
Abstract
A broad spectrum of chemical entities have been associated with drug-induced seizure (DIS), emphasizing the importance of this potential liability across various drug classes (e.g., antidepressants, antipsychotics, antibiotics, and analgesics among others). Despite its importance within drug safety testing, an understanding of the molecular mechanisms associated with DIS is often lacking. The etiology of DIS is understood to be a result of either a deficit in inhibitory (e.g., gamma aminobutyric acid) or an elevated excitatory (e.g., glutamate) signaling, leading to synchronous neuronal depolarization affecting various brain regions and impairing normal neurological functions. How this altered neuronal signaling occurs and how these changes interact with other non-brain receptor driven DIS-associated changes such as metabolic disturbances, electrolyte imbalances, altered drug metabolism, and withdrawal effects are poorly understood. Herein, we discuss important molecular mechanisms identified in DIS for several drugs and/or drug classes. With a better understanding of the molecular mechanisms associated with DIS, in vivo or in vitro models may be applied to characterize and mitigate DIS risk during drug development. Susceptibility stratification for DIS presents species differences in the following order beagle dogs > rodents and cynomolgus monkeys > Göttingen minipigs with a more than 2-fold difference between canines and minipigs, which is important to consider during non-clinical species selection. While clinical signs such as myoclonus, severe muscle jerks, or convulsions are often associated with abnormal epileptiform EEG activity, tremors are most of the time physiological and rarely observed with concurrent epileptiform EEG activity which need to be considered during DIS risk evaluation.
Introduction
Drug attrition rates for central nervous system (CNS) diseases are high when compared to other indications.1-4 In an attempt to better detect and ultimately mitigate adverse drug reactions (ADR), the International Conference on Harmonisation (ICH) S7A guidelines 5 were adopted which provided a framework under which drug safety pharmacology evaluations are to be conducted. Since the implementation of the ICH S7A guidelines (ca. 2001), no marketed pharmaceutical has been withdrawn due to seizurogenic potential and only one marketed pharmaceutical withdrawal has occurred as a result of a central nervous system (CNS) liability (i.e., Rimonabant for neuropsychiatric adverse events including suicide and depression). 6 However, drug attrition rates remain high during premarket CNS drug development with the likelihood of approval for neurological indications from Phase 1 trials onward at approximately 10%.4,7 This is due, in large part, to CNS ADR which include, among others, psychiatric (e.g., fatigue, depression, and suicidal ideation) and nervous system (e.g., neuroleptic malignant syndrome) disorders, neurotoxicity, neuropathies, and neurological impairment/dysfunction (e.g., seizures, learning and memory deficits).3,8,9
Drug-Induced Seizures
Drug-induced seizure (DIS) is a relatively well characterized adverse effect which can be observed in non-clinical studies but also in the clinical setting.1,10 DIS are seizures precipitated by the pharmacological and/or toxicological effects of a given chemical entity following acute or chronic exposure or withdrawal. Pharmaceutical agents targeting CNS disorders show the highest propensity for DIS given the high blood brain barrier permeability. However, many pharmacological classes and therapeutic areas (e.g., antibacterial, antivirals, cardiovascular agents, immunosuppressives, and NSAIDs) are known to possess, or be susceptible to, seizure liability risks11,12 highlighting that seizure risk is not restricted to CNS-targeted therapies.
Retrospective analyses highlight that the incidence of DIS within the clinical population is 0.8% to 1.7%13-16 and is characterized by unintentional (e.g., unforeseen drug or drug-interaction effects, inadvertent overdose) and intentional (e.g., recreational or deliberate suicidal overdose of stimulants and/or narcotics)17,18 risks. Phenotypically, DIS frequently present as generalized tonic–clonic seizures although partial seizures and convulsive and non-convulsive seizures or status epilepticus have also been reported.14,19 Drug-induced seizure (of any phenotype) may occur within patients with no prior history or risk of seizures and should be differentiated from epileptic seizures which result from a genetic and/or non-genetic neurological disorder. Nevertheless, there is a lack of discernable EEG characteristics which differentiate DIS from epileptic seizures in non-clinical models that are available early in drug development. A greater understanding of the pathophysiology of DIS is essential for the development and risk assessment of the next generation of pharmaceuticals.
