
Abstract
Development of toxicology-based criteria such as occupational exposure levels (OELs) are rarely straightforward. This process requires a rigorous review of the literature, searching for patterns in toxicity, biological plausibility, coherence, and dose–response relationships. Despite the direct applicability, human data are rarely used primarily because of imprecise exposure estimates, unknown influence of assumptions, and confounding factors. As a result, high reliance is often placed on laboratory animal data. Often, data from a single study is typically used to represent an entire database to extrapolate an OEL, even for data-rich compounds. Here we present a holistic framework for evaluating epidemiological, controlled in vivo, mechanistic/in vitro, and computational evidence that can be useful in deriving OELs. It begins with describing a documented review process of the literature, followed by sorting of data into either controlled laboratory in vivo, in silico/read-across, mechanistic/in vitro, or epidemiological/field data categories. Studies are then evaluated and qualified based on rigor, risk of bias, and applicability for point of departure development. Other data (eg, in vitro, in silico estimates, read-across data and mechanistic information, and data that failed to meet the former criteria) are used alongside qualified epidemiological exposure estimates to help inform points of departure or human-equivalent concentrations that are based on toxic end points. Bayesian benchmark dose methods are used to estimate points of departure and for estimating uncertainty factors (UFs) to develop preliminary OELs. These are then compared with epidemiological data to support the OEL and the use and magnitude of UFs, when appropriate.
Introduction
Protection of individuals from potentially toxic effects of chemical exposures is a critical responsibility imposed upon multiple jurisdictions and government agencies. Whether it is protecting the health of the worker, members of the general population, or other subpopulations, a keystone risk assessment component requires the development of an evidenced-based toxicity reference value (TRV) for human health applications. This premise is based on the threshold concept where at exposures below the TRV, there is a low potential to experience adverse effects given specific exposure conditions. The challenge remains accurately interpreting the available toxicity information and extrapolating it to humans in the derivation of the TRV.
Regulatory and voluntary/guideline TRVs are developed by many agencies in the United States and worldwide, including the Occupational Safety and Health Administration (OSHA), American Conference of Governmental Industrial Hygienists (ACGIH), Workplace Environmental Exposure Levels Committee, and the National Institute for Occupational Safety and Health (NIOSH). These values differ in their approach and application in that some are entirely health-based while others consider other aspects such as chemical properties (eg, flashpoint), technical feasibility, and cost. Some vary on the sensitivity of the intended worker population of interest (eg, sensitive but not particularly sensitive individuals), as such definitions can be confusing. Because procedures for TRV development and the intended application of the TRV differ among organizations, multiple TRVs may exist for a given chemical. An OSHA permissible exposure limit (PEL), which takes into consideration the technical and economic feasibility of an exposure limit, may differ substantially from a health-based threshold limit value (TLV)-time weighted average established by ACGIH. In Army workplaces, current industrial hygiene policy requires the use of the more stringent of the ACGIH TLV or OSHA PEL. 1 In the absence of a TLV or PEL, occupational exposure levels (OELs) derived from other sources could be used. The NIOSH recommended exposure limits are applied for exposures for specific engineered nanomaterials, such as carbon nanotubes and nanoscale titanium dioxide, 2 since neither OSHA nor ACGIH provide OELs for these materials. US Army OELs have been developed for chemical warfare agents; however, OELs are not available for many other military relevant compounds. 3 Additionally, not all values are regularly updated; hence, some may not be based on the most recent science or a product of more rigorous systematic review procedures (eg, see study by Rooney et al 4 ). As such, in cases where no applicable federal or other recognized criteria exist or when existing values are considered inapplicable given the specific target population considered, approach used, or availability of recent information, OELs may need to be derived to protect Department of Defense worker populations.
Here we provide a process for deriving and documenting health-based OELs when such values are lacking. Army specific OELs are intended to be health-based exposure guidelines for military relevant compounds that would protect worker populations (ie, soldiers, workers, and those potentially in nonlaboratory and office environments who may be exposed 8 hours per day, 5 days per week for a working lifetime including susceptible populations such as pregnant women and most workers with underlying medical conditions). Here we define a working lifetime as 30 to 45 years in duration. These OELs are intended to focus on the available scientific information as it pertains to probability for adverse health effects and does not consider other factors (eg, feasibility, chemical properties, economics, etc.) in their derivation. No policy decisions are implicit in their derivation.
The purpose of this effort is to clearly describe a procedure for the derivation of OELs when a need exists. This procedure includes documentation of the literature review, followed by interpretation and evaluation of the appropriate available scientific evidence supporting the derivation of the OEL. Given resource and time constraints, this document is not intended to be a systematic review of all literature available on the compound or a complete review of its toxicity. Rather, this document is intended to provide a narrative review and an integrated analysis of studies most relevant to the derivation of the OEL—to best use the available toxicity information in a holistic manner to develop a toxicity-based benchmark value. This procedure includes documentation of the factors considered in the literature review and analysis to provide a transparent record of the critical decisions and the associated rationale regarding the derivation of the OEL. The scientific quality and relevance of the information are assessed and rationales for the selection of key studies and toxic end points of concern are provided. Studies selected for dose–response analysis are evaluated and used to ascertain a point of departure (POD) for OEL development that will be corroborated with available mechanistic, read-across, and human experience information. A list of acronyms is provided in Table 1.
List of Acronyms.
Methods
Literature Search and Screening
The effort begins with problem formulation and establishment of review criteria. Based upon the establishment of clear criteria, literature searching and screening procedures are conducted to be specific to focus efforts on the selection of information most directly applicable to TRV/OEL derivation. Published studies are screened against the review criteria for tagging into appropriate bins based on inclusion/exclusion, type of study (ie, human epidemiological, controlled animal study, in vitro/mechanistic/mode of action [MOA]), and disease end points.
Chemical-specific data are located by conducting searches of computerized databases using keyword search strategies to identify literature of potential relevance. Methods for documenting literature searching and screening procedures have been described by others 4 ; however, full adoption of these procedures is resource intensive and likely not practical for narrative reviews. Documentation of search criteria provides reviewers with a record of rigor that may provide a level of confidence for the review. Important elements for the document include the database(s) searched and date range covered by the search, search terms used, and date(s) that the searches were performed. Archiving the number of citation retrieved, abstracts reviewed, and publications retrieved is desirable.
Considerable time and effort can be maximized by using other reviews as a starting point. Examples include state, federal, or international health agency assessment/reviews, if available (eg, ATSDR, NIOSH, NTP, OSHA, EPA, ECHA, EFSA, IARC, WHO, ACGIH; see Table 1). If an existing review is available, a new literature search should be conducted to gather any more recent information since date of last publication. For data poor compounds, it may be unnecessary to refine searches beyond using just the chemical name(s) and Chemical Abstracts Service registry numbers as this may return a reasonable number of studies. For data-rich compounds, search strings may be refined to target end points of interest identified in previous assessments or the initial literature search. Searches for supplemental information (eg, in vitro, mechanistic information) may be conducted at this stage or as needed later in the development of the review as critical end points are identified. Additional studies may be identified through review of reference lists in relevant papers and by searching for articles that cite or are cited by relevant work (ie, forward and backward searching). The use of these methods and their results should be documented.
The literature search should be conducted in multiple databases using a documented search strategy. Most common databases include but are not limited to PubMed, PubChem, Web of Science, National Technical Information Service, and US NIOSH Toxic Information Center 2. Databases containing government research such as Defense Technical Information System, Federal Research in Progress, and Chemical and Biological Defense Information Analysis Center should also be searched to identify technical reports and other literature not found in peer-reviewed journals.
