
Abstract
Palmitoylethanolamide (PEA) is an endogenous ethanolamine playing a protective and homeodynamic role in animals and plants. Prenatal developmental toxicity of PEA was tested following oral administration to pregnant female Wistar rats, from days 0 to 19 of gestation, at dosage of 250, 500, or 1,000 mg/kg body weight, according to Organisation for Economic Co-operation and Development Test Guideline No. 414. On gestation day 20, cesarean sections were performed on the dams, followed by examination of their ovaries and uterine contents. The fetuses were further examined for external, visceral, and skeletal abnormalities. Palmitoylethanolamide did not cause any alterations at any of the given dosages in the measured maternal parameters of systemic toxicity (body weight, food consumption, survival, thyroid functions, organ weight, histopathology), reproductive toxicity (preimplantation and postimplantation losses, uterus weight, number of live/dead implants and early/late resorptions, litter size and weights, number of fetuses, their sex ratio), and fetal external, visceral, or skeletal observations. Any alterations that were recorded were “normal variations” or “minor anomalies,” which were unrelated to treatment with PEA. Under the condition of this prenatal study, the no-observed-adverse-effect level of PEA for maternal toxicity, embryotoxicity, fetotoxicity, and teratogenicity in rats was found to be >1,000 mg/kg body weight/d. It indicates that PEA is well tolerated by and is safe to pregnant rats even at a high dose of 1,000 mg/kg body weight/d, equivalent to a human dose of greater than 9.7 g/d. This prenatal developmental toxicity study contributes greatly in building a robust safety profile for PEA.
Introduction
Herbal and naturally occurring endogenous derivatives as supplements are becoming a significant interest in global health debates. Therefore, world’s leading authorities such as US Food and Drug Administration (FDA) and World Health Organization have made substantial research and awareness investments in natural and botanical supplements for better quality of life. 1 One of the most crucial aspects of such research is to unveil the overall toxicity, if any, and form a robust safety profile of these naturally occurring products in their standardized form for potential use.
Palmitoylethanolamide (PEA) is an endogenous fatty acid derivative present nearly in every tissue of the human body. 2 It was identified more than 5 decades ago in egg yolk and subsequently in soy lecithin and peanut oil. 2,3 Therefore, it is an integral part of a basic balanced diet. It is a naturally occurring N-acyl ethanolamine, playing protective and homeodynamic roles in animal and plant kingdoms. Palmitic acid (C16:0) is the most common fatty acid in animals and is a precursor for the synthesis of PEA. Synthesis of PEA takes place in membranes of various cell types, is produced on demand, and acts locally. Primarily, it was shown to reduce allergic reactions and inflammation in animals and relieve influenza symptoms in humans. Recent interest in PEA has revealed a plethora of its properties, including analgesia, inhibition of peripheral inflammation and mast cell degranulation, anxiety reduction, relaxation as well as neuroprotective, nootropic, and antinociceptive effects in both animals and humans. 4,5 These are the results of over 50 years of research and more than 350 papers compiled in PubMed. Starting with 6 clinical trials focusing on PEA as a therapy against cold and influenza during 1960s, the major focus of PEA research has shifted to neuropathic pain states and mast cell–related disorders. The use of PEA on animal models of chronic inflammation and neuropathic pain indicated that PEA might contribute to the reduction in the recruitment and activation of mast cells, known as the origination of pro-inflammatory mediators, and endoneurial edema, therefore aid in pain and inflammation reduction. Additionally, it preserves the peripheral nerve morphology. 6 The most recent clinical studies have demonstrated PEA to be a promising analgesic and effective in treating neuralgia, 7 -9 carpal tunnel syndrome, 10,11 postoperative pain, 12 endometriosis, 13 osteoarthritis, anxiety, sleep, 5 autism spectrum disorder, 14,15 and cognition. 14 It has also been helpful in restoring gut microbiota changes in gut–brain axis 13 and as an adjunct therapy in Parkinson disease. 16 Moreover, there has been evidence that PEA has synergistic or additive effect in human chronic pain upon combining with subtherapeutic doses of carbamazepine, pregabalin, and oxycodone treatment. 5,17 Aside from mild side effects and non–dose-related incidents, no studies thus far have reported any adverse effects of PEA in human or various animal models, including rodents, dogs, and humans. 5,18 Since 2008, PEA is marketed as dietary supplements all over the Western world.
The most specific data pertaining to the safety of PEA are reported in a review by Nestmann. 19 Here, genetic and mammalian toxicology of a micronized PEA formulation was evaluated through Good Laboratory Practice (GLP) compliant toxicological investigations, which included the Ames test, in vitro mammalian cell micronucleus test, the Acute Oral Up-and-Down Procedure, a 14-day toxicity study, and a 90-day toxicity study. The results revealed no evidences of mutagenicity, cytotoxicity, or genotoxicity. Additionally, results of the Acute Oral Up-and-Down Procedure found the acute oral LD50 in rats to be >2,000 mg/kg body weight. Furthermore, the 14-day (acute) and 90-day (subchronic) oral toxicity studies conducted on rats revealed no treatment-related adverse events and determined the no-observed-adverse-effect level (NOAEL) of microPEA on rats was 1,000 mg/kg body weight /d, the highest dose tested. 19 The results reported by Nestmann are in line with those reported (unpublished) in a review by Masek. 20 An acute toxicity study was performed on 30 albino mice, where PEA dissolved in 5% Tween-80 was administered orally in doses of 1, 2, or 4 g/kg body weight. There were no deaths reported during the observation period of 7 days. The second study investigated chronic toxicity, on male mice. Palmitoylethanolamide was administered orally each day in doses of 100 and 500 mg/kg body weight for 6 months. Following interim and terminal sacrifice, histological examination of the liver, kidneys, spleen, lymph nodes, and lungs revealed no adverse occurrences due to treatment with PEA compared to the control. The third reported study was a 6-month chronic toxicity study in rats where PEA was administered in doses of 100 and 500 mg/kg body weight. Once again, histological examination showed no difference in the adverse events observed in treated groups and the control group. The fourth study investigated chronic toxicity of PEA in dogs, with PEA administered orally in doses of 100 or 500 mg/kg body weight daily for 2 months. However, there was an inability to draw sound conclusions from this study due to problems in study design: low sample size and background disease. Finally, an embryotoxicity study was conducted on pregnant female rats where PEA was administered at a dose of 50 mg/kg body weight for 12 days. This study revealed no teratogenic or embryotoxic effects. 20 Therefore, both reviews convey that microPEA lacks genotoxic potential and that even testing with high doses of PEA does not cause adverse events.
