
Abstract
From a micro to macro scale of biological organization, macromolecular diversity and biological heterogeneity are fundamental properties of biological systems. Heterogeneity may result from genetic, epigenetic, and non-genetic characteristics (e.g., tissue microenvironment). Macromolecular diversity and biological heterogeneity are tolerated as long as the sustenance and propagation of life are not disrupted. They also provide the raw materials for microevolutionary changes that may help organisms adapt to new selection pressures arising from the environment. Sequence evolution, functional divergence, and positive selection of gene and promoter dosage play a major role in the evolution of life’s diversity including complex metabolic networks, which is ultimately reflected in changes in the allele frequency over time. Robustness in evolvable biological systems is conferred by functional redundancy that is often created by macromolecular diversity and biological heterogeneity. The ability to investigate biological macromolecules at an increasingly finer level has uncovered a wealth of information in this regard. Therefore, the dynamics of biological complexity should be taken into consideration in biomedical research.
Keywords
Introduction
Variety’s the very spice of life that gives it all its flavor is an old and widely known proverb in English language. Scientific advances have provided a biological meaning to this proverbial literary expression. In the realm of biology, “variety” means biodiversity. From a macro to micro scale of life’s organization, biodiversity includes the diversity of species, habitats, genetics, as well as the diversity of biological macromolecules. 1 With the power to dissect and study the structure, function, and interactions of biological macromolecules at an increasingly finer level, understanding the diversity of biological macromolecules producing biological heterogeneity has become an important focus of molecular sciences—be it molecular genetics, or molecular glycobiology. Macromolecular diversity—whether in terms of structure, function, or abundance, is at the root of biological heterogeneity, which is an important determinant of an organism’s ability to respond to the environment, including toxicants.
Genetic Variations and the Changing Concept of Allele
Variations have been traditionally equated with genetic variations at the population level (genomic variations at the individual level). This is expected because genetic/genomic variations are at the root of most other variations leading to macromolecular diversity and heterogeneity in biological systems.
Genetic variations in a population are derived from a wide assortment of alleles. The classical concept of alleles is phenotype-based, that is, alleles are alternative forms of a gene (e.g., R vs r) that occupy the corresponding loci in homologous chromosomes and influence specific phenotypes. At the molecular level, an allele is a DNA sequence that codes for a protein and influences a phenotype. Alleles may show a clear order of dominance, codominance, or incomplete dominance. Codominant alleles create an intermediate phenotype in a heterozygous state, such as red and white colors producing pink color. In contrast, alleles with incomplete dominance create a mixed phenotype in heterozygous state, such as red and white colors producing a variegated pattern of red and white. If there are more than two possible alleles for a particular locus, such as A, a1, a2, a3, a4, then it creates a set of alleles called multiple alleles. The genotypes and the corresponding phenotypes could be determined by any combination of these alleles. These definitions of alleles are part of genetics textbooks; hence are not elaborated here any further.
This classical view of alleles has undergone major revision in the age of genomics. Any DNA sequence variant of a gene is now called an allele, including variants that do not influence the phenotype. 2 Alleles also refer to variants involving non-coding DNA sequences. 3 Because of this changing perspective from classical to molecular definitions, alleles are now defined in both ways—(i) as alternative forms of a gene controlling phenotype variations, which is the traditional textbook definition and (ii) DNA sequence variants of a gene. 4
The realization that alleles include DNA sequence variants that do not affect the phenotype or are non-coding, has essentially put the classical allele concept in a somewhat fluid state.
Generation of New DNA Sequence Variants and Their Relevance to Health and Disease
DNA sequence variants among genomes create polymorphism (genetic differences) in the population. Hence, alleles are polymorphic DNA sequences. Polymorphisms in the human genome are caused by single nucleotide polymorphisms (SNPs; pronounced snips); copy number variations (CNVs), also called copy number polymorphisms (CNPs); small insertions and deletions (indel mutations); and rearrangements. 5 Many of these variants are associated with disease phenotypes. Not all classes of variants occur with equal frequency, nor are they equivalent with respect to their contribution to disease.
Single nucleotide polymorphisms in health and disease
A single nucleotide polymorphism (SNP) is a point mutation affecting a single base in the DNA strand (hence a single base pair in the DNA double helix); the change could be due to a substitution mutation or an indel mutation. For a variation to be considered a SNP, it should occur in at least 1% of the population. This frequency of occurrence distinguishes SNPs from rare point mutations. The median number of autosomal SNPs in most populations is around 3.5 million. 6 The genomes of two individuals are between 99.5 and 99.9% identical; hence, the number of genetic variations between any two individuals is between 0.5 and 0.1%. 7
A well-known example of a SNP determining specific disease susceptibility is in the human apolipoprotein E (ApoE) gene. Two SNPs in the ApoE gene produce three possible alleles termed E2, E3 (wild type), and E4. The corresponding protein product of each gene differs by one amino acid. The SNP in the E2 allele produces one amino acid substitution (R158C; the 158th amino acid in the wild type (E3) ApoE protein is arginine, which is replaced by cysteine in the E2 allele encoded ApoE protein). Likewise, the E4 allele has one SNP (C112R; the 112th amino acid in the wild type (E3) ApoE protein is cysteine, which is replaced by arginine in the E4 allele encoded ApoE protein). Inheriting two E4 alleles increases the susceptibility to developing Alzheimer’s disease. The C112R SNP in the E4 allele is thought to alter the interactions between the N-terminal and the C-terminal domain of the protein resulting in major functional differences and increasing the risk of disease development. 8
The occurrence of certain SNPs may predict the toxicity of certain chemotherapeutic drugs. For example, fluoropyrimidines, such as 5-fluorouracil (5-FU), capecitabine (prodrug for 5-FU) and tegafur are used to treat gastroenteropancreatic cancers. The enzyme dihydropyrimidine dehydrogenase (DPYD) in the liver metabolizes ∼80% of 5-FU into its inactive metabolite 5,6 dihydroxy-5-FU. If DPYD function is compromised, severe toxicity of 5-FU ensues that often includes fever, mucositis, stomatitis, nausea, vomiting, and diarrhea, and neurologic abnormalities in some patients. Some of the most reliable SNP markers of fluoropyrimidine toxicity are DPYD*2A (rs3918290), DPYD-c.2846 A>T (rs67376798), DPYD-Hap-B3 (rs56038477) and DPYD*13 (rs55886062). 9 The “rs” prefix (e.g., rs3918290) in the SNP designation (SNP accession number) is an acronym for RefSNP (rs).