Primary studies which provide sufficient detail to allow evaluation of the study methods are required for derivation of an OEL. Documentation of these studies may be found in peer-reviewed literature, grey literature, unpublished industry reports, and government reports (unclassified and classified). Unpublished industry reports will be used if proprietary information (confidential business information) do not prohibit publication of summary information. Classified government reports will not be used; limited distribution information will be used only as needed and will be determined on a case-by-case basis.
Literature search results may initially be screened at the title and abstract level, without regard to study quality, to remove irrelevant documents and identify sources of information relevant to the toxic effects of the compound of interest in animals and humans, as well as information on the chemical/physical properties, absorption, distribution, metabolism, and elimination (ADME), toxicokinetics, toxicodynamics, and MOA. Studies retained after initial screening will be evaluated at the full-text level.
Data Acquisition
Types of Data
Several types of information are evaluated to develop health-based OELs, including human clinical and epidemiologic studies, acute to chronic animal toxicity studies, in vitro toxicity or mechanistic studies, ADME or toxicokinetic studies, in silico model estimates, read-across extrapolations, and genotoxicity studies. Data on the physical and chemical properties of the substance are also evaluated to support toxicity conclusions. All observed toxic effects should be considered (Figure 1), assuming that the end points measured have a clear link to a toxic effect.
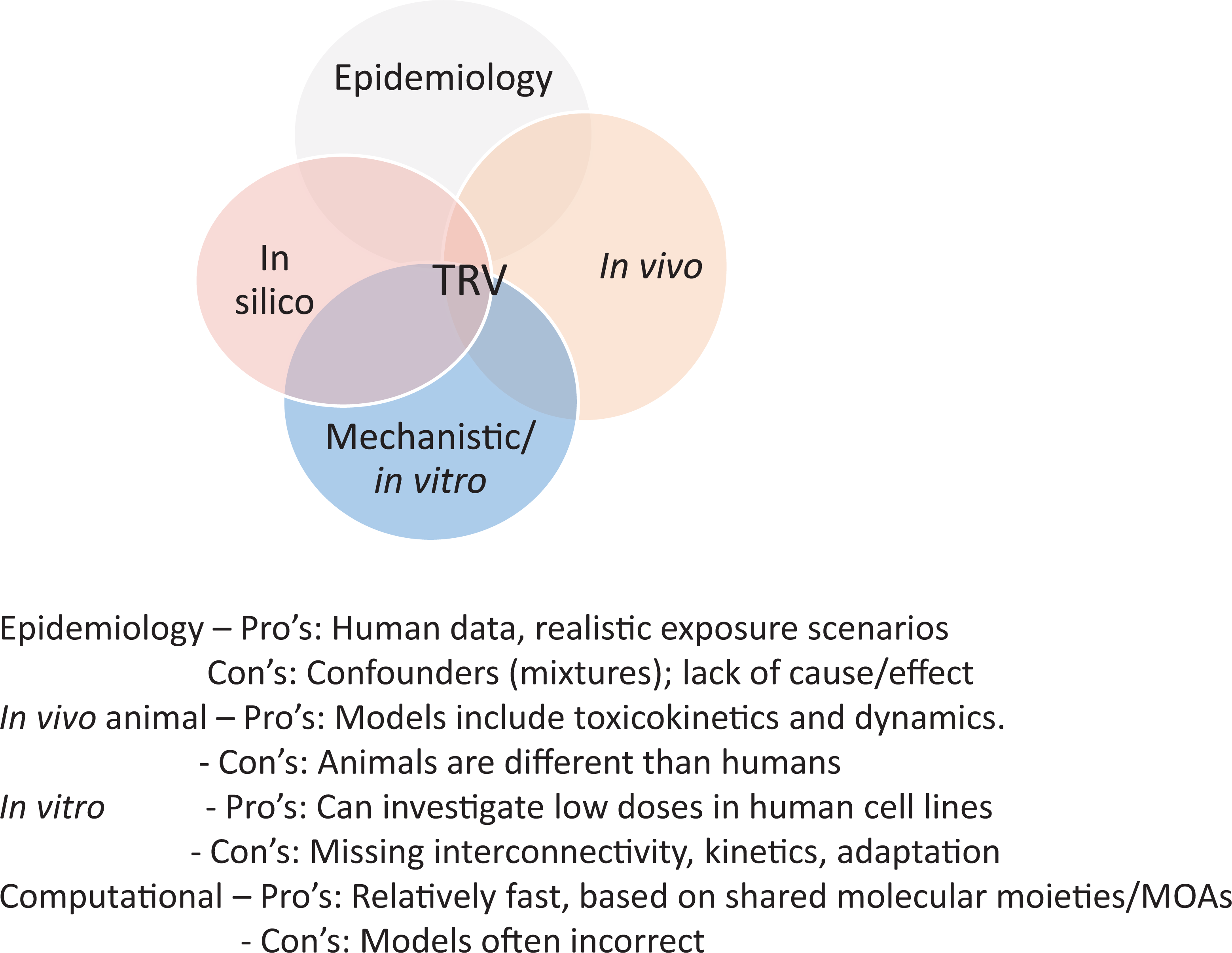
Utility of various lines of evidence in toxicity reference value (TRV) derivation.
Human studies
Human toxicity data may be obtained from epidemiologic and occupational exposure studies, case reports, or controlled exposures. Use of human studies eliminates uncertainties associated with relevance of animal studies to humans and may also provide insight on individual variability. Blinded randomized controlled trials conducted in a human population of interest are often considered to be the gold standard of evidence and are a staple of pharmaceutical medicine. However, this evidence is rare in the field of toxicology, with human data being limited to observational studies. Human observational studies may provide information that is most direct to the population of concern; however, these studies have several limitations. Data usually lack precision in characterization of exposure and health outcome(s) and are typically limited by the presence of known and unknown coexposures and confounders. Additionally, most human observational studies demonstrate associations rather than causative relationships (eg, cross-sectional designs).
Animal studies
Although human studies are preferred when available, most assessments rely on data from controlled laboratory animal studies, either alone or in combination with human studies. Controlled animal studies may overcome many of the limitations inherent to observational studies; however, anatomy, physiology, and metabolism differ between species, and animal studies may provide results that do not predict human responses or may not accurately extrapolate from one species to another. Confidence is increased when multiple species exhibit similar toxic effects, and available information (eg, ADME, mechanistic) do not indicate that effects will be different in humans. Animal studies are most useful when exposure occurs via relevant routes and over relevant durations so that uncertainties associated with extrapolation are limited. Although physiologically based pharmacokinetic (PBPK) modeling may aid in extrapolation between species and/or exposure routes, the reliability of these models varies based on accuracy of the parameters and assumptions used in its derivation.
In vitro studies
In vitro experiments can be used to assess MOA/mechanistic questions or be used to understand key events or develop PODs through in vitro/in vivo extrapolation (IVIVE) methods. 5,6 Since in vitro methods rarely provide data to assess disease outcomes, kinetics, or interorgan/system relationships; extrapolation of these in vitro mechanistic responses to health-related effects of an in vivo organ system or multiorgan system that results in a disease state can be useful but challenging. By itself, in vitro evidence is rarely considered to be a sufficient basis from which to derive an OEL, but can be used to help corroborate candidate values.