However, a niche exists for comprehensive research on assessment of safety of PEA in animal models and humans, especially with regard to the prenatal developmental toxicity. In the light of this fact, our current study evaluates the effect of PEA, when daily administered to pregnant female rats by oral gavage at different dose levels from the beginning of their gestation period (gestation day 0) until 1 day prior to their expected day of parturition (gestation day 20). This GLP compliant study was aimed to build a more robust safety profile of PEA encompassing information on the potential hazards likely to be caused to the unborn rats, which may arise from exposure of the mother during pregnancy.
Methodology
Animals
All the procedures in this study was in compliance with the “Principles of GLP” enforced by Organisation for Economic Co-operation and Development (OECD) and in accordance with the conditions recommended by the “Committee for the Purpose of Control and Supervision of Experiments on Animals Guidelines for Laboratory Animal Facility” (Gazette of India, 1998) and “Guide for the Care and Use of Laboratory Animals” (National Academy Press, Washington, 2011). The study protocol was in accordance with INTOX (INTOX Pvt Ltd, Pune, India) Study Plan No. P/18069/PDTS-R, approved mutually by the sponsor, Gencor Pacific Limited (Hong Kong). Healthy, nulliparous, and nonpregnant female Wistar rats (age 12-14 weeks) were procured from Charles River Laboratories Inc (USA). Thirty-five male Wistar rats, age >16 weeks and proven to be fertile, were procured from a breeding colony of Wistar rats in INTOX. At least 7 days after acclimation in the animal house of INTOX (in groups of 3 per sex), on each day, upon requirement, 30 to 40 female rats were cohabited with male rats (male:female ratio of 1:2) overnight. Finding of spermatozoa in vaginal smears of the female rats was considered day “0” of successful gestation, and thereafter the pregnant female rats were housed individually. They were housed in solid bottom polypropylene cages with stainless steel grill tops, with facilities for food and water. The bedding was made of clean and sterilized paddy husk. The cages were suspended on stainless steel racks. The rats were housed in air-conditioned rooms with 10 to 15 air changes per hour, between 19 °C to 25 °C, humidity of 30% to 70%, and illumination cycle set to 12 hours of light and dark, respectively. “Altromin” brand pellet rat feed (M/s Altromin Spezialfutter GmbH & Co. KG) and potable filtered water were provided ad libitum. The successful “0” day pregnant rats were randomly assigned to the control and treatment group following mating.
Chemicals
White crystalline powder of the test item, PEA with the active ingredient to be >99% (wt/wt), was characterized by and supplied by sponsor (Gencor Pacific Organics India Pvt Ltd). The test item was then characterized by the test facility of INTOX. As PEA has been reported and verified to be insoluble in water, it was thus formulated as an aqueous suspension using Tween-80 (employed at about 0.2% [wt/vol]; Sigma Aldrich, batch number BCBG4547) as a wetting agent. Three dosing formulations were prepared with test item concentrations of 50, 100, or 200 mg/mL for the dose levels of 250, 500, and 1,000 mg/kg body weight/d, respectively. Samples of the dosing formulation of PEA were prepared freshly and were subjected to verification of concentration via chromatography for their PEA content twice, in the first and last weeks of dosing. On both occasions, the formulation was found to be adequately stable with respect to content of PEA (>99% [wt/wt]).
Method
The study plan followed the recommendations made in OECD Guideline for the Testing of Chemicals No. 414, “Prenatal Developmental Toxicity Study,” adopted on June 25, 2018. It was also in compliance with the principles of GLP as set forth in OECD, 1998: OECD Series on Principles of GLP and Compliance Monitoring, Number 1, “OECD Principles on GLP” ENV/MC/CHEM (98) 17. The doses for the main study were determined on the basis of a dose-range finding (DRF) study conducted at INTOX Pvt Ltd on pregnant rats.
Dose-Range Finding Study
Male and female Wistar rats were cohabited overnight in 1:2 ratio, and in the following mornings, all females under mating were examined for the presence of spermatozoa in their vaginal smear. Upon successful detection of sperm-positive female rats, it was considered to be their day “0” of gestation. Groups of ten “0-day pregnant” female rats (targeted to yield a minimum of 7 pregnant rats per group at the end of gestation period) were randomized into 4 different groups. The treatment groups were administered with test item, PEA, by oral gavage, daily from day 0 of pregnancy to day 19, at the doses of 250 (low-dose), 500 (mid-dose), or 1,000 mg/kg (high-dose) body weight. Female rats in the control group were administered with vehicle only, that is, analytical grade water with Tween-80 (0.2% wt/vol), also by oral gavage daily in volumes of 5 mL/g. From gestation days 0 to 19, the dams were observed for systemic toxicity signs along with monitoring of their body weight and food consumption. On gestation day 20, the dams were sacrificed and the fetuses were removed by a cesarean section and examined for any external abnormalities as well as their anogenital distance (AGD). Necropsy examination of dams was conducted wherein their ovaries and uterine contents were closely examined for corpora lutea and implants. The DRF study concluded that PEA did not induce any systemic maternal toxicity, developmental toxicity, and any incidence of external abnormalities in fetuses at and up to the highest tested dose of 1,000 mg/kg body weight/d. Since this limit dose did not produce any observable maternal and fetal toxicity, the doses determined for the main prenatal developmental toxicity study were 250, 500, and 1,000 mg/kg body weight/d. Also, since there were no dose dependent and statistically significant differences in the incidence of preimplantation losses in both control and treated groups, it was concluded that PEA did not induce any preimplantation losses and hence the dose administration for the proposed definitive study be initiated on day 0 of gestation in accordance with the OECD Test Guideline No. 414.