The pathophysiology of DIS is broadly characterized by patient- and drug-related factors. 20 Patient-related factors include age, hepatic or renal function, genetic susceptibility to seizure, and/or the seizurogenic potential of drugs. Likewise, drug-related factors associated with DIS include blood brain barrier penetration, on target and off-target pharmacological activity on brain receptors/proteins, drug class, dose level, route of administration, and lipid solubility, among others. Drug-related factors are of particular interest during drug development, since early attrition is preferable over late-phase attrition. Drug attrition rates related to neurological safety evaluations are relatively low preclinically 3 and seizure liability risks may be investigated in early drug development. 21
The etiology of DIS is largely understood to be a result of either a deficit in inhibitory (e.g., gamma aminobutyric acid ((GABA)) or elevation in excitatory (e.g., glutamate) signaling. How this altered neuronal signaling occurs and how these changes interact with other non-brain receptor driven DIS-associated changes such as metabolic failure, electrolyte disturbances, altered drug metabolism and withdrawal effects, as applicable, remain poorly described for most drugs. Accordingly, we aim to highlight molecular mechanisms involved in DIS of several known drugs and/or drug classes. Interestingly, the timing of seizures can help to identify the putative mechanism of action underlying the DIS. For instance, DIS due to acute disruption to synaptic neurotransmission or ion channel function typically occur when the concentration of the pharmacological compound is highest (Tmax), as is the case with clozapine.22,23 Conversely, DIS originating from toxicity to neurons and cellular constituents can present delayed onset and is less predictable.24,25
Altered Ion Channel Function/Neurotransmission
As there is no standard study design or guideline for evaluating preclinical seizure liability, a science-driven CNS safety testing strategy is typically used and comprehensive mechanistic investigations for potentially altered ion channel functions or neurotransmitter interferences are often considered a starting point in DIS investigations. The mechanisms underlying the origin of seizures may involve the regulation of neuron excitability as seen in some forms of epilepsy. 26 These can be broadly described as defects with either synaptic signaling 27 or intrinsic excitability.28,29 At the synaptic level, changes to excitatory glutamatergic (e.g. α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/AMPA, N-Methyl-D-aspartate/NMDA, and kainate receptor) or inhibitory (e.g. GABA or glycine) synaptic signaling underlies the most frequent mechanistic targets for the origins of seizures. 27 Additionally, intrinsic excitability involves modulation of the functional properties of the voltage gated sodium (Na+), potassium (K+), and calcium (Ca2+) ion channels which are critical for action potential generation and propagation. 30 Thus, there is a large list of potential molecular targets which have been implicated in DIS and may be screened for the development of safe therapeutics.
One of the main causes of DIS is a reduction in the efficacy of inhibitory GABA signaling which can be associated with limiting presynaptic GABA neurotransmitter release or reducing postsynaptic GABA receptor function or expression. GABAergic neurotransmission involves the activation of inhibitory postsynaptic GABA receptors which are classified into two families based on their functional properties: ionotropic GABAA and metabotropic GABAB receptors. 31 Adding further diversity to GABAergic signaling is that GABAA receptors have two distinct pharmacologically relevant sites; the GABA binding site and the benzodiazepine binding site. 32 The GABAA receptor benzodiazepine binding site is only found on a subset of GABAA receptors expressed regionally throughout the CNS. 33 Antagonism of GABAA receptors has been reported as the cause of antibiotic induced seizures 34 as seen with the β-lactam family consisting of penicillin G, 35 cephalosporins, 36 and carbapenems.37,38 Meanwhile, fluoroquinolones are known to cause seizures and likely do so through antagonizing GABAB receptors and potentiating excitatory NMDA receptor currents.39,40 Other mechanisms antagonizing GABAA receptors and causing seizures include blocking the GABAA receptor pore (picrotoxin), 41 blocking the benzodiazepine binding site (flumazenil), 42 or acting as GABAA receptor negative allosteric modulators (bicuculline and gabazine). 43 Notably, drugs blocking the benzodiazepine site are challenging clinically as benzodiazepine administration is the preferred antiseizure treatment. 44 Finally, DIS can be elicited by synthetic opioids such has meperidine which cause a reduction in the synaptic release of GABA. 45