In silico/read-across
In silico (eg, quantitative structural activity relationships [QSAR]) models or read-across techniques estimate the toxicity of compounds based on structural similarities to known chemicals. They can be used to estimate animal data (eg, rat subchronic no observed adverse effect levels [NOAELs]/lowest observed adverse effect levels [LOAELs]) from which to develop PODs or used qualitatively for corroboration with other PODs. Some models provide estimates of confidence and other methods can be used to determine the suitability of using read-across qualitatively and quantitatively. 7,8
Mechanistic studies
Mechanistic studies, which may include data collected from specific in vitro and in vivo studies and some in silico models, provide information on effects of compounds at the cellular or subcellular level. These studies provide information about the biological or chemical events associated with exposure-related phenotypic effects but are not generally considered to be adverse outcomes (apical effects). Molecular evidence supporting mechanism or mode of toxicological action can be generated relatively quickly using high-throughput data and new approach methods and can be the most abundant and diverse evidence stream. 6,9 Mechanistic evidence can inform key events responsible for biological effects and provide biological plausibility that supports a MOA observed in animal studies. Measurement of upstream molecular, cellular, or physiologic interactions can implicate disruptions leading to adverse effects and provide alternate PODs (eg, nonlinear cancer assessments).
Information from mechanistic studies, in vitro tests, in silico models, and read-across extrapolations can be used as supporting evidence, however, the type of summary data and synthesis will depend on the needs of the assessment and can vary from a high level summary of potential mechanisms of action to a synthesis to answer specific questions (eg, applicability of the animal evidence to humans, mutagenicity, and applicability of linear low-dose extrapolation). Evaluation of individual studies that address mechanistic end points will only be pursued as needed to address issues that impact interpretation of key studies, hazard conclusions, and assumptions about the dose–response. If issues are addressed in prior assessments or reviews, individual studies need not be specifically evaluated.
Absorption, distribution, metabolism, elimination and toxicokinetic studies
Studies on the ADME of a compound provide information useful in relating measured end points to internal doses and affect the dose–response relationship. The ADME information, including uptake, disposition, half-life, protein binding, and metabolic activation/deactivation at low and high exposure levels, is reviewed as supporting information and may support extrapolation of animal data to humans. The ADME studies may provide information pertinent to interspecies and route-to-route extrapolations and may clarify discrepancies between in vivo and in vitro results. Given assumptions inherent in all PBPK models, we generally value data from similar routes of exposure greater than data from dissimilar routes.
Genotoxicity studies
Genotoxicity studies, including studies on DNA adduct formation, DNA strand breaks, gene mutations, chromosomal aberrations, aneuploidy, and changes in DNA methylation, should be critically reviewed with respect to relevance for determining a mutagenic mechanism of action for cancer. Genotoxicity tests differ in ways that may affect their relevance to humans and ability to assess a mutagenic mechanism of action, including whether they measure direct genetic effects or associated processes (eg, unscheduled DNA synthesis), whether the measured effects are permanent or may be repaired (eg, DNA damage), whether or not they include potential active metabolites, if the study was conducted in vitro or in vivo, and whether in mammals or in prokaryotes/lower eukaryotes 10,11
Physical and chemical properties
Physical and chemical properties are reviewed as supporting information. Physical and chemical properties of a compound influence the absorption, distribution, metabolism, and excretion from the body and the relationship between dose and time to response.
Study Evaluation
Study evaluation at the full-text level is conducted to determine the quality and relevance of the study dose–response data for identifying PODs for OEL derivation. Studies are qualitatively evaluated at the individual end point level in terms of strength of the methods that reflect on the quality of the results used to measure the response and the biological relevance of the response (see Table 2), 12,13 but see Klimisch et al. 14 Studies should be assessed at the end point level because different end points in the same study may have different strengths and limitations. The purpose of the strength of methods assessment is to evaluate the methodological conduct of studies, irrespective of the study findings, to determine the likelihood that the results are a reliable and accurate representation of an adverse response. Assessment of biological relevance (ie, whether the change measured is biologically meaningful in the manifestation of disease) serves to evaluate the likelihood that study findings are relevant to expected exposures and adverse outcomes in humans. The evaluation criteria presented in the current analysis were developed based on several published methods for assessing risk of bias and biological relevance in epidemiological and animal studies for narrative and systematic reviews. 15 -18 The evaluation criteria are meant to be guidelines to assist in selecting the most relevant and highest quality studies applicable to OEL development. The criteria are not intended to be a rating scheme or checklist and should be flexible so as to allow the requisite professional judgment.
During evaluation, studies may also be determined to have serious flaws in design or execution that would preclude use of the study for POD determination and subsequent OEL derivation. Fatal flaws that would make epidemiological studies unacceptable for use include biased participant selection; large loss (cohort studies)/exclusion of subjects (case-control and cross-sectional studies); incomplete outcome data (cohort studies); exposure groups or cases and controls that were not similar; lack of temporality in exposure and response; exposure levels too low for detection or to cause an effect; exposure measured with flawed methods; and covariates, confounders, and/or coexposures that differed between exposure groups (cohort and cross-sectional studies) or between cases and controls (case-control studies). Fatal flaws that would exclude animal studies for dose–response assessment and POD derivation would include poor or inadequate characterization of test compound; no (or inappropriate) concurrent negative control group or data from controls not reported; positive control group not included when necessary or responses not acceptable; biased allocation of animals to treatment groups; no information on preparation or storage of test compound; method used in inhalation studies to generate test substance concentration; exposures that differ between treatment groups; exposure frequency/duration not reported or not appropriate for the study type; dose/concentration range not appropriate (eg, all lethal or no effects observed); species/strain/sex/number of test animals not reported; end point assessment not sufficiently sensitive (eg, only mortality assessed); and end point assessment that differs between controls and treatment groups.
It is important to note that although fatal flaws may preclude the use of such data in POD derivation, those data and information should be retained as they may provide corroborative support for other questions involved in OEL derivation.
Study Evaluation Parameters.

Evidence Analysis/Synthesis
Exposure levels that intended to be safe from noncarcinogenic and likely many carcinogenic compounds are based on the premise that adverse health effects do not occur below some threshold of exposure and that safe exposure levels can be established through the application of uncertainty factors (UFs) to establish exposure levels that ensure the safety of the specific population of interest. For carcinogenic effects resulting from nongenotoxic mechanisms, a threshold response may be considered. However, for mutagenic carcinogens, a linear no threshold dose is often assumed. Exposure levels and mechanisms for cancer and noncancer effects can be considered simultaneously during evidence analysis and integration; however, PODs will be developed from noncancer end points with ranges of cancer risk being compared with resultant human equivalent concentrations (HECs; or candidate OELs). Because derivation of an OEL relies on dose- or concentration-dependent effects (ie, changes in measured end points) in study populations at relevant exposure durations, effects that may serve as the POD in OEL derivation, or candidate critical effects, must be identified within each line of evidence (LOE; evidence that is corroborative for the critical effect). Candidate critical effects are those end points demonstrating a statistically and/or biologically significant response attributable to exposure to the compound under study. Critical effects can range from lethality and severe or irreversible effects (eg, tissue injury) to reversible responses and upstream effects or biomarkers of effects (eg, clinical chemistry changes, adverse outcome pathway alterations, and organ/body mass changes). The effect under evaluation for deriving the POD should be considered adverse; yet the measured end point may be a nonadverse, precursor event, or defined biomarker of the adverse effect. Determination of adversity requires professional judgment; however, consideration should be given to control incidence, severity, and correlation with relevant changes in target organ/tissue appearance and/or function. 19 -22
Candidate critical effects may be identified by summarizing standard study information and comparing overall study NOAEL/LOAEL/Benchmark dose—low (BMDLs; ie, lowest NOAEL/LOAEL for all end points in each study selected and compared across studies). Candidate critical effect summary information should include species, strain, gender, exposure duration, dose/concentration–response information including LOAEL/NOAEL/BMDL, description of effect including relevance to health effect, severity or magnitude of effect, recovery information if available, correlates providing supporting evidence of coherence, and data quality issues. The LOAELs/NOAELs identified by study authors should be verified by the review author as they are subject to bias as well as policy and scientific standards that are subject to change. Summaries may be narrative but should also be graphical (scatter diagram) to show the variation in the end point-specific potential PODs. However, narrative summaries are required for all studies pertinent to derivation of the OEL.