Main Study
0-Day pregnant female rats were divided into 4 groups, with each group containing 30 rats in order to yield a minimum of 20 pregnant rats per group by the end of the gestation period. Palmitoylethanolamide was administered by oral gavage daily in volume of 5 mL/kg (in doses of 250, 500, or 1,000 mg/kg body weight) from gestation days 0 to 19, following which rats were sacrificed on gestation day 20. Rats in the control group were administered only with analytical grade water with Tween-80 (0.2% [wt/vol]), the vehicle. Female rats were observed for signs of ill health, behavioral changes, or adverse reactions to treatment daily during the study period. The body weight of females was recorded on days 0, 5, 8, 11, 14, 17, and on day 20 of gestation, prior to their terminal sacrifice. Body weight changes were calculated between each subsequent check and also between gestation days 0 and 20, and gestational weight change was corrected, after accounting for the weight of the gravid uterus. Consumption of food was calculated for gestation days 0 to 5, 5 to 8, 8 to 11, 11 to 14, 14 to 17, and 17 to 20 and also between gestation days 0 and 20. The food consumption was measured in gram per animal per day. On the morning of gestation day 20, blood samples were collected from pregnant females in order to measure the levels of thyroid hormones thyroxine (T4), triiodothyronine (T3), and thyroid-stimulating hormone (TSH). Analysis of hormones was performed using Multiskan Go Microplate and Cuvette Spectrophotometer (ELISA reader, Thermo Scientific) with commercially available ELISA kits manufactured by Elabscience. The experimental design did not include toxicokinetics assessment of plasma for its PEA content.
All female rats were sacrificed on day 20 of gestation by CO2 asphyxiation, and cesarean section was performed for the immediate removal of fetuses. Gross pathological changes were investigated by necropsy examination. The uterus from each female was examined for the following details: uterus weight, the number and placement of implantation sites, the number of live and dead fetuses, the number of early and late resorptions, and any abnormalities of the uterus or embryonic sac. In addition to this, the ovaries of each female were investigated for the number of corpora lutea. Any dam that had undergone premature delivery was also subject to investigation of the above parameters in addition to examination of gross pathological changes. Any uterus that did not contain any visible implantation sites or contained one horn pregnancies were stained with 10% ammonium sulfide solution in order to determine the presence of implantation sites that may have resorbed very early. The thyroid gland from each dam was extracted and preserved in 10% neutral-buffered formalin. Its weight was measured approximately 48 hours after preservation. Thereafter, the fixed thyroid glands were processed by serial dehydration through grades of alcohol, clearing in xylene and embedding in paraffin wax. These were then sectioned on a Leica microtome at 7 µm, stained with hematoxylin and eosin, and evaluated for microscopic pathology by a board-certified pathologist. Microscopic examination of the thyroid gland was conducted by a board-certified pathologist, and any findings were allocated into classifications of “minimal,” “mild,” “moderate,” and “severe.”
Following delivery by cesarean section, fetal investigations were carried out. This included for each litter, the total number of fetuses, number of abnormal fetuses, number of dead fetuses, number of live fetuses, number of male fetuses and their individual weights, number of female fetuses and their individual weights, sex ratio, and AGD (in mm). Each fetus was subjected to a comprehensive external examination, to take note of any external abnormalities. These were classified into “normal variants,” “minor abnormalities,” or “major malformations.”
The AGD of live fetuses was measured using a Dial caliper (Mitutoyo dial caliper), in mm. With respect to male rats, AGD was measured from the anterior edge of the anus to the base of the anogenital aperture, and for females, it was measured from the anterior edge of the anus to the base of the urinary aperture.
Fetuses were sacrificed using barbiturate anesthetic (sodium thiopentone). From each litter, around half of the fetuses were randomly chosen for visceral examination by modified Wilson’s technique for soft-tissue development assessment. The other half of fetuses were subjected to skeletal examination using Alizarin Red S staining method. Once again, any findings were classified into “normal variants,” “minor abnormalities,” or “major malformations.” As per the standard operating procedures at the test facility, a “normal variant” is an incidence distributed throughout a population in consistently large percentage, including the ossification variations such as delayed, incomplete, poor, or scrambled ossification; a “minor abnormality” is a malformation that would not be expected to directly affect the survival of the fetus; while a “major malformation” is a malformation of teratological significance and/or which would be life-threatening to the fetus.
Statistical Analysis
The IBM SPSS Statistical Software (version 23) was used to perform statistical analysis. One-way analysis of variance, following Levene test for homogeneity, was used to compare the data pertaining to maternal body weights, body weight gain, food intake, thyroid weight, thyroid hormone assessment, uterus weights, fetal weights, preimplantation and postimplantation loss (%), and AGD of fetuses. The data were subjected to log-transformation when required. Dunnett test was conducted on data with homogeneous intragroup variances when the “F” value was significant. The Kruskal-Wallis 1-way analysis was conducted on data with heterogeneous intragroup variances.
Data pertaining to the litter size, the number of male and female fetuses, sex ratio, the number of corpora lutea, the number of implantations, the number of live and dead fetuses, and the number of early and late resorptions were evaluated using nonparametric statistical tests such as Kruskal-Wallis test and Mann-Whitney U test.