Potentiating the signaling of excitatory neurotransmission is another well-known cause of DIS. In non-clinical settings, direct activation of glutamatergic signaling by either kainic acid 46 or NMDA 47 can cause seizure activity. Clinically, clozapine is a widely prescribed antipsychotic medication used in the treatment of schizophrenia which exhibits strong relationship between serum concentration levels and seizure development risk.22,23 Clozapine increases neuronal activity by promoting the glial release of D-serine, a co-agonist of excitatory NMDA receptors. 48 Similarly, opioid metabolites such as morphine-3-glucuronide have proconvulsant activity by acting indirectly to activate NMDA receptors. 49 In some cases, individual drugs can act on multiple ion channels concurrently to cause the onset of seizures through both short- and long-term effects. For example, many of the physiological effects of alcohol are due to acute and long-term effects on both GABA and NMDA receptors. 50 While acute alcohol ingestion inhibits NMDA receptor function, 50 chronic alcohol exposure increases the expression of NMDA receptors throughout the brain.51,52 Moreover, GABAA receptor expression is changed during chronic alcohol consumption with a reduction of synaptic α1 containing GABA receptors (the principal synaptic GABA receptor) and trafficking of α4 containing receptors out of the extrasynaptic space. 53 Ultimately, withdrawal from chronic alcohol exposure induces seizures due to circuit hyperexcitability through increased NMDA receptor expression and altered signaling properties of GABA-associated inhibition. 54
DIS can also arise from hyperexcitability due to changes in the properties of voltage gated ion channels. Unsurprisingly, broad spectrum K+ blockers such as 4-aminopyridine (4-AP), which is used to treat multiple sclerosis, must be used with caution since, although it is generally well-tolerated, 55 high concentrations have been known to cause seizures. Moreover, the seizurogenic properties of 4-AP are regularly used in fundamental research to induce seizure activity within animal models. Similarly, the local anesthetic lidocaine functions as a voltage gated Na+ channel blocker at lower concentrations (<10 μM) but, at high levels, is associated with increased seizure risk by selectively blocking inhibitory mechanisms.56-62 Monoethylglycinexilidide (MEGX) and 2,6-glycine xylidide (GX) downstream metabolites of lidocaine have also been reported to lower seizure threshold63,64 and potentiate lidocaine-induced seizures. Specifically, lidocaine has been shown to inhibit both the inhibitory action of cerebellar neurons on the vestibular nuclei, 65 as well as the L-glutamate-induced transcallosal inhibition of contralateral neurons in a GABA-independent manner. 66 Warnick et al. 66 (1971) also proposed that the proconvulsant effects of lidocaine is likely not via synaptic action, but may be due to its central local anesthetic action on the inhibitory fibers that are involved in direct cortical inhibition. Recent evidence suggests that the seizure inducing properties of lidocaine have been reported to be in part due to a dose dependent block of two-pore domain K+ channels (K2P) (IC50 207 μM) 67 but the concentrations that are used clinically are lower and this would require dose levels in the toxicology range. Other local anesthetics such as bupivacaine, levobupivacaine, and ropivacaine have also been observed to inhibit K2P channels in a similar dose dependent manner and are known to possess seizurogenic properties. 68
Neuronal Death And/or Injury
A large body of evidence supports the notion that spontaneous seizures can be brought on by the death of vulnerable neurons in the hippocampus and amygdala, which can alter the balance of excitation/inhibition in the limbic network.24,25 Therapeutics which are commonly given at doses close to the toxic range, and/or have slow/impaired clearance, may cause higher rates of neuronal damage due to increased exposure as seen in several human case studies. For instance, many cancer therapies have been shown to induce seizures via neuronal damage, especially when given at high doses or in the presence of renal or hepatic disorders, where clearance of the drug is impaired.