Copy number variations in health and disease
Copy number variations (CNVs) represent structural variations caused by the gain or loss of segments of genomic DNA, which is produced by sequence duplications (gain) or deletions (loss). Copy number variations can result in variations in the number of gene copies, thereby influencing gene dosage, magnitude of gene expression, and sometimes disease phenotypes. 10 The definition of the minimum length of CNVs has changed over time. Earlier studies defined the minimum length as 1000 bp, which was subsequently revised to 100 bp. Currently, the size range of CNVs is defined from 50 bp to 5 Mbp (5 Mega bp, i.e., 5 million bp).5,11 Operationally, the smaller CNVs are distinguished from insertions and deletions (indels) based on length—the indels being between 1 and 49 bp. 5 Other structural variations include rearrangements like inversions and translocations.
Unlike SNPs, CNVs have not been as extensively studied for their contributions to functional genomic variations and chemical toxicity. However, there are some well-known examples that demonstrate the effect of CNVs (functional gene dosage) in determining drug metabolism phenotypes. Cytochrome P450 (CYP) 2D6 is the most well studied polymorphic CYP enzyme in humans. Individuals with ultrarapid metabolizer (UM) phenotype have multiple copies of the CYP2D6 gene, such as among many individuals from Ethiopia and Saudi Arabia.12,13 The allele is termed CYP2D6*2XN (N= 2, 3, 4, 5 or 13). The UM phenotype has been reported in other populations as well. A CYP2D6 genotyping study to analyze the functional gene dosage among a group of Japanese subjects showed that about 46% of the subjects had 2 copies, 35% had 3 copies, 9% had 4 copies, and 1% had 5 copies. 14 The impact of gene dosage on CYP2D6 drug metabolism phenotypes is further discussed later in the text.
CNVs can be utilized for predicting toxicity outcomes of many drugs, particularly when the CNVs involve deletions. Several studies documented examples of CNVs of various CYP alleles involving loss-of-function deletions and their frequencies in specific populations. For example, a major study 15 reported CNVs of various CYP alleles involving loss-of-function deletions, such as CYP2C19 in Finns, CYP2B6 and CYP1A2 in Africans, and CYP4F2 in East Asians. The allele frequencies of many such loss-of-function variants show >10-fold difference among populations, demonstrating that these deletions are highly population-specific. 15 Another example is CYP3A4, which is an important drug metabolizing enzyme in humans. The CYP3A4*20 allele encodes a non-functional enzyme because the protein is truncated; however, this is a rare allele. In some regions in Spain, this allele is present at a high frequency (up to 3.8%) and is responsible for adverse drug reactions during paclitaxel therapy.15,16
For a sexually reproducing population, new genetic variations are created by mutation, recombination, and gene flow through migration. These processes create both phenotype-influencing and phenotype-neutral DNA sequence variants (allelic variants).
Sequence Evolution
A base substitution point mutation in the open reading frame (ORF) of DNA can have three types of mutational events—a missense (or nonsynonymous) mutation that changes an amino acid in the protein; a nonsense mutation that creates a premature stop codon, thereby truncating the polypeptide product; a silent (or synonymous) mutation that does not change any amino acid in the protein. The mutations in the DNA sequence, whether SNPs or rare mutations, that are discussed here in the context of health and disease are missence or nonsynonymous mutations.
Effects of specific mutations in a codon on the nucleotide and amino acid sequence
Except for methionine (Met) encoded by the ATG codon (AUG in mRNA), and tryptophan (Trp) encoded by the TGG codon (UGG in mRNA), all other amino acids are encoded by multiple codons. It should be emphasized that in the nucleotide databases the mRNA sequences are reported as in the sense strand of DNA. Hence there is no uracil (U) in the sequence. For example, AUG in mRNA is represented as ATG. The same rule is followed in this text.
All the codons encoding an amino acid differ at the third base (wobble base); the first two bases are invariant. A mutation involving the first or the second base changes an amino acid in the polypeptide, but a mutation in the third base can have several types of consequences depending on the codon. For some amino acid codons, the third base mutation creates a stop codon, such as TGG (Trp) → TGA (Stop). For the ATG codon, the third base mutation results in a missense mutation. All the possible new codons due to third base mutation (from ATG → ATA, ATC or ATT) cause Met → Ile substitution in the polypeptide. For all other amino acid codons, the third base mutation does not change the amino acid in the polypeptide. Therefore, mutations causing nucleotide sequence variations within an ORF may or may not affect the amino acid sequence depending on the base mutated.
The specific base mutation of a codon (first or second, vs third) determines the extent of nucleotide vs protein identity. For example, the cannabinoid receptor CB1 binds endocannabinoids. The human and rat CB1 nucleotide sequences are 90% identical, whereas the amino acid sequences are 98% identical. 17 Less identity in nucleotide sequence than protein sequence for the CB1 receptor is because many of the codons have third base mutation resulting in no change in the amino acid sequence in the protein but reducing the nucleotide sequence identity. In contrast, if the mutations involve either the first or the second base of the codon, it will cause a change in amino acid; as a result, the amino acid sequence identity will be reduced but the nucleotide sequence identity will remain higher.