The χ2 test was used to compare incidence of malformations, the number of females with resorptions, and the number of females with confirmed pregnancies. For data pertaining to the fetus, the basic sampling unit used was the litter. The variance was evaluated at 5% level of significance.
Results
Maternal Observations
Daily examinations during the period of study did not reveal any treatment-related abnormal clinical signs and death in any of the dams, irrespective of treatment or control groups. Two dams (1 of each from control group and 1,000 mg/kg/d) were found to deliver healthy, normal, and well-grown litters on gestation day 13 and hence were sacrificed prior to the scheduled sacrifice date. A comparison between the females in the control and treatment groups of widespread pregnancy parameters showed no significant differences (P > 0.05) indicative of any maternal toxicity due to the treatment with PEA. All other dams survived till the determined sacrifice on gestation day 20 (Table 1). In necropsy at gestation day 20, no gross pathological alterations were reported in any of the dams, from any of the dose groups. An isolated incidence of dead and undersized fetuses was observed in a dam at mid dose (500 mg/kg/d). However, it was considered to be unrelated to the treatment in the absence of any causative observation. Evaluation made to assess thyroid gland functions of dams sacrificed on day 20 of their gestation revealed that PEA did not induce any adverse effects in treated dams, with respect to organ weight and microscopic appearance of their thyroid glands, and also the serum levels of thyroid hormones, namely, T3, T4, and TSH.
Maternal Observation Upon Treatment With Different Doses of PEA.a
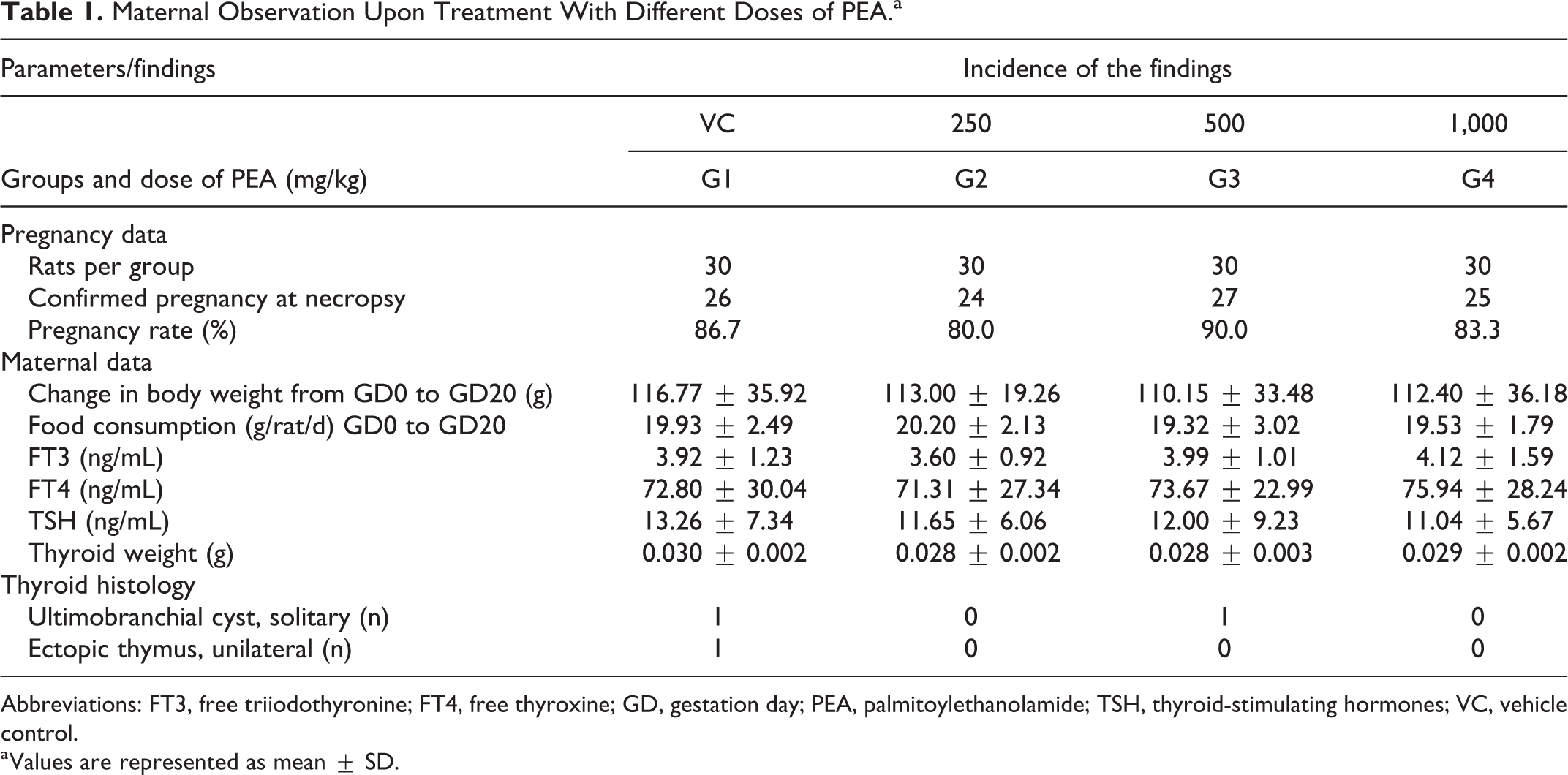
Abbreviations: FT3, free triiodothyronine; FT4, free thyroxine; GD, gestation day; PEA, palmitoylethanolamide; TSH, thyroid-stimulating hormones; VC, vehicle control.
a Values are represented as mean ± SD.