Cisplatin, an effective and widely used antineoplastic agent in the treatment of solid tumors, has been demonstrated to induce encephalopathies and seizures in rare cases. 69 Cisplatin chemotherapy has been linked to the occurrences of two types of CNS disorder: posterior leukoencephalopathy syndrome, where patient exhibit abnormalities predominantly in the occipital lobes,70-74 and focal neurological deficits.75-78 It is proposed that these cisplatin-associated encephalopathies may be caused by vascular events that are generally not seen in cerebral imaging—a notion supported by studies where cisplatin was found to have high vascular toxicity, and was able to induce cerebrovascular ischemic events. 79 The mechanism of this vascular toxicity is still unknown, but may include cisplatin-induced hypomagnesemia or altered platelet aggregation. 69 In rare cases, oxaliplatin, which is an alkylating agent commonly used in the treatment of colorectal and gastric cancers, have also been shown to cause seizures either directly 80 or as a result of posterior reversible leukoencephalopathy syndrome (PRES), 81 possibly via vascular effects.82,83 In addition, cyclosporine, a commonly used immunosuppressive agent, can cause a form of posterior reversible encephalopathy syndrome, similar to those caused by cisplatin. 84 Seizures due to cyclosporine neurotoxicity have been proposed to result from a variety of causes such as endothelial damage and the release of endothelin-1, a potent vasoconstrictor,85-87 or a direct effect on neuronal excitability. 88
Similarly, intrathecal and intravenous treatment with methotrexate, an antimetabolite that is a competitive inhibitor of the enzyme dihydrofolate reductase, has also been shown to cause seizures and subcortical encephalopathy that may be ischemic in origin.89-92 Mineralizing microangiopathy were shown in pathological specimens obtained on autopsy of those who received methotrexate alone (along) or in combination with radiotherapy, which supports the theory that methotrexate-induced encephalopathy has a vascular origin. 92 As well, interferon-α treatment has been reported to induce seizures in 1–4% of patients.93-95 Its toxicity has been attributed to mechanisms such as the disruption of the blood brain barrier and subsequently inducing vasogenic brain edemas. 96
A list of known drugs with risk for the development of DIS and their targets.
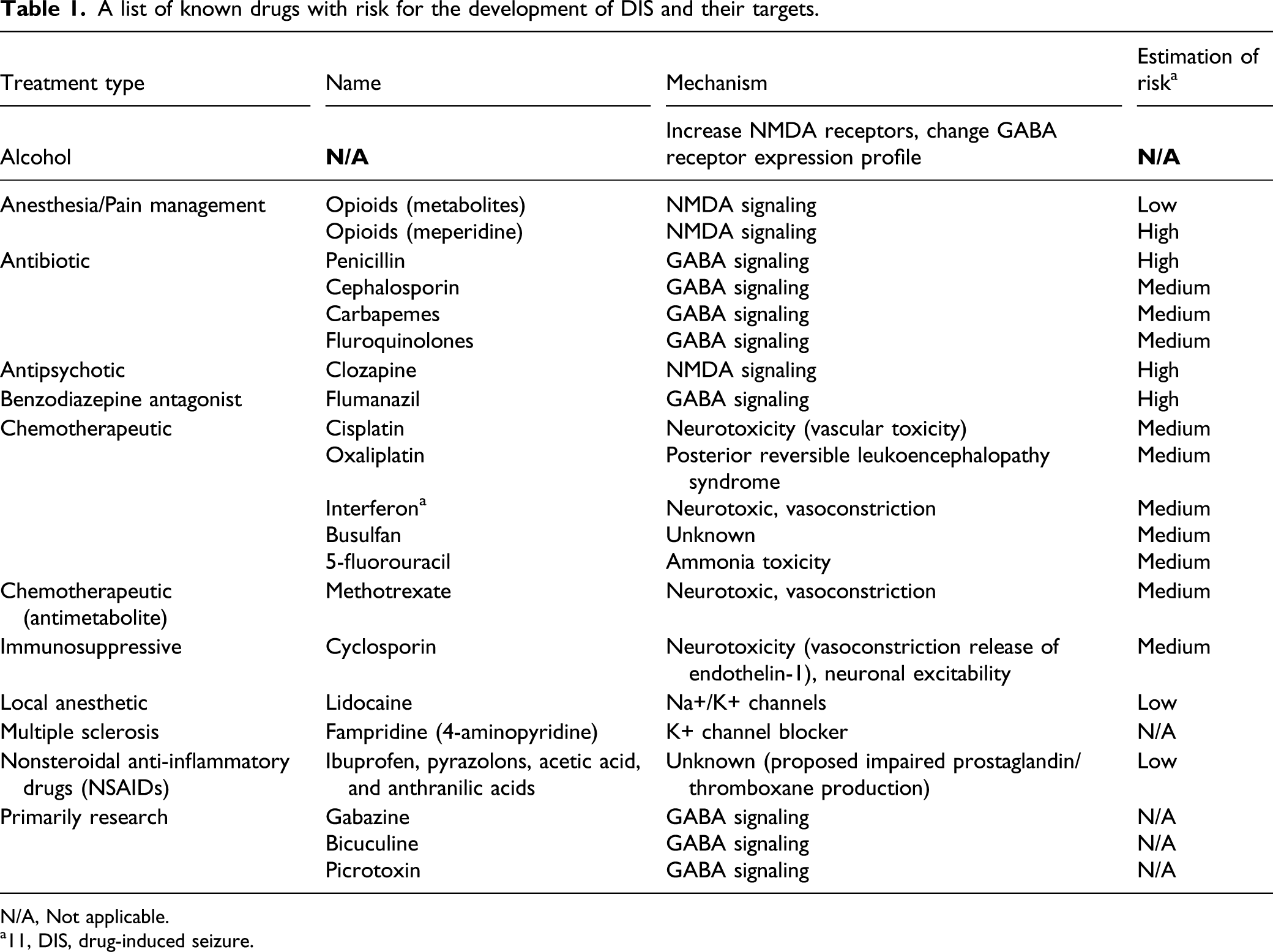
N/A, Not applicable.
a11, DIS, drug-induced seizure.