Instead of selecting data from a single critical study based on the end point representing the lowest LOAEL/NOAEL across all studies, all end points representing LOAELs/NOAELs from studies of relevant duration(s), and end points should be considered as candidate critical effects. Only studies containing fatal flaws need not be considered. More than one candidate critical effect may be identified for evaluation of the body of evidence for hazard identification and use in the dose–response/POD assessment.
If multiple studies document effects for the critical end point or multiple critical end points are being considered, the data should be expressed in a consistent manner, preferably internal dose units (such as plasma concentrations), to facilitate comparisons across data sets. If daily oral exposures/unit body mass are not reported for compounds delivered via drinking water or food, study data (body weights, ingestion rates) or other demographic information such as published default values may be used to calculate these parameters. 23
After selection of the critical effect(s), the previously reviewed studies, for which the study LOAEL/NOAEL was based on a different end point, may be screened again for any additional information (ie, negative data or effects at higher doses) on the identified critical effect(s). Furthermore, a targeted literature search may be conducted to identify additional studies related to the critical effect(s).
Evidence Integration
Evidence integration occurs in 2 primary phases. Evidence is first integrated within the human and animal LOEs separately, followed by integration across LOEs. Mechanistic evidence is also integrated within the LOE using a similar process and is then used as supporting evidence during overall evidence integration (Figure 2).
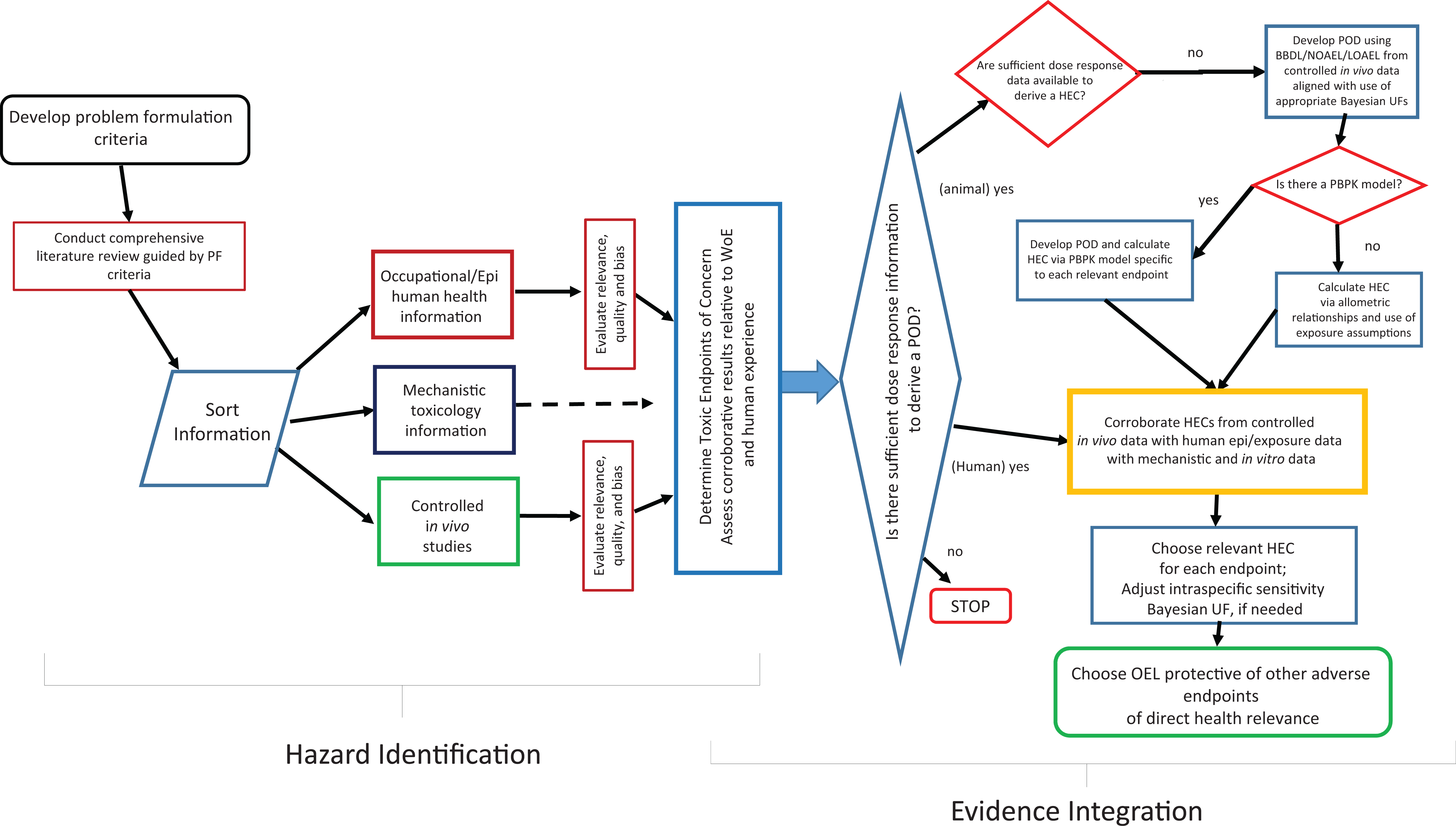
Process schematic for conducting a narrative review, weight of evidence, evidence integration used in the development of an occupational exposure level.
Evidence Integration Within LOEs
The body of evidence is then evaluated, within each LOE, for overall evidence of an effect for each critical toxicological end point. The evidence from animal studies is evaluated as a whole to determine the strengths and weaknesses of the evidence based on criteria developed by Bradford-Hill 15 including strength of effect, consistency, temporality (cause and effect), biological gradient (dose response), plausibility, and coherence. Additionally, directness and precision of end points, elements of the Grading of Recommendations Assessment, Development and Evaluation, and National Toxicology Program Office of Health Assessment and Translation (OHAT) evidence integration tools may also be considered. 4 -24 These criteria are not meant to be a grading scheme or a check list, but rather a guide or framework for evaluating and integrating a body of evidence. The criteria should be used to develop a narrative synthesis of the animal LOE that discusses the evidence for the critical effect across studies, indicating strengths and limitations of the evidence.
Evaluation of candidate critical effects across studies may be aided by evidence visualization. Scatter plots (including stem-and-leaf plots) in which each candidate critical effect being evaluated is plotted on a scatter diagram that displays the normalized effect response (ie, treatment group response divided by control group response), doses/exposures tested for each study may aid in identification of effects that occur at low concentrations/doses in multiple studies and/or have large effect sizes (Figure 3). Additionally, plots of candidate critical effects may be grouped by related sets of end points, model species, or exposure route to facilitate comparisons.