Functional constraints and the differential evolution of coding and noncoding sequences with some examples relevant to toxicology
In the gene, sequences that are under selection pressure because of functional constraints tend to be conserved during evolution, whereas sequences that are not under selection pressure tend to diverge by accumulating mutations. Hence, the 5′- and 3′-untranslated regions (UTRs) between two functionally related mRNAs are more likely to show lower sequence identity than the ORF. As a rule, functionally important regions of biological sequences are under strong purifying selection and they evolve more slowly. 18
Mammalian metallothionein (MT) proteins have highly conserved metal-binding function and they protect against Cd-induced hepatotoxicity by altering the subcellular distribution of Cd, particularly limiting its distribution to endoplasmic reticulum and mitochondria. Most of the Cd is sequestered in the cytosol by MT. 19 MT genes originated through a series of duplication events during evolution, which generated four major MT genes- MT-I though MT-IV. Of these, MT-I and MT-II code for ubiquitous proteins performing similar metal-binding functions, whereas MT-III and MT-IV evolved to perform specific roles in brain and epithelium, respectively. 20 Using the Needleman–Wunsch global alignment algorithm (EMBOSS Needle), a pairwise alignment of the nucleotide sequence of the ORFs of mouse MT-I mRNA and MT-II mRNA shows 80% identity with no gaps introduced. In contrast, the nucleotide sequence of the 5′- and 3′-UTRs shows approximately 51% identity with 23% gaps introduced, and 52% identity with 17% gaps introduced, respectively. Extensive gaps introduced to attain the highest possible alignment score clearly indicates that the 5′- and 3′-UTRs have significantly diverged in sequence during evolution. In contrast, the coding sequence maintains significant identity because of the functional constraints.
Organic anion transporting polypeptides (OATPs) 1B1 and 1B3 in humans are involved in the uptake transport (in the liver) of various clinically important drugs, such as statins, endothelin receptor antagonist (e.g., bosentan), certain angiotensin converting enzyme (ACE) inhibitors (e.g., enalapril) and certain angiotensin II receptor blockers or ARBs (e.g., telmisartan), certain chemotherapeutic drugs (e.g., docetaxel), and the antidiabetic drug repaglinide.21,22 These transporters also transport endogenous substrates like bilirubin, bile acids, coproporphyrins, and conjugated hormones, as well as environmental toxins, such as the mushroom toxin phalloidin and the cyanobacterial toxin microcystin-LR. 23 OATP1B1 and 1B3 polypeptides are encoded by SLCO1B1 and 1B3 genes, respectively. In primates, SLCO1B1 and 1B3 arose by gene duplication after divergence from rodents. 23 Using the Needleman-Wunsch global alignment algorithm, a pairwise alignment of human SLCO1B1 and 1B3 mRNAs shows that the coding sequence (ORF) of these 2 transporters maintain 86% identity with 1.6% gaps introduced. In contrast, the 5′-UTR shows 35% identity with 57% gaps introduced, and the 3′-UTR show 57% identity with 28% gaps introduced. Extensive gaps introduced to attain the highest possible alignment score clearly indicates that the 5′- and 3′-UTRs have significantly diverged in sequence during evolution. In contrast, the coding sequence maintains marked identity because of the functional constraints. Therefore, both MT-I and II genes in mice and SLCO1B1 and 1B3 genes in humans clearly show differential evolution of the coding and noncoding sequences.
Evolutionary fate of duplicated genes with some examples including those relevant to toxicology
Gene duplication and functional divergence of the duplicated genes provides an important means of genome evolution. Duplicated gene sequences can diverge to have one of three fates; they may acquire new function (neofunctionalization), or become nonfunctional (pseudogenization), or retain the function, in part or whole, of the ancestral gene (subfunctionalization) (Figure 1). Neofuntionalization, subfunctionalization, and pseudogenization (nonfunctionalization) following duplication of a gene. Sequence divergence of the cis-regulatory modules following duplication demonstrates the three possible fates of the original sequence. (The figure is reproduced, with slight modifications, from Figure 2.4 of Bioinformatics for Beginners: Genes, Genomes, Molecular Evolution, Databases and Analytical Tools (Author: Supratim Choudhuri), 2014, with permission from Elsevier).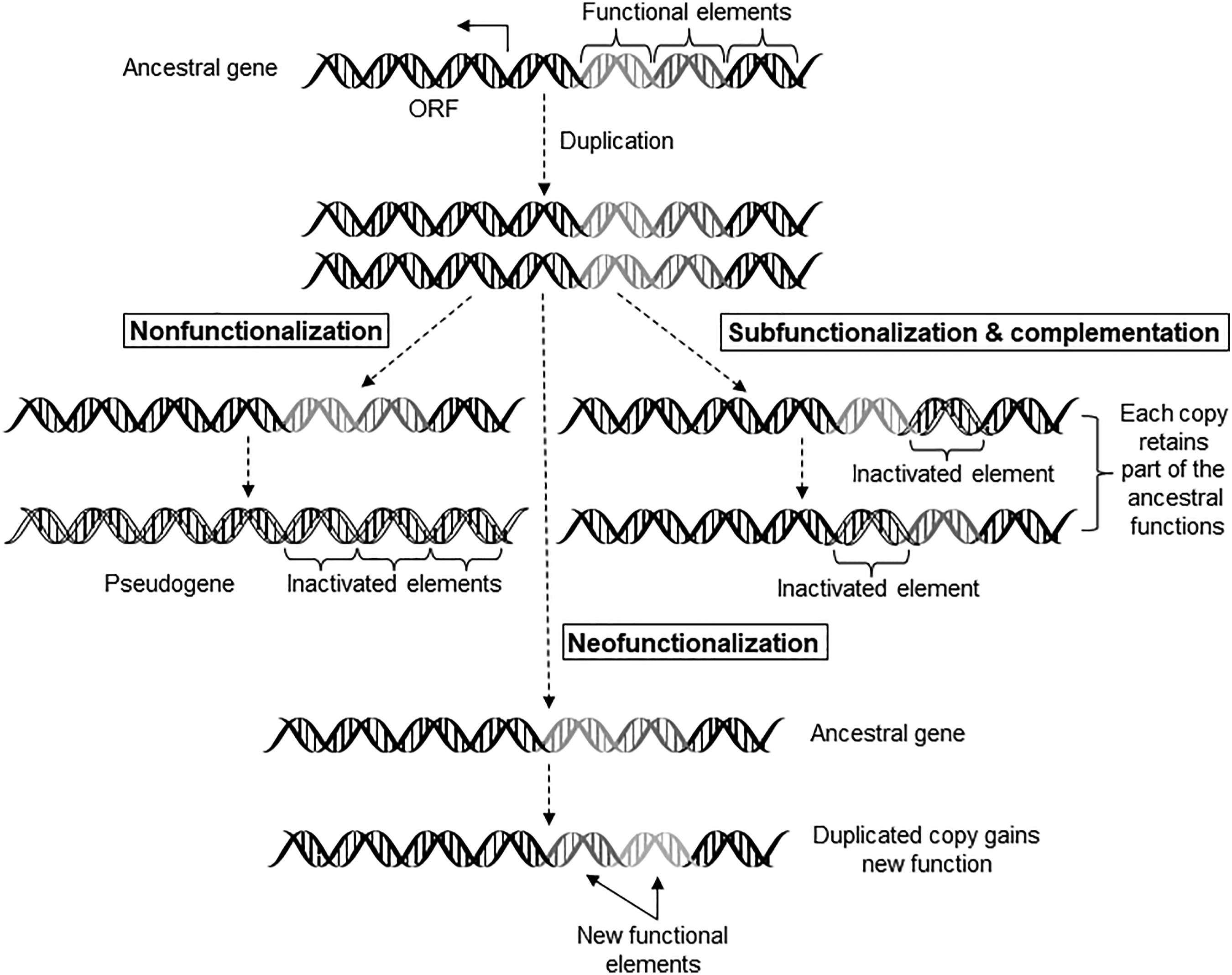