5-fluorouracil is an antimetabolite that is widely used as a chemotherapeutic agent in the treatment of various cancers. High-dose administration of 5-fluorouracil has been previously reported to cause encephalopathy—report, in some cases, to be up to 5.7% of patients—with severe symptoms of stupor, coma, and/or seizures concurrently with hyperammonaemia and lactic acidosis. 102 Although direct evidence is lacking, it is postulated that high 5-fluorouracil dosage may induce the accumulation of ammonia (an end product of 5-fluorouracil metabolism). This ammonia would be normally cleared by the ATP-dependent urea cycle, but fluroacetate, the intermediate product of 5-fluorouracil, is known to inhibit the ATP producing Kreb’s cycle and results in lactic acidosis and the impairment of the ATP-dependent urea cycle. 103 Accordingly, a transient hyperammonaemia state with encephalopathy may arise. 102 Additionally, 5-fluorouracil toxicity has been linked to an inhibited deficiency of the enzyme dihydropyrimidine dehydrogenase, due to genetic mutations in the protein of which at least 23 different polymorphisms have been discovered. 104 As this enzyme in involved in the breakdown of 5-fluorouracil, its deficiency can lead to toxic accumulations of the drug. In one report, a polymorphism of dihydropyrimidine dehydrogenase was associated with multiorgan failure, seizures, myoclonus, and encephalopathy after 5-fluorouracil treatment. 105
Other Mechanisms Associated With Drug-Induced Seizures
In rare cases, the ingestion of therapeutic doses of nonsteroidal anti-inflammatory drugs (NSAIDs) such as propionic acid derivatives (ibuprofen), pyrazolones (phenylbutazone), acetic acid derivatives (diclofenac and indomethacin), and anthranilic acids (mefenamic acid and meclofenamate) have been associated with seizures.106,107 While the exact mechanism by which NSAIDs induces seizures is unknown, the ability of NSAIDs to interfere and inhibit cerebral arachidonic acid metabolism—which lowers the seizure threshold—has been proposed as a possible explanation. 106 In support of this theory, changes in the levels of prostaglandin and/or thromboxane, both products of the arachidonic acid metabolism pathway, have been implicated in the appearance of convulsions and seizures.106,108 Neuronal prostaglandin, in particular, has been previously shown to be essential for seizure suppression and the regulation of sleep that follows seizures via the activation of prostaglandin D receptor (DP1R).109-111
Hypoglycemia was historically identified as a risk factor for seizures but more recent data suggests that seizures induced by hypoglycemia are rare. 112 Hyperthermia is known to cause febrile seizures in a proportion of the population 113 most likely by acting on NMDA and GABA related pathways. 114 Electrolyte disturbances such as hyponatremia, hypocalcemia, and hypomagnesemia are also well characterized changes associated with seizures and may be identified by clinical chemistry investigations when conducting non-clinical neurotoxicity studies. 115
Considerations Related to Drug Development and Seizure Risk Characterization