Furthermore, the test item did not have any adverse effect on the mean maternal body weights, body weight gains, and food consumption, with no significant differences between the control group and the treatment groups, up to 1,000 mg/kg/d (P > 0.05), throughout the study period (Table 1). The mean maternal body weight changes during the study period (gestation days 0-20) were 117, 113, 110, and 112 g in control group, dose 250, 500, and 1,000 mg/kg/d, respectively.
Uterine and Embryo–Fetal Observations
The gravid uteri of females sacrificed on day 20 of gestation revealed no remarkable alterations indicative of adverse effects of PEA. There were no significant differences (P > 0.05) in any of the parameters evaluated, such as relative uterus weight, total implants (live and dead), preimplantation and postimplantation loss (%), resorption (early and late), and the number of corpora lutea across all the dams, throughout the study period. The test subject had no interference with the implantation process, and the group mean values of these parameters were comparable between the treatment groups and the control group (Table 2). The litter data of the females that were sacrificed on gestation day 20 also showed that there were no remarkable alterations indicative of any adverse effect of PEA (Table 2). Group mean values of the parameters such as litter size (13.39, 11.74, 13.30, and 13.77 at control, 250, 500, and 1,000 mg/kg/d, respectively), the number of fetuses (live and dead), fetus sex ratio (% of male and female and male fetuses per litter), average weights of male fetuses (4.26, 4.19, 4.19, and 4.19 g in control, 250, 500, and 1,000 mg/kg/d, respectively), average weight of female fetuses (3.99, 3.94, 3.72, and 3.96 g in control, 250, 500, and 1,000 mg/kg/d, respectively), and AGDs did not differ significantly (P > 0.05) between the treatment groups and the control group (Table 2).
Uterine and Embryo–Fetal Observation Upon Treatment With Different Doses of PEA.a
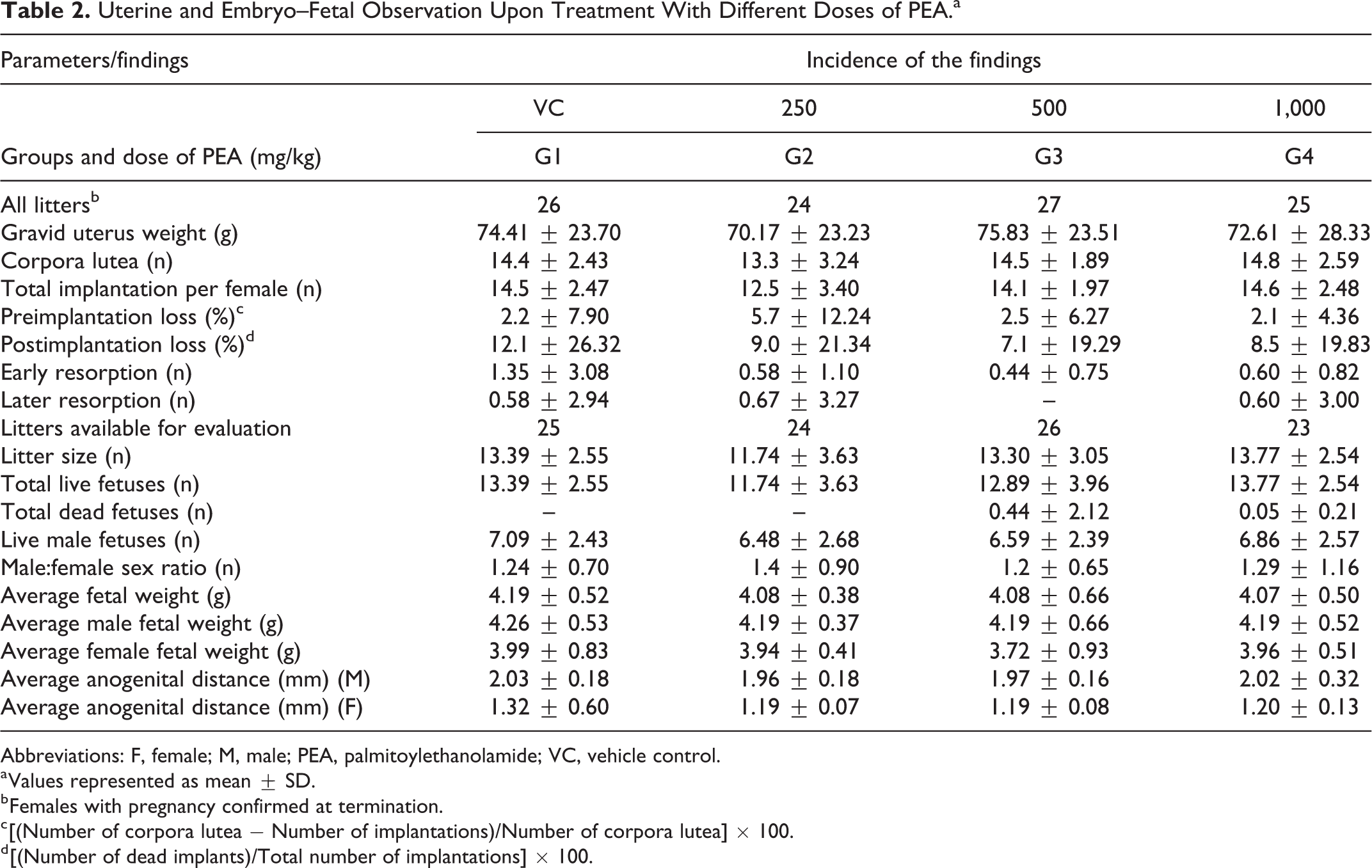
Abbreviations: F, female; M, male; PEA, palmitoylethanolamide; VC, vehicle control.
a Values represented as mean ± SD.
b Females with pregnancy confirmed at termination.
c [(Number of corpora lutea − Number of implantations)/Number of corpora lutea] × 100.
d [(Number of dead implants)/Total number of implantations] × 100.