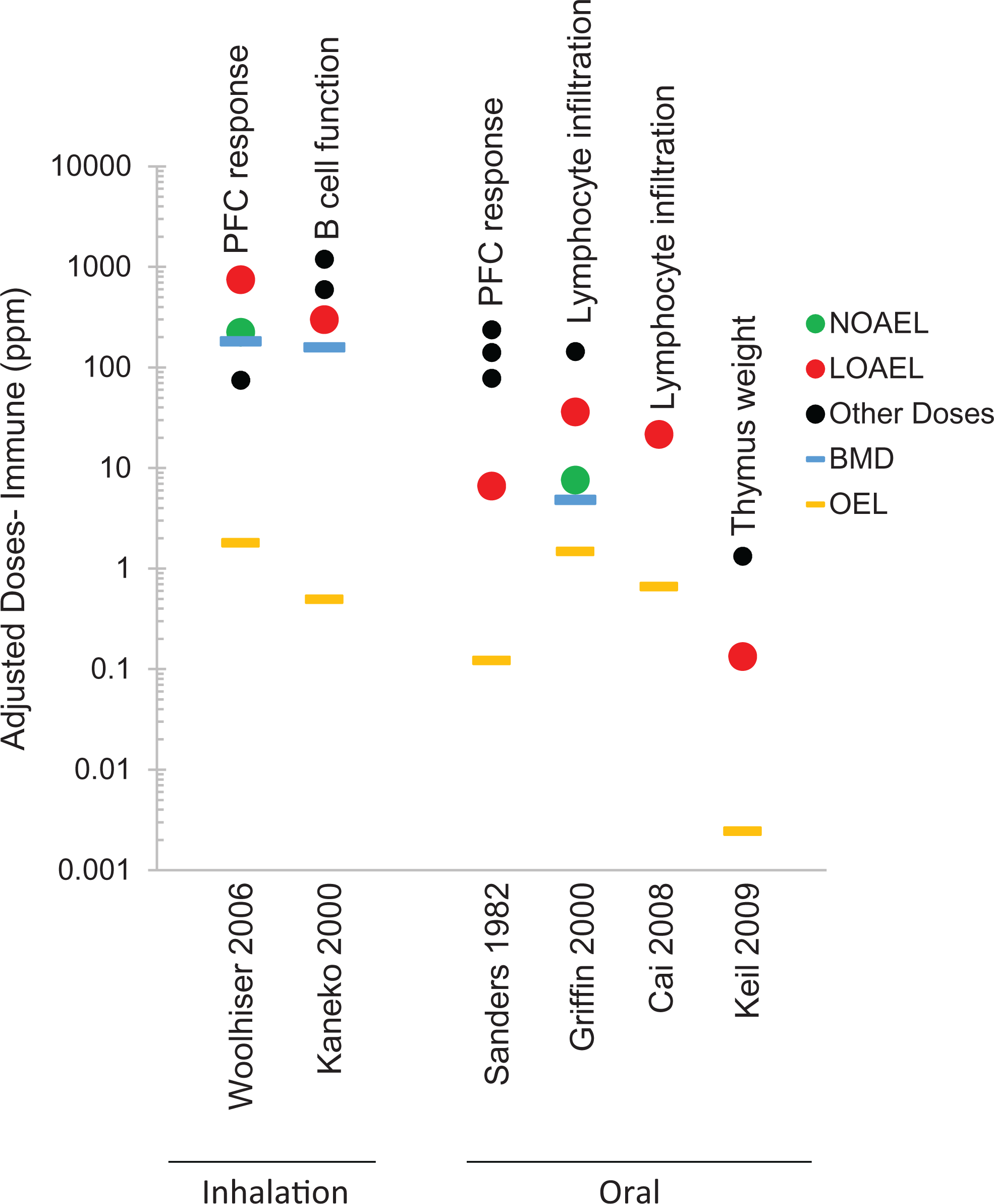
Example scatter plot of experimental studies included in assessment of immunological end points in a trichloroethylene (TCE) assessment. All exposure concentrations tested in each study (adjusted to 40 hours/wk) are plotted, including those concentrations determined to be the NOAEL (green circles) and LOAEL (red circles), as well as other exposure concentrations used (black dots). Results of dose–response modeling (Bayesian Benchmark Dose-Low, blue lines), if conducted, and candidate OELs (orange lines) developed for each study are indicated. Annotations within the graph indicate the critical effect. LOAEL indicates lowest observed adverse effect level; NOAEL, no observed adverse effect level; OELs, occupational exposure levels.
The LOE should be assigned a qualitative hazard characterization of “sufficient evidence of hazard” or “insufficient evidence of hazard” based on the strength of the evidence. Publication bias (ie, the incentive to publish information that is statistically significant and of toxicological importance) increases the probability of reporting and discovering outlier data. Positive outlier data can be useful if measuring an end point previously not identified or using an otherwise sensitive method. However, recognition of type I errors (false positive) is important in OEL development if the end point is of toxicological importance to the population of interest. Here, other LOEs (eg, corroborative evidence of other studies, plausible mechanistic information, and negative data from similar well-conducted studies) can provide support for inclusion or discarding outlier data points.
Sufficient evidence of hazard is generally provided by a set of high quality studies demonstrating consistent effects. However, sufficient evidence of hazard may come from a single high quality study, with or without supporting evidence, provided that no conflicting data are evident in other high quality studies. If supporting evidence is available, it should provide consistent and coherent evidence. Insufficient evidence of hazard should be assigned to LOEs where no or only low quality animal studies are available or if the end points are not informative for health effects in humans. An LOE with data from a high quality study(ies) may also be deemed insufficient evidence if conflicting evidence is available from studies of similar quality and no supporting evidence of coherence is available to increase confidence in the strength of the overall conclusions (Table 3).
Line of Evidence Evaluation Criteria for In Vivo Studies.a

Abbreviation: LOE, line of evidence.
a Adapted from Hill, 1965. 15
Overall Evidence Integration to Develop Hazard Conclusion
Overall evidence integration requires an evaluation of the consistency and coherence of effects across the LOEs, taking into account the relevance of the animal evidence based on the available mechanistic and other relevant evidence. If both human and animal LOEs for the critical effect(s) demonstrate insufficient evidence of a hazard, then an overall designation of insufficient evidence of hazard is determined and an OEL is not developed. If either the human or animal LOEs demonstrate sufficient evidence of hazard then the evidence is integrated across LOEs. Integration across LOEs, rather than following prescribed rules about which LOEs are preferred under given circumstances, follows an alternative explanations approach. 25
The pattern of human, animal, and mechanistic evidence is examined, explanations for the pattern considered, and the logic for selecting one explanation over another explained. For example, if the animal data provide consistent evidence of exposure-related toxicity but human data do not, explanations might include that there are interspecies differences in toxicity due to metabolic differences or that the human evidence derives from low quality studies that were insufficient for detecting effects. Competing explanations would be explored and evidence supporting and refuting each considered (Figure 2).
The justification for critical effect should be presented in a concise statement regarding the conclusions from the LOEs, explaining reasoning for weighing of evidence and citing studies pivotal in reaching conclusions.
Selection of POD
A POD is a data point (empirical or derived) that is used as the starting point for subsequent extrapolation to develop an OEL. Point of departures are derived from high quality studies demonstrating a dose–response. If sufficient ADME data or mechanistic/MOA data exist, then it may be useful to consider IVIVE to a POD that could be used in a corroborative manner. Dose–response assessment should not be conducted for critical effects with insufficient evidence for hazard. Rather than selecting the lowest LOAEL/NOAEL/BMDL as the POD for each critical effect, information in the hazard identification synthesis statements and attributes of the studies are reviewed to select studies with the most appropriate POD for extrapolation to the human health effect of interest.