Neofunctionalization of duplicated genes provides an important mechanism for the genome to diverge both structurally and functionally. The gene encoding type III antifreeze protein evolved from a sialic acid synthase gene. The sialic acid synthase (SAS) gene in the Antarctic zoarcid fish duplicated and one of the copies lost the N-terminal SAS domain-encoding segment. Further optimization of the C-terminal domain-encoding segment in the same copy produced type III antifreeze protein gene. 24 Many of the blood coagulation proteins are related to each other because of gene duplications and divergence that occurred early in vertebrate evolution. The 11 blood coagulation factors in humans have been identified across all vertebrates, suggesting that they appeared in the first vertebrates around 500 million years ago. 25 Employing a residue-by-residue comparison during sequence alignments and analyzing amino acid substitutions, the authors identified signatures of positive selection for fibrinogen (factor I), factors III, VIII, IX, and X in mammals, reptiles, and birds. In primates, signatures of positive selection (conserved residues) were identified in at least nine of the 11 coagulation proteins, suggesting that the coagulation pathway was under considerable adaptive constraints. In general, the disease-causing mutations primarily affect the highly conserved residues.
Neofunctionalization is also observed in drug metabolizing enzyme and transporter genes. The breast cancer resistance protein (BCRP) is an efflux transporter encoded by the ABCG2 gene in humans and rodents. It transports a broad range of endogenous substances (e.g., various sulfate conjugates) and drugs (chemotherapeutics like topotecan and irinotecan). 26 Both ABCG2 and ABCG3 in rodents were derived from the common ancestral sequence. ABCG3 diverged to perform different functions, which is yet to be clearly defined. ABCG3 is primarily expressed in murine hematopoietic cells and contains no ortholog in the human genome. 27 Some other examples of neofunctionalization include the metazoan copper transporters Ctr1 and Ctr2, 28 and the cytochrome P450 CYP6ER1. 29 Duplication and neofunctionalization of the CYP6ER1 gene, which involves its high expression, led to the evolution of the insecticide imidiacloprid resistance in the brown planthopper. 29
Unlike neofunctionalization, subfunctionalization occurs when the duplicated copies (paralogs) retain the function, in part or whole, of the ancestral gene. This is because natural selection does not distinguish which paralog of the duplicated gene pair should be under selection and which paralog should be free from selective constraint. A common example is the normal human hemoglobin, which is composed of two α-chains and two β-chains (α2β2) encoded by the α-globin and β-globin genes, respectively. The α- and β-globin genes are products of gene duplication and subsequent subfunctionalization. 30 Duplication events creating SLCO1B1 and 1B3 genes in humans, discussed above, is another example of subfunctionalization.
Promoter dosage controlling gene expression in health and disease
The promoter dosage (functional copy numbers of specific promoter sequence), like gene dosage itself, may also determine the health and wellness of individuals by modulating gene expression. In humans, the X-linked monoamine oxidase A (MAOA) gene has four exact repeats of a 30-bp sequence located at −1142 to −1262 bp position relative to the translation initiation codon ATG. This 30-bp sequence is often referred to as the upstream variable number tandem repeat region of the MAOA gene (MAOA-uVNTR). The four complete repeats are followed by a half repeat of the first 15 bp of the 30-bp sequence at −1141 to −1127 bp position relative to the ATG. The 30-bp sequence itself consists of 5 repetitions of the core sequence ACC(A/G/C)G (C/T). Expressing firefly luciferase reporter gene under the control of MAOA gene promoter containing different numbers of the promoter repeat sequences in three different cell lines showed that the 3.5 or 4 repeats of the 30-bp sequence drove significantly higher levels of expression. This suggests that an optimal length of the repeat region drives the highest expression. 31 MAOA and MAOB are mitochondrial enzymes and they have overlapping substrate specificities as well as tissue distribution. They inactivate neuroactive amines like serotonin, dopamine, and norepinephrine. 32 Thus, individuals with suboptimal/lower expression of the MAOA gene due to fewer number of promoter repeats (MAOA-uVNTR) may fail to adequately metabolize the neuroactive amine substrates.