Once a seizure risk is identified often in toxicology or safety pharmacology studies, characterization of this liability may be an important part of the neurotoxicology investigation. The use of a range of non-clinical models can help predict safety margins of the new chemical entity (NCE) in humans and this integrated risk assessment is pivotal in drug development. Drugs with safety margins based on exposure ≥10X generally progress through development with minimal regulatory concerns since the dose dependence of seizurogenic effects is a widely accepted concept. Non-clinical animal models (i.e., not genetically modified) that are used to evaluate the potential for a NCE to cause seizurogenic activity differentiate between simple epileptiform activity and the spectrum of limited to severe DIS (unpublished data) but the severity observed in these non-clinical models may not translate to the DIS liability observed clinically. Sensitivity to seizurogenic effects differ between species in the following order beagle dogs > rodents and cynomolgus monkeys > Göttingen minipigs with a more than 2-fold difference between canines and minipigs.11,21,116,117 Understanding the clinical signs that may precede seizures can help develop a monitoring strategy for clinical trials. While premonitory clinical signs often differ between drugs, the clinical sign profile (types of clinical signs and time/exposure from clinical sign onset to seizure) tends to be similar for a given NCE with common trends also observed between species. 118 Abnormal EEG morphologies (e.g., interictal epileptiform activity) are often observed in the absence of concurrent clinical signs but most animals presenting drug-induced interictal activity will also present typical clinical signs and epileptiform EEG morphologies, simultaneously. The seizure risk associated with specific clinical signs such as myoclonus, myoclonic jerks, and tremors also differs greatly. Myoclonus and myoclonic jerks in conscious/awake animals are frequently associated with abnormal epileptiform EEG activity during the conduct of non-clinical neurotoxicology studies. 118 In contrast, myoclonus or myoclonic jerks during sleep (especially during sleep onset) are often physiological and are typically not associated with abnormal EEG activity. 119 Low amplitude tremors can be observed in normal healthy animals and are not associated with abnormal EEG activity in most cases. It remains that tremors may be associated with drug-induced abnormal epileptiform EEG activity in some cases and video-EEG recorded by telemetry can be used to characterize background CNS clinical signs from premonitory changes. 118 A wide range of DIS assays can be considered from early screening panels 120 to ex-vivo electrophysiology 121 and ultimately EEG models sometimes with seizure threshold tests.118,122 Some programs will only require minimal DIS investigations with a single assay while others will require a more comprehensive characterization of this potential liability based on a perceived higher risk or regulatory interactions.
