirmed that silencing NUSAP1 enhances fludarabine or ibrutinib sensitivity in chronic lymphocytic leukemia through the DNA damage repair signaling pathway. Similarly, this study showed that NUSAP1 was significantly up-regulated in HCC, and that silencing NUSAP1 could accelerate HCC sensitivity to cisplatin chemotherapeutic agents. These results provide new insight into studying chemoresistance and may help reverse cisplatin resistance in HCC therapy.
This research found the upstream regulatory molecule E2F8 of NUSAP1. E2F8 remains an important member of the E2F transcription factor family and participates in cell development, including regulation of the cell cycle and DNA synthesis in mammalian cells.32–35 Previous studies have identified E2F8 as an oncogene promoting tumor progression in cervical cancer, 33 ovarian cancer, 36 colon cancer, 37 and HCC. 38 In the current study, we confirmed a binding relationship between E2F8 and NUSAP1. E2F8 could activate the transcription of NUSAP1 and was strikingly up-regulated in HCC cells. Our current results implied that overexpressed E2F8 resulted in abnormal cell cycle progression, leading to a prominent decrease in cell number in G0/G1 phase and an evident increase in cell number in the S phase. It is consistent with the findings of Ye et al., 34 which shows that E2F8 transcriptionally up-regulated CCNE1 and CCNE2 by directly interacting with their respective gene promoters, thereby accelerating the G1 to S phase transition in breast cancer cells. Notably, we also found that overexpression of E2F8 inhibited DNA damage. Previously, Zalmas et al. 35 reported that E2F8 underlies the cell cycle response to DNA damage. Sun et al. 39 observed that E2F8 is highly expressed in breast cancer and YTHDF1 induces cisplatin resistance in breast cancer cells by promoting DNA replication and DNA damage repair via E2F8. The findings of experiments in this study established that E2F8 overexpression could repress γ-H2AX expression and DNA damage, and promoted cisplatin resistance in HCC. The rescue experiment suggested that knockdown of NUSPA1 reversed the inhibitory effect of E2F8 overexpression on DNA damage and cisplatin sensitivity. No previous studies have reported the specific mechanism by which E2F8 regulates NUSPA1 expression to affect HCC progression and cisplatin resistance. However, this study provides a new understanding of the mechanism of cisplatin resistance in tumors, suggesting that the E2F8/NUSPA1 axis may become a target to defeat cisplatin chemoresistance in HCC.
To conclude, we found for the first time that silencing NUSPA1 could increase DNA damage and then enhance cisplatin sensitivity in HCC, suggesting that NUSPA1 is not only an oncogene, but also a target for overcoming cisplatin resistance in HCC. In addition, the function of NUSPA1 in DNA damage repair was regulated by the transcription factor E2F8. The results preliminarily validated that E2F8/NUSPA1 may be a DNA damage-associated resistance gene, providing a reference basis for clinical guidance of cisplatin treatment for patients. However, there are still shortcomings in our work. The role of the E2F8/NUSPA1 axis in cisplatin resistance in HCC has not been verified in an animal model. In addition, we did not explore the downstream regulatory mechanism of this regulatory axis. We intend to make a deeper exploration in the future to enrich the regulatory mechanism in the progression of HCC. In summary, this study not only enriches the knowledge of molecular regulatory mechanisms affecting HCC progression, but also provides a novel insight into improving tumor cisplatin resistance mechanisms in clinical practice.
Footnotes
Author Contributions
Jianqiao Kong contributed to conception and design, drafted manuscript, and critically revised manuscript; Song Xu contributed to conception and design, drafted manuscript, and critically revised manuscript; Peng Zhang contributed to analysis and interpretation and drafted manuscript; Yi Wang contributed to acquisition and drafted manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.