Critical Mutations; Adaptive, Maladaptive and Disease Phenotypes; and Heterogeneity in Biological Systems
The practice of classical genetics relied on obtaining mutations creating new phenotypes to study gene function. With the development of DNA sequencing, the specific molecular change (SNP, CNV, and Indel) can be traced back to the DNA sequence. The critical point mutations discussed in this section are not necessarily all SNPs (SNPs occur in at least 1% of the population).
Single amino acid substitutions with pronounced effects on protein structure, function, stability and consequent effects on health and disease
If the point mutation in the ORF causes an amino acid change such that it induces significant structural and functional change of the polypeptide, or alters its expression and stability, then a single mutation may affect the phenotype, including inducing a disease phenotype. In proteins, amino acid substitutions involving similar amino acids, that is, amino acids with similar physicochemical and functional properties are called conservative or similar substitution. Conservative substitutions are not expected to alter the protein’s structure and functions around the substitution sites. Examples include substitution of arginine and lysine by one another (positively charged R group in both), substitution of aspartic acid and glutamic acid by one another (negatively charged R group in both), substitution of isoleucine, leucine, and valine by one another (aliphatic, hydrophobic). 33
Generally, the substitution of hydrophobic amino acids by polar amino acids, and the substitution of histidine, cysteine, and proline, by any other amino acids may result in harmful mutations. 34 Substitution of cysteines that participate in disulfide (-S-S-) bond formation to stabilize the structure and function of a protein (e.g., Insulin and RNAse A), or participate in metal-binding through the formation of metal-thiolate bonds (e.g., metallothionein) can result in a profound alteration of protein structure and function.
Proline throws a rigid kink in the polypeptide chain, and thus facilitates the folding of many proteins by introducing rigid turns in the peptide chain. Proline substitutions can stabilize sections of the polypeptide chain and/or the entire polypeptide chain in both cytosolic and transmembrane (TM) proteins. Proline-mediated kinks in the helix of membrane proteins aid in the tight packing of TM helices. Depending on the location, a proline substitution in a TM helix can essentially repack the segment due to a swivel movement. This results in an overall TM complex stabilization, as demonstrated by proline-induced integrin αIIbβ3 stabilization. 35 Like other integrins, integrin αIIbβ3 is expressed at a high level on the cell surface, and it plays an important role in platelet aggregation, hemostasis, and arterial thrombosis. Integrin αIIbβ3 also helps in cancer metastasis. 36
By comparing the disease spectrum to the spectra of amino-acid mutation frequencies, non-disease polymorphisms in human genes, and substitutions fixed between species, it was observed that about 30% of genetic diseases are caused by mutations at arginine and glycine residues together, whereas random mutations at tryptophan and cysteine have the highest probability of causing disease. 37 Similar findings were reported by other authors 38 who investigated the effects of missense mutations on disease using proteins with experimentally defined secondary structure. They found that arginine and glycine showed the highest relative mutability (25%), followed by that for cysteine and tryptophan. Therefore, both studies observed that arginine and glycine are among the most mutated residues associated with human genetic diseases.
The authors noted that high mutability of arginine could be explained by the spontaneous deamination rate. Arginine is encoded by six codons:
Using solvent accessibility of amino acids in the protein it was further demonstrated that disease-causing random mutations (usually with much lower frequency of occurrence than SNPs) are most likely to be found in the interior of the protein. In contrast, SNPs are most likely to be found on the surface of the protein. 37 This observation is consistent with the suggestion that disease-causing mutations damage protein function by decreasing protein stability, as opposed to mutations of the active-site residues that are usually located on the surface of the protein.
Mutations of the splice donor and acceptor sites in various human diseases
One more source of variation by a single base random substitution is mutation of the splice donor or acceptor site. The vast majority (∼98%) of the exon/intron boundary sequences in genes contain the dinucleotides GT and AG at the 5′ and 3′ ends of the intron, respectively. In other words, the splice donor site at the 5′ end of the intronic DNA has GT (hence GU in the pre-mRNA) and the splice acceptor site at the 3′-end has AG. A single base mutation involving the splice donor or acceptor site will cause a splicing defect and produce an aberrant mRNA and protein. Consequently, many splicing mutations tend to have serious consequences and have been implicated in human disease, such as neurofibromatosis type I, cystic fibrosis, Duchenne muscular dystrophy, xeroderma pigmentosum, and many others. 39
Silent mutations can also impact macromolecular function
Even silent mutations in the ORF that do not change the amino acid sequence may alter protein activity by altering conformation. The SNP C3435 T (C at position 3435 replaced by T in exon 26) in human Multidrug Resistance 1 (MDR1) gene results in the production of a P-glycoprotein (P-gp) of reduced functionality that is characterized by an altered ability to interact with drugs and inhibitors. 40 The authors observed that this mutation did not change the levels of mRNA or protein expression but created a rare codon. The authors hypothesized that this rare codon affected the timing of cotranslational folding and insertion of P-gp into the membrane, thereby altering the structure of substrate and inhibitor interaction sites.
It is well-documented that there is a codon usage bias in genomes. Synonymous codons (different codons encoding the same amino acid) are recognized with different efficiency by their cognate tRNAs, which correlates with the levels of cognate tRNAs in the cell. Therefore, the placement of a rare codon impacts the timing of ribosomal movement along the mRNA template. This, in turn, results in variable rates of polypeptide emergence from the ribosome influencing the folding toward the native state.41,42
A critical mutation producing a phenotype that can be adaptive or maladaptive depending on the environment
Linus Pauling and coworkers 43 first demonstrated that at neutral pH, sickle cell hemoglobin (HbS) has a different electrophoretic mobility and greater net positive charge compared to the normal hemoglobin (HbA). Subsequent studies by Vernon Ingram44,45 demonstrated that the glutamic acid residue in HbA (amino acid number 6 at the N-terminal end) is replaced by valine in HbS. Later, DNA sequencing showed that a GAG → GTG mutation (second base mutation) in the sixth codon of the hemoglobin β-chain gene results into Glu → Val substitution in HbS.