Although studies are prescreened to select only high quality studies, variation may still exist in particular aspects of study quality and preference may be given to data from studies that are supported by other lines of evidence (eg, studies that are corroborative or report the same level of effect at the same higher quality or more relevant studies). Although human data may generally be preferred due to greater relevance and reduced need for extrapolation, issues identified in the hazard identification synthesis statement (eg, confounders, low sample size) may indicate that the animal data are preferred for POD derivation. Additionally, the human LOE, although relevant and deemed sufficient for hazard identification, may not provide data suitable for dose–response assessment. Administration by a route relevant to known or suspected human exposure route(s) is preferred. Route-to-route extrapolation, preferably using a validated PBPK model, may be conducted; however, higher priority should still be given to data from the same intended route of exposure. Route-to-route extrapolation is contraindicated for irritants. Studies of chronic or subchronic duration are preferred over acute studies for OEL derivation as they provide data applicable to a working exposure lifetime. Studies of shorter duration may be used for compounds that exhibit effects that do not increase in severity and/or incidence with duration of exposure or if the critical effect is a precursor or biomarker that regresses or is not expected to progress with increased exposure periods (eg, irritants). Additionally, critical effects that require exposure/measurement during critical windows would necessitate use of studies of appropriate exposure timing and duration (eg, reproductive and developmental studies). Studies using dose/concentration ranges near reported human environmental exposures are preferred. Preference is also given to studies measuring end points considered more direct measures of adverse health effects.
When multiple studies measuring the same end point/critical effect are determined to be of sufficient quality and adequate for dose–response modeling, candidate PODs are derived for each study. Human equivalent concentrations are derived from PODs using PBPK or default assumptions from animal data when human data are available for comparison purposes. Use of Bayesian benchmark dose (BBMD) methods to derive PODs is preferred over selection of LOAEL/NOAEL from study data points. The BBMD may be derived if not explicitly stated in the study. 26 However, aspects of the data set may impact the ability to conduct BBMD modeling. 27 Optimal data sets suitable for BBMD analysis have: at least 1 exposure level near the benchmark response (BMR) to reduce low dose extrapolation, large sample sizes, and at least 3 treatment levels including a control. Unsuitable data sets include those in which all exposure groups have effects in excess of control including a high response rate in the lowest exposure group and data sets in which only the highest exposure group demonstrates effects in excess of control (unless the response is near the BMR). Rationale for selection of studies for BBMD analysis should be provided.
(Bayesian) Benchmark dose modeling
Benchmark dose (BMD) modeling is widely accepted as preferred over the traditional NOAEL/LOAEL method for dose–response assessment because it takes the entire dose range into consideration and is not restricted to study-defined doses when determining a POD. If BMD analyses were not conducted by study authors and the data are determined to be suitable for BMD modeling, analyses should be conducted to derive a BMD for the exposure that is expected to cause a specified BMR incidence. The type of low-dose extrapolation used, linear or nonlinear, is determined by the MOA. A linear approach is used for extrapolation of cancer end point response to compounds (or their metabolites) that have direct mutagenic activity and are DNA reactive. Linear extrapolation is also used if data are insufficient to establish the MOA or if the MOA is known and does not support a threshold nonlinear approach. 28 A nonlinear approach is used for noncancer effects and for cancer effects if the MOA can be determined and it is not linear at low doses. For some carcinogens, both linear and nonlinear approaches may be used if multiple MOAs are identified and may be operational at different exposure levels. For nonlinear threshold carcinogens, PODs are developed based not on tumor incidence but on a key precursor event leading to cancer (eg, cellular proliferation, cytotoxicity, inhibition of apoptosis, immune suppression, and estrogenic activity). 29 Precursor events may be more evident in shorter term studies which may also be more frequently conducted and more amenable to dose–response modeling due to the dose range used.
For linear-low dose extrapolations, 10% extra risk may be used for cancer bioassays and 1% for epidemiologic cancer data. For both animal and human studies, if there are multiple tumor types, composite or overall risk should be used to characterize the risk of developing a tumor in at least one site. If common dose metrics apply to all tumor types, the multistage model (eg, MS-Combo) may be suitable while the Markov Chain Monte Carlo methods may be used to estimate a POD for overall tumor risk if different dose metrics apply to some tumor types. 30 For dichotomous data, a BMR of 10% extra risk is generally used, however, a BMR of 5% may be used for serious or frank effects (eg, developmental effects). Lower BMRs can be selected for effects such as developmental malformations or for severe effects. 27
For continuous data, 1 standard deviation (SD; note 1) from the mean control response is used unless data are available to establish a definition of biological significance (eg, 10% reduction in body weight). Point of departures derived based on BMRs of 10% extra risk for dichotomous data and 1SD for continuous data should be reported, regardless of selection of BMR, to facilitate comparisons across end points. Model inputs and results should be documented. 27 If data are not suitable for BMD modeling (eg, due to substantial low dose extrapolation or lack of model fit), the traditional NOAEL/LOAEL approach may then be used to determine a POD.
When modeling human cancer data, cumulative exposure and incident cases are preferred (see Environmental Protection Agency Guidelines for Carcinogen Risk Assessment for further guidance). 28 Modeling of human noncancer data is performed in a manner similar to that of laboratory animal data. Modeling of both cancer and noncancer end points, however, often requires specialized methods to account for covariates.
Analyses may be conducted using one of the widely available models including Benchmark Dose Software (BMDS) 27 and BBMD. 26 Regardless of choice of software, use of Bayesian dose–response modeling (limited to dichotomous variables in BMDS currently) and model averaging are preferred over point estimates and individual/hierarchical model selection, since Bayesian approaches consider prior knowledge regarding the distributions of data types that improve predictions and model outputs. 26
Duration Adjustment and Extrapolation
Occupational exposure levels are developed for typical 8 hour/d, 40 hour/wk exposures over the working life-time of an individual. Because experimental exposures sometimes use discontinuous exposure protocols (eg, 6 hours/d, 5 days/wk for 90 days), time weighted averaging may be necessary for chemicals that are nonirritants and act through systemic toxicity. For inhalation exposures, this may require initial duration extrapolation to a 40-hour/wk exposure equivalent. The relationship between toxic effects and a specific toxic response may be expressed as a function of cumulative exposure (Cn × t = k; where C = concentration, n = time scaling exponent, t = time, and k = constant) based on Ten Berge et al. 31 If sufficient data are available to examine the compound-specific relationship between exposure concentration and time, a compound-specific value for the time scaling exponent (n) may be empirically derived. If sufficient data are not available, a default value of n = 1 may be used when extrapolating from shorter to longer exposure durations and a default value of n = 3 when extrapolating from longer to shorter durations. 32 However, the validity of any extrapolation should be carefully considered in light of any available supporting or opposing data (eg, differences between other exposure durations suggest metabolic saturation/induction). For oral studies, variable treatment protocols may require adjustment to standard exposure (eg, 5 days per week). Exposures via diet and drinking water should be converted to daily doses (mg/kg-d) prior to duration adjustment. Adjustment to work week exposure may then be conducted based on the following:
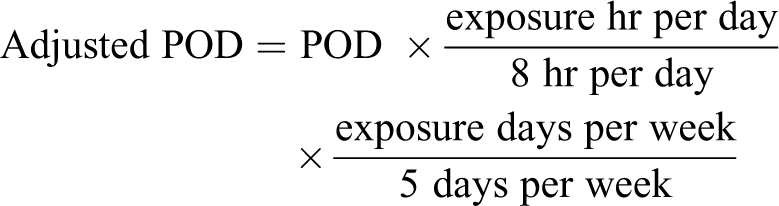
Route-to-Route Extrapolation
Extrapolation from one exposure route to another may be necessary if data from the appropriate/preferred exposure route are very limited or not available. The reviewer should determine the appropriateness of such extrapolations based on the degree to which similar results would be expected due to differences in biological processes (eg, metabolism and distribution) among exposure routes. Considerations should include portal of entry effects (eg, irritation), differential absorption and metabolism, first-pass effects, and route-specific toxicity. In general, confidence is increased when similar systemic effects are observed and absorption is similar via routes of exposure (or data are available on absorption and metabolism at point of entry and target of toxicity occurs via systemic circulation). If route-to-route extrapolation is deemed appropriate, PBPK modeling is generally the preferred method. If data are not sufficient for PBPK modeling, extrapolation may be performed via other methods (eg, using default values, structural analog data, in vitro uptake data, and physical/chemical data); however, these methods should be clearly described. For compounds requiring an OEL despite a limited data base, extrapolation may be performed using default assumptions as long as absorption, metabolism, and distribution by both routes may be reasonably assumed. If data from multiple exposure routes are available, route-to-route extrapolation may be used to understand how exposure route affects the dose–response.