Fetal Malformations and Variations
The fetal abnormalities were identified, and the “litter incidence” of such abnormalities (ie, the number and % of litters in a group having the abnormality) for each group was determined. External examination of the fetuses showed that PEA administration at doses of 250, 500 or 1,000 mg/kg body weight did not cause any major malformation in any of the litters of the treated dams (Table 3). The external examinations pertained to length, cranium, eyes, palate, limbs, tail, genitals, sex, and so on, which revealed no “minor anomalies” or “major malformations.” The only findings included undersized or underdeveloped fetuses noted in 1 litter, each in control (G1: 4.17%), low- (G2: 4.17%), and mid- (G3: 3.70%) dose level groups, and were classified into “normal variants.” There were no external malformations observed in any litter from the high-dose group. In the mid-dose group, there was the presence of a dead fetus from 1 litter with an accumulation of excess amniotic fluid (hydroamnios; G3: 3.70%). These isolated findings, with no dose-related incidence, were concluded to be spontaneous alterations, unrelated to the treatment.
Results of Fetal External and Visceral Evaluations.
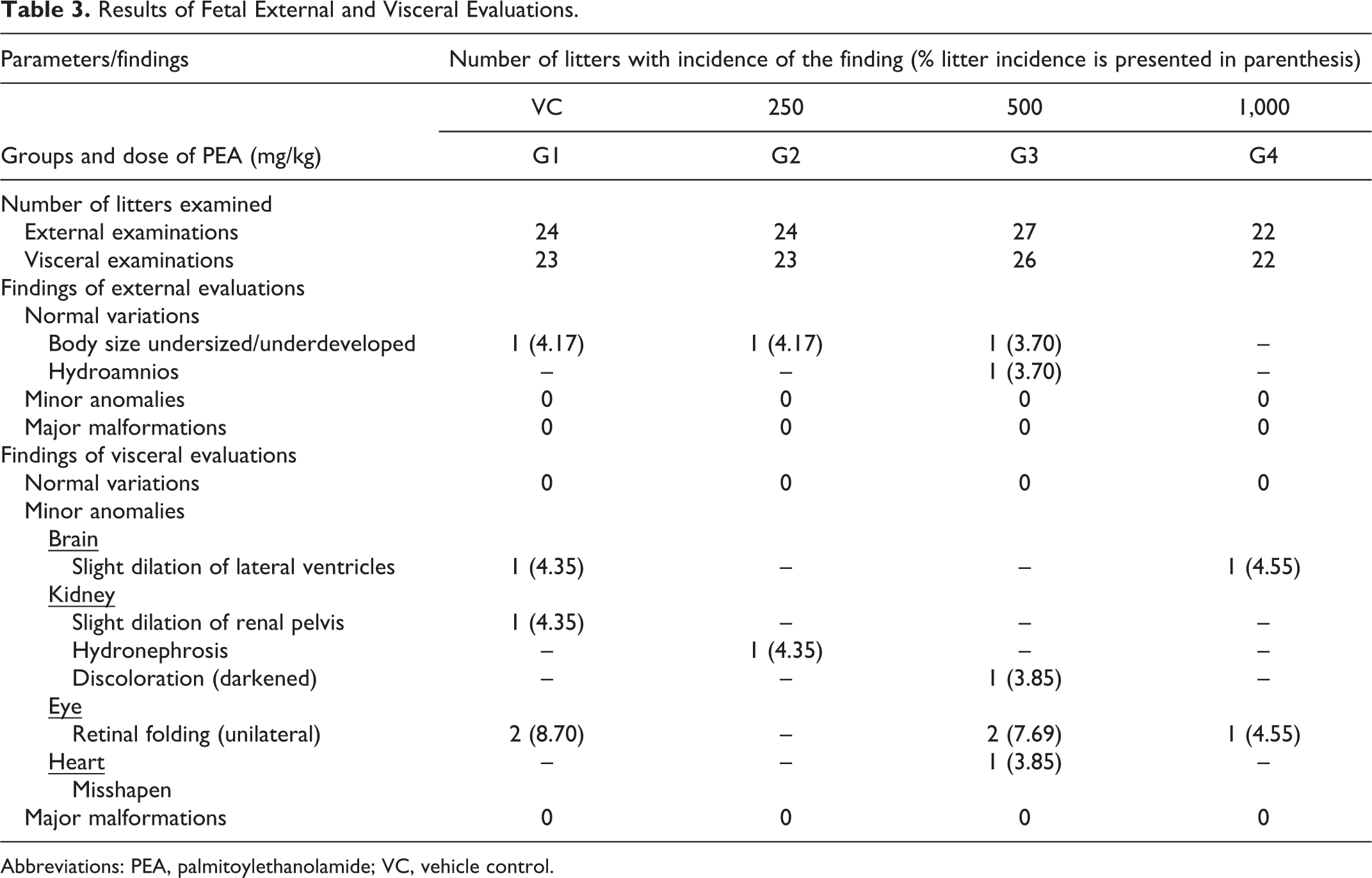
Abbreviations: PEA, palmitoylethanolamide; VC, vehicle control.