Individuals homozygous for HbS (HbSS) experience severe vascular complications and (hemolytic) sickle cell anemia that can lead to early death. In contrast, heterozygous individuals (HbAS) do not experience severe vascular complications and are partially protected against severe malaria caused by the parasite Plasmodium falciparum. Therefore, the homozygous condition produces the sickle cell disease, whereas the heterozygous condition produces the sickle cell trait, which is a benign condition.
The sickle cell trait confers adaptive advantage in areas where P. falciparum is spread through mosquito bites. Nevertheless, the presence of sickle cell trait makes heavy activity and sports at high altitude (with low oxygen tension) risky because these individuals would be vulnerable to hypoxemia, exercise-induced rhabdomyolysis, splenic infarction, abdominal pain, and other adverse symptoms. Thus, the same character that confers adaptive advantage in one environment becomes a maladaptive character in a different environment. 46
In addition to the sickle cell trait, there are several other heritable traits that offer protection against malaria, such as hemoglobin disorder (e.g., thalassemia), erythrocyte surface antigen polymorphism (e.g., Duffy-null blood group), and enzymopathies (e.g., glucose-6-phosphate dehydrogenase (G6PD) deficiency, and pyruvate kinase deficiency). All these characters are maladaptive because they have adverse health effects, but in the context of malaria infection they confer survival advantage to the carrier, thus acting as adaptive traits. 47
Nutritional adaptation during evolution producing biologically relevant heterogeneity
There is a direct correlation between the AMY1 gene copy number and salivary amylase content; amylase helps digest starch. High-starch consuming populations (e.g., agricultural populations of European Americans, Japanese, and Hazda hunter gatherers) have a higher diploid AMY1 gene copy number (median 7) compared to the low-starch consuming populations (e.g., Biaka and Mbuti rainforest hunter-gatherers, Datog pastoralists, and Yakut pastoralist and fishing society) (median 5), suggesting that high-starch diet resulted in the expansion of AMY1 gene copy number. Because of its selective advantage, this character has undergone positive selection and has been fixed in the population. 48
The ADH1B*47His allele of alcohol dehydrogenase (ADH) has higher activity and causes increased metabolism of ethanol. This allele is dominant among East Asians but rare in Europeans and Africans. Molecular dating suggests that the emergence of this allele coincides with the origin of rice domestication in East Asia. The regionally restricted enrichment of this allele in southern China and the adjacent areas suggests positive selection. 49 It has been proposed that the rise of ADH1B*47His allele frequency in East Asian populations was an adaptation to rice domestication and the subsequent production and consumption of fermented foods and beverages.
CYP8B1 is a 12α-hydroxylase that is responsible for the production of the primary bile acid cholic acid (3α,7α,12α-trihydroxylated). The other primary bile acid is chenodeoxycholic acid (3α,7α-dihydroxylated). Expectedly, the absence of a functional CYP8B1 results in the absence of cholic acid. In naked mole rats the CYP8B1 gene is deleted, whereas in elephants, the CYP8B1 gene has accumulated multiple inactivating mutations. 50 Shinde and coworkers 51 screened the CYP8B1 gene sequence of more than 200 species to identify the signatures of relaxation of selection. The authors concluded that the loss of CYP8B1 gene in certain species indicates recurrent changes in the selection landscape that is associated with changes in dietary lipid content.
Drug metabolism phenotypes—polymorphism of one enzyme causing differential metabolism of substrates vs overlapping substrate specificity of multiple enzymes
A single amino acid substitution (S180C; Ser at position 180 replaced by Cys) determines the polymorphism of the male rat-specific cytochrome P4502C13 (CYP2C13) by altering protein stability. 52 This polymorphism results in an increase in half-life of the CYP2C13 protein from 53 min to 156 min and a 20–40-fold difference in the hepatic content of CYP2C13 between the high and the low expressor phenotypes. Rat CYP2C13 is an ortholog of human CYP2C19. 53 Hence, the use of the S180 C variant of rat CYP2C13 as a surrogate for human CYP2C19 to study the metabolism of various CYP2C19 substrates may lead to an incorrect conclusion about the kinetics of metabolism.
CYP2D6 is the most well studied polymorphic CYP enzyme in humans. Over 130 different CYP2D6 star alleles have been cataloged. 54 Individuals can be classified as ultrarapid metabolizers (UM), extensive metabolizers (EM), intermediate metabolizers (IM), and poor metabolizers (PM) of CYP2D6 substrate drugs. Depending on the metabolizer phenotype of the individual, which is reflected in the plasma concentration of the CYP2D6 substrate drugs, the dose of the drugs may need to be adjusted. The UM phenotype is caused by the presence of multiple copies of the CYP2D6 gene in the individual, such as among many individuals from Ethiopia and Saudi Arabia.12,13 The allele is termed CYP2D6*2XN (N= 2, 3, 4, 5 or 13). EMs possess at least one fully functional CYP2D6 allele and are phenotypically normal. IMs have two reduced function or one reduced and one non-functional allele, and PMs have two non-functional alleles. Many different mutations cause the PM phenotype. Two well-known examples are CYP2D6*4 (G1934A) and CYP2D6*10 (C188T). The G1934A mutation is in the splice acceptor site of intron 3 of the CYP2D6 gene, and it results in a splicing defect producing a defective enzyme, whereas the C188T mutation produces an unstable enzyme.55,56 For some populations, the frequency of these mutations is high, but for other populations it is low. For example, the frequency of CYP2D6*4 (G1934A) is high among the Swedes (22%) but low among Chinese (≤1%); in contrast, the frequency of CYP2D6*10 (C188T) is high among Chinese (50%) but low among Caucasians. 55 These examples demonstrate the importance of a crucial single base mutation in determining the drug metabolizing phenotype. Two other highly polymorphic and well-studied CYP enzymes in humans are CYP2C9 and CYP2C19.