Extrapolation to Human Equivalent Exposure Levels
Exposure levels from animal studies may be converted to human exposure levels before or after PODs analysis, depending on the nature of the relationship between animal and human external and internal doses. If dose response relationships are expected to be linear, interspecies extrapolation may be conducted after POD analysis. In these cases, the animal PBPK model is used to estimate an internal dose for the animal POD and the human PBPK model, when available, is then used to estimate an HEC/human equivalent dose (HED), in terms of external dose, for the POD. When relationships are nonlinear, animal PBPK models may be used to estimate the internal animal dose for each external animal dose. The internal doses are then used in a dose–response analysis (eg, BBMD) to determine a POD for the animal data. The human PBPK model is then used to estimate an HEC/HED at the POD. When PBPK models are not available, bolus (oral gavage) doses in animal studies are allometrically scaled using mg/kg3/4-day. 33,34 Inhalation exposures and oral exposures delivered via diet and drinking water are not allometrically scaled because intake, and therefore exposure, is dependent on basal metabolic rate and scales according to body size. 35
Derivation of Candidate OELs
Noncancer and Nonlinear Threshold Effects Related to Cancer
Candidate OELs are estimates of exposure that are expected to be without appreciable risk of adverse health effects to workers exposed during a working lifetime. An OEL is then derived from the HECs, typically after application of UFs, if needed, to account for uncertainty associated with the use of animal toxicity data to predict human health effects (interspecies; UFA), individual variability expected within the worker population (intraspecies; UFH), extrapolation across exposure durations (subchronic to chronic; UFS), and extrapolation to below the threshold for adverse effect (LOAEL to NOAEL; UFL). If few data are available, a UF to account for lack of information may be considered (database; UFD). Values applied to these UFs typically range from 1 to 10, with default values of generally 10 or equivalent to 1 log unit. However, default values are increasingly replaced with chemical-specific or data-derived values. 36,37 Although selection of UFs is specific for each compound under consideration and is based on all available data and expert judgment, general guidance on the application of UFs is available. 36 -38 Application of interspecies UFs will depend upon the use, corroboration or other human experience information, and expert judgment and could be used to increase or decrease the candidate OEL depending upon the data. Human data should be compared with HECs and UFs may be adjusted accordingly.
Interspecies uncertainty factor (UFA)
In the absence of data to the contrary (eg, human workplace information), humans are typically considered to be more susceptible than animal species and a default UFA of 10 is applied for PODs derived from animal studies. The UFA may be subdivided into toxicokinetic and toxicodynamic components for application of data-derived UFs. 39 This factor may be divided into equal component parts of 3 (ie, 10½) and default values reduced based on supporting data. The toxicokinetic component may be reduced based on the use of PBPK modeling and the adjustment of animal exposure levels to HEDs/HECs. Both components may be reduced and a value less than 1 used when data indicate that humans are less sensitive than the animal model(s).
Intraspecies uncertainty factor (UFH)
The UFH may similarly be broken into component parts to account for toxicokinetic and toxicodynamic variability among humans. Default factors of 3 may be used if the POD represents effects in a sensitive human population or if the effects are not expected to differ among individuals (ie, direct contact irritation). The UFH may be reduced if the OEL is to be applied to a population with reduced variability (eg, a subset of the population).
Subchronic to chronic exposure extrapolation (UFS)
A default value of 10 is typically applied to the UFs factor to account for uncertainty in extrapolating responses observed following subchronic exposure to develop an OEL relevant for a chronic exposure duration. This value may be decreased when the end point under consideration is not expected to increase in incidence or severity with increasing duration of exposure (eg, irritant effects, no increase in effect observed between subacute and subchronic studies, or no increase probability for adverse effect for increasing exposure duration given etiology of disease).
Lowest observed adverse effect level to NOAEL/BBMDL extrapolation (UFL)
The UFL addresses the uncertainty of extrapolating from an LOAEL rather than an NOAEL or BMDL. In contrast to other UFs, the default factor for UFL (4.5) is empirically derived based on the ratio of LOAEL/NOAEL from 175 chronic studies. 40
Database deficiency (UFD)
If the toxicity data for a compound is very limited, then an adjustment is sometimes made to account for the uncertainty regarding whether additional studies might identify more sensitive effects. Examples include a UF of 3 for lack of systemic toxicity data in a second species and a value of 3 for lack of reproductive toxicity information. There is flexibility in using this UF and final decision should be based on professional judgment.
Bayesian application of UFs
Traditional application of UFs relies on multiplicative compounding of individual UFs, which may result in an overly conservative composite UF. An alternative approach, which was recommended by the National Academy of Sciences 25 and others, 41,42 is to use Bayesian methods to apply UFs. Bayesian approaches incorporate an estimate of the appropriate adjustment based on prior knowledge as well as a level of uncertainty in that estimate, which are reflected as the log-normal distributions of the geometric mean (µ) and geometric SD (σ) of the composite UF. Simon et al 40 provided a refinement of the method recommended by the National Academy of Sciences, which incorporates the µ and σ for each individual UF, rather than only considering these parameters for the overall composite UF. Our approach is adapted from the methods described by Simon et al, with the following formula for applying UFs to derive a candidate OEL:
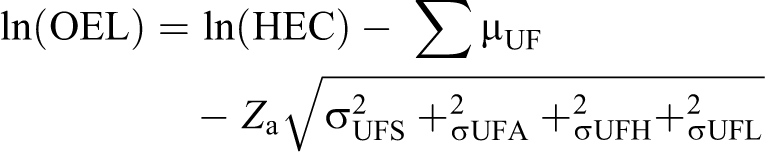
Where: Zα is the Z -score, which for the 95th percentile is 1.645.
The geometric means for all UFs except for UFL are assumed to equal 1 (µ = 0 for a log-normal distribution), indicating that these UFs address uncertainty only. When µ = 0, σ is calculated as the ln(UF)/Zα. Thus, at the 95% CI, a UF of 1 corresponds to σ = 0, a UF of 3 corresponds to σ = 0.668, and a UF of 10 corresponds to σ = 1.4. Other confidence levels may be selected to reflect the state of knowledge or confidence in the sources of uncertainty (see Table 1 in Simon et al 40 ). As described by Pieters et al, 43 the geometric mean and SD of the LOAEL/NOAEL ratio from 175 chronic studies are 4.5 and 1.7, respectively (µ = 1.504 and σ = 0.531 on log-normal scale). Thus, these values are used for UFL instead of those adopted for the other UFs. As a result, the sum of µUF in this analysis is either 1.504 or 0, depending on whether or not the HEC was derived from an LOAEL.
This formula differs from that used by Simon et al in 2 key ways: When the HEC was derived from BBMD analysis, Simon et al used the BBMD, rather than the BBMDL, as the basis for the HEC derivation and added a separate operator to account for the variance between the BBMD and BBMDL. Our current method uses the BBMDL as the basis for the HEC derivation and thus does not incorporate this additional measure of variance. The use of the BBMDL as the basis for the HEC will generally result in a slightly more conservative OEL compared to the method employed by Simon et al, although the ratio of the BBMD/BBMDL can vary substantially based on a number of factors including the BMR level, the BBMD software model, the number of animals in each dose group, the variance of the data set, and how close the BMR is to the actual data. Simon et al weighed the merits of applying the UFA either before or after incorporating PBPK modeling to derive the HEC. Their analysis indicated that this decision had a modest impact on OEL derivation. Simon et al applied the UFA prior to derivation of the HEC, while our current approach applies UFs after derivation of the HEC. However, it is recommended that UFA be applied to internal dose for accuracy when a PBPK model is available or when nonlinearity is expected.