With respect to visceral examinations, there were no remarkable or major soft-tissue alterations in any litter of the treated dams at any dose of PEA (Table 3). There was the presence of a minor anomaly, namely, slight dilation of lateral ventricles of the brain, observed in 1 litter from the control group (G1; 4.35%) and 1 litter from the high-dose group (G4; 4.55%). In the kidneys too, there were isolated instances of minor anomalies, namely, a slight dilation of the pelvis in a fetus from control group (G1; 4.35%), hydronephrosis in a fetus from the low-dose group (G2; 4.35%), and discolored (darkened) kidneys in a fetus from the mid-dose group (G3; 3.85%). Two litters each from the control (G1; 8.70%) and mid-dose groups (G3; 7.69%) and 1 litter from the high-dose group (G4; 4.55%) exhibited unilateral retinal folding. In addition to these observations, another minor anomaly, misshapen heart was observed in 1 litter at mid-dose (G3; 3.85%). These isolated and minimal incidences of minor anomalies were not dose dependent and occurred in all groups, including the vehicle control group. Therefore, these anomalies were regarded as incidental and unrelated to the treatment.
In terms of skeletal abnormalities, there was no incidence of any malformation in any litter that could be described to be of teratological significance and/or which would be life-threatening to the fetus (major malformation; Table 4). There were incidences of normal variants and minor anomalies in most of the litters of the control and treatment group dams. In skull, 1 litter each from the low-dose (G2; 4.35%) and mid-dose (G3; 3.85%) groups and 2 litters from high-dose group (G4; 9.09%) exhibited scrambled, poor, or incomplete ossification patterns, which are classified as normal variants. Five litters from the control group (G1; 21.74%), 3 from the low-dose group (G2; 13.04%), 19 from mid-dose group (G3; 73:08%), and 13 from the high-dose group (G4; 63.64%) exhibited poor, incomplete, or asymmetric ossification patterns of the sternebrae, also classified as normal variant. It may be noted that though these had higher incidence in the mid- and high-dose groups, it was not dose dependent; moreover, it was comparable to the historical control incidence at the test facility for such variations, which is 59.2%. Hence, this variation was not considered to be related to exposure to PEA. Variations in the pattern of ossification are considered “normal variation.” These findings are commonly seen in 20-day-old rat fetuses. Incomplete or reduced ossification in fetal skeletons has been listed as having low level of concern. 21
Results of Fetal Skeletal Evaluations.
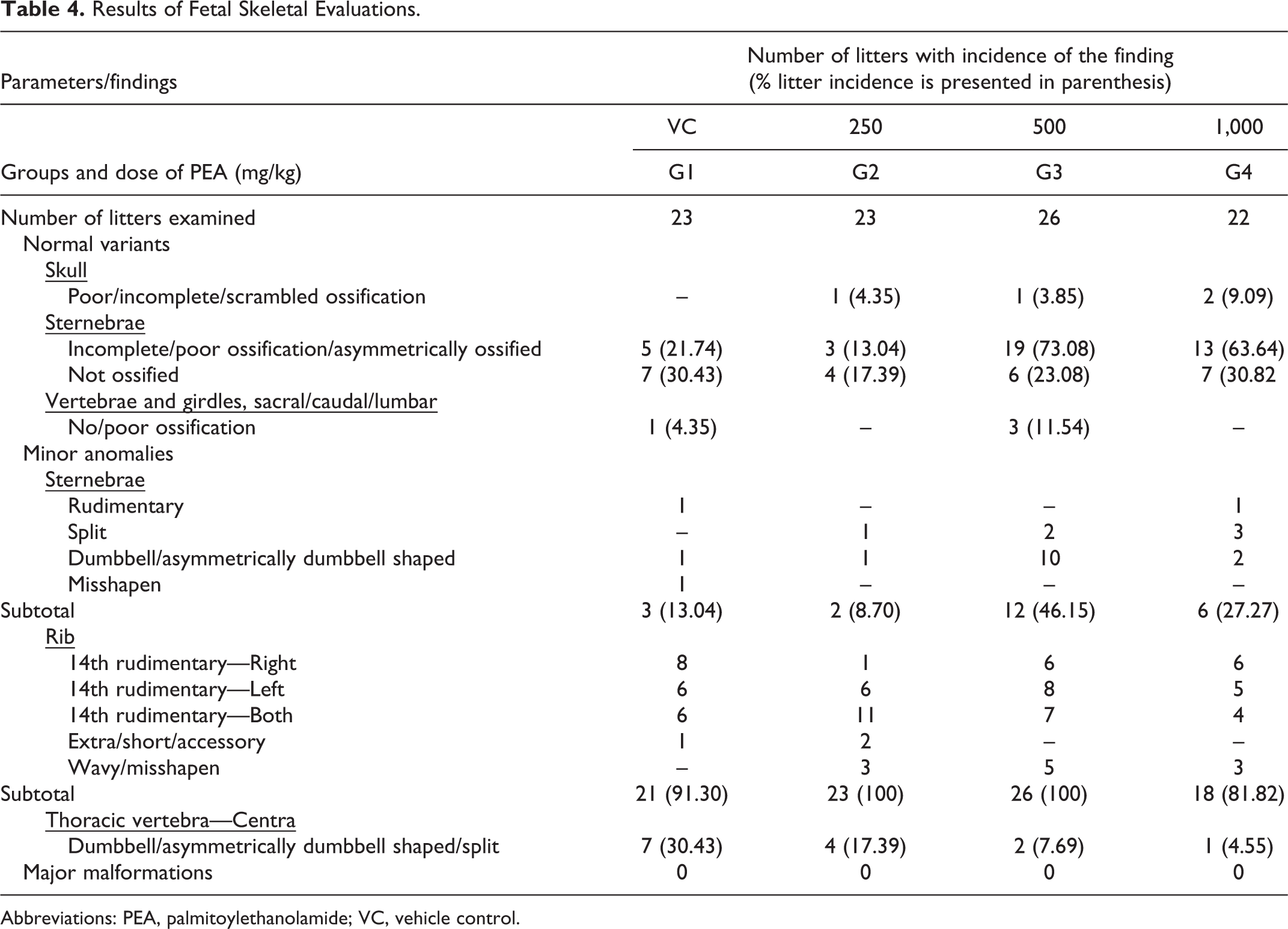
Abbreviations: PEA, palmitoylethanolamide; VC, vehicle control.