In contrast to the polymorphism of one enzyme causing differential metabolism of substrates, more than one drug metabolizing enzyme (DME) may metabolize a specific drug resulting in an overlapping substrate specificity for some DMEs. This includes many drugs that humans are exposed to. Thus, drugs may be metabolized by only one DME (e.g., metoprolol by CYP2D6) or by multiple DMEs (e.g., warfarin [Coumadin] by CYP1A2, CYP2D6, and CYP3A4). 57 When a DME catalyzes a unique reaction that is not catalyzed by any other DME, that reaction can be used as a probe for that specific DME. In rats, phenacetin-O-deethylation and 7-ethoxyresorufin-O-deethylation are catalyzed by both CYP1A2 and CYP2C6; benzyloxyresorufin-O-dealkylation and pentoxyresorufin-O-dealkylation are catalyzed by CYP1A2 and CYP2B1; bufuralol 1′-hydroxylation is catalyzed by CYP2D2, CYP2C6 and CYP2C11; p-nitrophenol-2-hydroxylation and chlorzoxazone-6-hydroxylation are catalyzed by CYP2E1, CYP1A2, and CYP3A1. 58
Normally, the DME with the most favorable kinetics of metabolism for a substrate (ideally low Km and high Vmax) preferentially metabolizes the substrate but if this enzyme is non-functional, other enzymes can take over the metabolism of the same substrate. Such examples demonstrate the existence of a necessary redundancy of metabolic pathways/mechanisms in living systems to deal with the exposure to a diverse array of environmental chemicals, and to compensate for the loss of activity of one or more DMEs.
Heterogeneity of human milk oligosaccharides
Another example of redundancy and heterogeneity is provided by the human milk oligosaccharides (HMOs). HMOs are indigestible carbohydrates that are thought to play a role in the blocking and sequestering of various pathogens and toxins by mimicking cell surface entry receptors in the infant gut. 59 There is also evidence to suggest that HMOs play a role in supporting the developing mucosal and systemic immune system. 59 At present, about 200 molecular species of HMOs have been identified in human milk, and there is a great degree of biological variation in specific HMO content in mothers’ milk. In addition to environmental factors such as diet, mothers’ genetics including the functional status of the FUT2 and FUT3 genes play an important role in determining the composition and variation of the HMO content in the milk. 60 Mutations in the secretor gene (FUT2) encoding fucosyltransferase-2 (FUT2), and the Lewis gene (FUT3) encoding fucosyltransferase-3 (FUT3) contribute in a large part to the variations in the HMO composition. Mothers with an active FUT2 enzyme are secretors, whose milk contains high amounts of α1-2-fucosylated HMOs that are among the abundant HMOs in human milk. In contrast, mothers with an inactivated FUT2 are non-secretors and their milk has almost no α1-2-fucosylated HMOs.
Heterogeneity of protein glycosylation
A great example of macromolecular heterogeneity in living systems is protein glycosylation. Glycosylation is an important post-translational modification of proteins. Protein glycosylation can be N-linked glycosylation (on Asn) and O-linked glycosylation (mostly on Ser and Thr); the N-linked glycosylation is the most prevalent in human cells. 61
The pattern of glycosylation varies from tissue to tissue, and species to species due to the varying expression of hundreds of glycosyltransferases and glycosidases, thereby creating heterogeneity. Heterogeneity is an inherent feature of glycosylation and it often produces a variety of glycan structures on separate molecules of the same protein; these are known as glycoforms.
62
Two types of heterogeneity of glycosylated proteins are macroheterogeneity and microheterogeneity. Macroheterogeneity represents the structural diversity due to the presence or absence of glycans at specific glycosylation sites, which is caused by inefficiency in the initial transfer of glycans to proteins.
63
Microheterogeneity concerns the variations of glycan structure at a specific site (Figure 2). Heterogeneity of glycosylation arises because glycosylation is not a template-directed process.
64
Both macro- and micro-heterogeneity can influence the physical and biochemical properties of a protein, such as protein folding, stability, protein–protein interactions, and cell–cell recognition.