Conclusions
Neurotransmitter mediated effects, ion channel alterations, cell damages and necrosis are only a few of the potential mechanisms involved with DIS (Figure 1). While characterization of molecular mechanisms involved with DIS can help during risk assessment and can further define translational considerations, the complexity of the neurotoxicity underlying DIS often precludes from clearly defining the pathway(s) involved with DIS. Although a perturbation of the excitation-inhibition balance is a commonality across several mechanisms, a definitive understanding of the root causes of DIS is often difficult to ascertain due to the complex nature of the neuronal network functions. In vitro electrophysiological investigations may provide an early stage screen for DIS risk owing to their ability to help define these alterations in the neuronal excitation-inhibition balance. However, these early stage in vitro assessments possess technical limitations which typically need to be complimented with in vivo investigations to characterize the DIS risk. For instance, long-term in vivo studies employing EEG telemetry may be required to identify potential seizurogenic liabilities resulting from prolonged drug exposure that cannot be plausibly assessed through acute in vitro exposures or via monosynaptic assessments of in vitro drug effects. Likewise, DIS caused by long-term changes to brain function, neuronal injury or cell death may also necessitate extensive toxicology studies highlighting the need for a wide array of both in vitro and in vivo seizurogenic risk assessments. In many cases, defining safety margins, precursor clinical and EEG signs, reversibility, dose dependence, and the nature of the neurological effects is adequate to complete an acceptable safety assessment. Several approved drugs present a DIS liability and a range of testing strategies can be considered to conceive an integrated risk assessment plan adapted to the specifics of the program. Drug-induced seizure investigations, like most other safety testing areas, benefits from scientifically driven study designs and multi-disciplinary analyses. An illustration highlighting the mechanisms of drug-induced seizure. (A) Impacts to metabolic pathways. (B) Changes to synaptic neurotransmitter release. (C) Cell death due to neurotoxicity. (D), (E). Altered function/expression of synaptic receptor proteins. (F) Altered function/expression of voltage gated ion channels.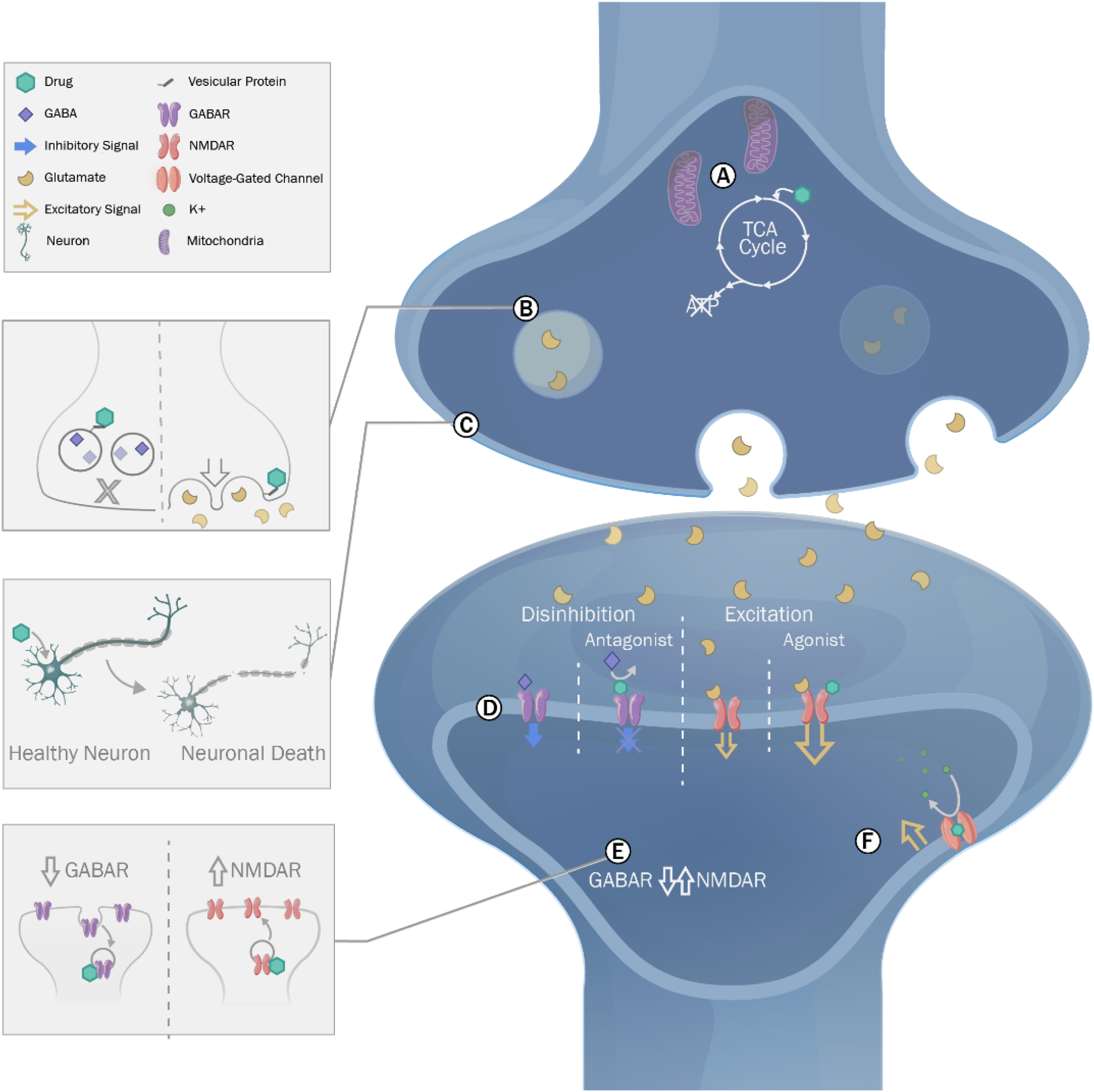
Footnotes
Declaration of Conflicting of Interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.