In addition to this, a comparable incidence of completely unossified sternebrae (normal variant), observed in 7 litters from G1 (30.43%), 4 from G2 (17.39%), 6 from G3 (23.08%), and 7 from G4 (31.82%), was considered to be unrelated to the treatment with PEA and of no toxicological significance. There was also the presence of minor abnormalities such as rudimentary, split, dumbbell shaped, asymmetrically dumbbell shaped, and misshapen sternebrae in 3 litters from G1, 2 from G2, 12 from G3, and 6 from G4. In addition to this, 1 litter in the control and 3 litters in the mid-dose group showed the presence of unossified or poorly ossified patterns of sacral, caudal, or lumbar vertebrae (classified as normal variant).
Dumbbell shaped, asymmetric dumbbell shaped, or split centra of thoracic vertebrae, which are considered minor anomalies, were observed in all the groups with an incidence of 7 in G1, 4 in G2, 2 in G3, and 1 in G4. There were also minor anomalies in the ribs, which included rudimentary, wavy, extra, accessory, nodulated, short, or misshapen ribs observed in all groups with litter incidence of 21 in G1 (91.30%), 23 in G2 (100%), 26 in G3 (100%), and 18 in G4 (81.82%). Since the above skeletal observations were normal variants or minor anomalies, which were not expected to directly affect the survival of the fetus, they were deemed to be incidental or of no toxicological importance.
Discussion
Palmitoylethanolamide was administered by oral gavage daily to pregnant female rats, during which period clinical and physical assessments were conducted. With respect to fetal observations, there were no external, visceral, or skeletal abnormalities observed that could be classified as “major abnormalities.” Anomalies that were observed were considered “minor anomalies” or “normal variants,” which are known to be distributed throughout a population in consistently large percentages and not expected to directly affect survival of the fetuses. Incidence of these observations was not significantly different between the control and treatment groups and was not dose dependent. Thus, it is shown that PEA failed to cause adverse events in terms of teratogenicity.
As for maternal and embryo–fetal observations, there were no treatment-related alterations encountered during this study in the parameters of survival, clinical signs, body weight, food consumption, pregnancy data, thyroid weight and related hormones, and thyroid histopathology and gross pathology, at the dose levels of 250, 500, and 1,000 mg/kg body weight. Similarly, the parameters evaluated for uterine observation did not reveal any remarkable alterations indicative of adverse effect of PEA.
The litter data such as the litter size, number of fetuses, sex ratio, fetal weights, and AGD of the litters of females sacrificed on day 20 of gestation also did not reveal any alterations, indicating that PEA has no adverse effect on the litter. Total number of litters examined were 24, 24, 27, and 22 from G1 to G4, respectively. Thus, it was evident that PEA failed to cause adverse events in rats in terms of maternal toxicity, embryotoxicity, and fetotoxicity.
Several efficacy studies on PEA, done on different cell types in vitro and in vivo on rodents and dogs, have also indicated that PEA did not exhibit any adverse effects, explicit behavior, and body weight changes, which are allusive of tolerability of PEA. 22,23 In some studies, intracerebroventricular administration of PEA in mice reported no change in locomotor activity or electroencephalogram parameters. Long-term topical use of PEA for the management of atopic eczema reported PEA to be safe in a multicenter cohort study, with only 3.4% of all cases reporting poor tolerance. 24 Similarly, the safety and tolerability of PEA are evident through the lack of adverse reactions observed in a multitude of human studies conducted to investigate its efficacy. 10,11,25 However, the safety of any food supplement must be ensured during prenatal exposure to attain a complete picture of its safety.
Under the conditions of this prenatal study, there was no evidence of developmental toxicity of PEA in Wistar rats administered 250, 500, or 1,000 mg/kg in the absence of overt maternal toxicity. The NOAEL of PEA for maternal toxicity, embryotoxicity, fetotoxicity, and teratogenicity was found to be greater than 1,000 mg/kg body weight. According to the FDA, 26 based on body surface area computations, this value translates to a human equivalent dose (HED) of greater than 9.7 g/d. Considering most studies 10,27,28,29 demonstrated PEA to be effective in doses of 600 mg/d, our study has shown the safety of administration of oral doses greater than the known effective dose. Therefore, our present study evaluating the effect of PEA on prenatal development toxicity is pertinent to all PEA food supplements and is unprecedented in the research on PEA.
It is, however, recognized that the OECD Test Guideline No. 414, followed for this study, aims to evaluate maternal toxicity and any prenatal developmental toxicity in terms of fetal death, fetal malformation, and effects on their growth. The study is not designed to evaluate functional deficits, which have to be evaluated in litters born to dams exposed to a test item (F1 generation).
Conclusion
This prenatal developmental toxicity study contributes greatly in building a robust safety profile for PEA. It indicates that PEA is well tolerated by and is safe to pregnant rats even at a high dose of 1,000 mg/kg body weight/d (NOAEL). This value translates to an HED of greater than 9.7 g/d.
Footnotes
Author Contributions
Narendra S. Deshmukh contributed to conception and design; contributed to acquisition, analysis, and interpretation; critically revised manuscript; gave final approval; and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Shailesh Gumaste contributed to design, contributed to acquisition and analysis, selects item, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy. Silma Subah and Nathasha Omal Bogoda contributed to conception, select item, drafted manuscript, critically revised manuscript, gave final approval, and agrees to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The study was conducted at INTOX Private LTD., 375, Urawade, Tal. Mulshi, Maharashtra India. SS and NOB are associated with Gencor Pacific Limited. NSD and SG are associated with Intox Private LTD.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was conducted under a grant from Gencor Pacific Limited, North Plaza, Discovery Bay, Lantau Island, Hong Kong.