64
The existence of macro- and microheterogeneity of glycosylation of the same protein molecules demonstrates that glycosylation cannot be used as a part of the molecular identity of glycoproteins; the molecular identity of proteins should be determined by the primary structure of the protein, that is, the amino acid sequence. Macroheterogeneity and microheterogeneity in protein glycosylation. The figure shows macroheterogeneity with the missing glycan chain on one Asn (N) residue. Microheterogeneity is demonstrated by variations in glycan structure; the middle glycan chain has two residues that are different from the corresponding positions of the other glycan chains, as well as two additional residues that are not present in other glycan chains.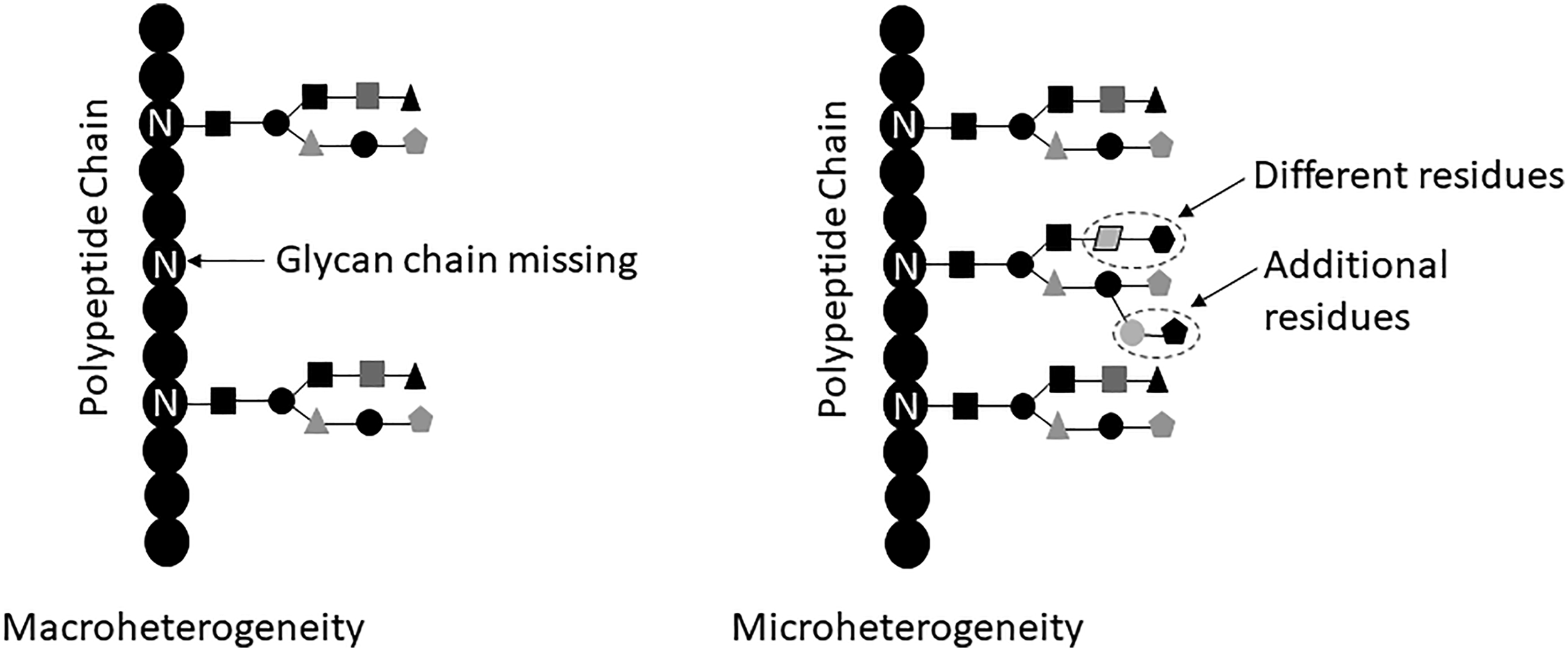
Glycoproteins are critical for normal cell–cell interactions that includes leukocyte trafficking. During cellular trafficking to the site of infection and inflammation, cells roll on the vascular endothelium to reach their destination. Such rolling on the vascular endothelium is facilitated by the action of the selectins (glycoprotein adhesion molecules) and other glycoproteins on the vascular endothelium. Expectedly, glycosylation can also promote cancer cell metastasis. 61 Glycosylation modulates inflammatory response, enables viral immune escape, and regulates apoptosis. Genetic defects in glycosylation are often embryo lethal. 65 Innate immune response relies on identifying the pathogen-associated molecular pattern (PAMPs). Many PAMPs are glycan-containing molecules, such as lipopolysaccharides, lipoteichoic acid, mannose-rich glycans, and many other molecules. Glycans are important in the adaptive immune response as well. Many cell surface proteins involved in the adaptive immune response are also glycoproteins, such as CD43, CD45, galectin-1, glycoproteins associated with T- and B-cell receptors, and major histocompatibility molecules.61,65 Changes in the complexity of N- and O-glycans have been associated with several human diseases. 61
Conclusion
The ability to investigate biological macromolecules at an increasingly finer level has uncovered a wealth of information on several aspects of life, such as the extent of macromolecular diversity, inter-individual variability, and biological heterogeneity that exists in all living systems. This is helping us rethink and revise many traditional concepts and definitions. For example, it is now becoming clear that except for situations like the primary structure of proteins (amino acid sequence), the identity of complex/conjugated macromolecules may not be as static as once thought.
The gene-centric view of biological heterogeneity is further complicated by epigenetic modifications of the genome. Epigenetic modifications of the genome begin right after birth and is shaped by the immediate environment that includes parental care. Epigenetic modifications, particularly DNA methylation, increases as a function of age but global and site-specific DNA methylations of the genome differ amongst individuals. 66
Robustness in evolvable biological systems is conferred by functional redundancy that is often created by macromolecular diversity and biological heterogeneity. For example, recent evidence shows that bile acids are involved in the regulation of various metabolic processes, such as triglyceride, cholesterol, glucose, and energy homeostasis. Consequently, bile acid homeostasis is of crucial importance and is maintained by the participation of multiple uptake transporters in the liver and intestine, such as sodium (Na+)-taurocholate cotransporting polypeptide, as well as multiple organic anion transporting polypeptides (OATPs), and apical sodium-dependent bile acid transporter. 67 The multiplicity of bile acid uptake transporters ensures that >90% bile acids absorbed from the intestine and carried through enterohepatic circulation are transported back into the hepatocytes. Multiple OATPs can transport bile acids in humans; however, evidence for human OATP polymorphism affecting endogenous bile acid uptake is sparse, 68 and this view is consistent with the presence of redundancy of hepatic bile acid uptake systems.
Macromolecular diversity and biological heterogeneity in biological systems provide the raw materials for microevolutionary changes and inter-individual variability, and it ultimately reflects changes in DNA sequence and allele frequency that occurs over time in a population. As Sir William Osler observed in 1892 “If it were not for the great variability among individuals, medicine might as well be a science and not an art.” 69
Footnotes
Author Contributions
The opinions expressed in this article are the author’s personal opinions and they do not necessarily reflect that of FDA, DHHS or the Federal Government.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.