Although Pieters et al 43 also determined the ratio of subchronic/chronic NOAELs based on 149 studies (geometric mean and geometric SD are 1.7 and 5.6, respectively), these values are not employed in the derivation of UFS for either the current assessment or in the analysis by Simon et al. 40 Our rationale for not using these values over a default UFS is that the relatively high geometric SD reported by Pieters et al 43 indicated that there is a great deal of uncertainty in this estimate, which limits its utility. Similar estimations of data-derived ratios for subchronic-to-chronic and LOAEL-to-NOAEL extrapolations as those derived by Pieters et al 43 have also been determined by others, 44 and the use of either set of published values may be appropriate.
Cancer Risk Values
Cancer risk values are predictive risk estimates. When linear low dose extrapolation is used, the result is a slope factor or a unit risk derived by assuming a linear relationship from the POD to the background response. Oral slope factors (mg/kg-day) are converted to inhalation unit risk (IUR; mg/m3) as needed using standard breathing rates and body weight for humans (ie, 20 m3/d and 70 kg). Risk is determined as:
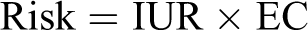
Where IUR = inhalation unit risk; EC = exposure concentration (mg/m3)
Exposure concentration is determined as:
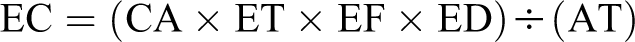
Where EC = exposure concentration (mg/m3); CA = contaminant concentration or OEL (mg/m3); ET = exposure time and for a worker it is assumed an 8 hour work day; EF = exposure frequency and for workers we are conservatively assuming 250 work days per year; ED = exposure duration and we are assuming a 30- to 45-year working career; AT = averaging time (hours) and is the lifetime (70 years) × 365 days per year × 24 hours per day.
Occupational exposure levels are initially developed primarily from noncancer effects. Cancer risk, considering working lifetimes of 30 and 45 years are developed at the carcinogenic risk of 1 × 10−4 to 1 × 10−6 levels and compared with noncancer candidate OEL values. Relative confidence associated with cancer risk estimates are then used to select the most appropriate OEL proposed for use.
Selection of the OEL
If multiple candidate OELs have been developed, the candidate values should be evaluated individually and as a group. Rather than simply defaulting to the lowest POD or HEC, selection of organ or organ system value is based on study quality/relevance considerations, patterns in the data, as well as overall confidence in the candidate OEL. Preference is given to candidate OELs that are based on BMDL rather than NOAEL/LOAEL extrapolations, higher quality and more relevant studies, studies that do not require extrapolation or use data-driven extrapolation rather than default UFs, and end points with strong evidence of causal effect. This decision can be strengthened by corroborative data where a toxicity end point is consistently observed at similar exposures across controlled animal studies and human experience and is supported by other lines of evidence (eg, mechanistic, in vitro, IVIVE, read-across, and QSAR). An organ/organ system OEL may be derived using a single study determined to be the most appropriate or may be a derived as a composite value from a group of studies identified as representing a pattern of response. Outliers should be rigorously scrutinized.
To help support this evaluation and to allow for clear visualization of reported adverse effects, each candidate OEL and its supporting POD are plotted on a scatter diagram (Figure 3). This scatter plot displays the study identifier, critical effect, doses/exposures tested, the PODs, and candidate OEL. The plotted critical effects may be arranged based on related sets of end points that pertain to a single critical health effect or may be created for individual end points if sufficient data are available. If studies differ in duration or route of exposure, duration adjustment, route-to-route extrapolation, and determining HECs (if compared with available human experience) should be conducted prior to graphing to standardize data for comparisons. Presentation of PODs in this manner assists in identifying patterns of consistency and aids in review.
The final OEL for the compound is selected from among any candidate OELs representing different organ or organ system toxicities. The lowest OEL may typically be selected, however, consideration may be given to the confidence in each value.
Expression of Values
Occupational exposure levels for inhalation exposures should be expressed in units of mass/volume (mg/m3), however, if the substance exists as a gas or vapor at normal room temperature and pressure, the OEL may be expressed in parts per million. All OELs should be expressed using 2 significant figures; however, rounding should only be performed on the final OEL value, not on interim calculations. If data are determined to be insufficient for OEL derivation, the OEL will be presented as “No value established” and the reason(s) for not developing an OEL clearly stated.
Discussion
A critical tenet in toxicology is that development of adverse effect is dependent upon magnitude of exposure. Exposure is an expression of time and concentration which can be proportional at the level where adverse effects occur. Defining that threshold level of effect where toxicity occurs in humans is the challenge where data are not always published with that purpose. Here we describe a process that integrates quantitative and qualitative toxicity information from a variety of data streams in a holistic manner to derive OELs to be protective for workers. This process deviates from other methods where all of the data can be integrated and used in a professional judgment approach toward OEL development. Studies are evaluated based on quality, relevance to OEL development, and applicability to demonstration of adverse effect or disease state. Qualitative information is used to support interspecies extrapolation, interpretations of the possible influence of error, and corroboratively support OEL derivation.
There is logic in choosing the most sensitive level where adverse effects could conceptually occur from which to develop an OEL. There is also value in using a very prescriptive approach that could easily be repeated by others. Unfortunately, as more data become available, the probability for correctly choosing a single study to represent an exposure that would be predictive for adverse effect decreases as error rates increase with a greater number of end points measured. Furthermore, as more variable types of information is developed, it becomes more important to integrate that information as it informs the selection of a critical effect from which to base an OEL. We submit that a flexible narrative approach is needed to integrate as much of this information as possible. We know of no procedure-driven method that could be done without professional judgment to derive an OEL.
Fedak et al 45 recently suggested how new molecular techniques in toxicology could further enhance the strength of association between environment and effects. However, specific methods to make these associations stronger have not been described or demonstrated. Here we describe a method that combines the Bradford-Hill criteria as well as those suggested by Fedak et al to provide a corroborative process for interpreting causation across all streams of evidence.
Ultimately, the challenge of deriving a value based on all available information and clear logic is dependent upon the rationale used at each decision point and the accurate interpretation of the toxicity information. This risk increases proportionally with the amount of pertinent studies available. We recognize that bias is likely present in all efforts and the greatest challenge comes from demonstrating that all pertinent information has been reviewed and that an objective rationale is provided for the OEL derivation. We also propose that values are peer-reviewed to reduce the probability for subjective decisions and increase confidence in OEL derivation.
Footnotes
Acknowledgments
The authors would like to express their gratitude to Mr. John Seibert and Ms. Laurie Cummings for support and helpful suggestions on this manuscript.
Author Contributions
Johnson, Mark S. contributed to conception and design, contributed to acquisition, analysis, and interpretation, and critically revised manuscript; Lent, Emily May contributed to conception and design, contributed to acquisition, analysis, and interpretation, and drafted manuscript; Leach, Glenn J. contributed to design, contributed to acquisition and interpretation, drafted manuscript, and critically revised manuscript; Sussan, Thomas contributed to conception and design, contributed to analysis, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The views expressed in this publication are those of the authors and do not necessarily reflect the official policy or position of the Department of the Army, Department of Defense, nor the US Government. Drs. Mark S. Johnson, Emily May Lent, and Thomas Sussan were employees of the US Government in the conduct of this work. The preparation of this publication was part of their official duties. Title 17, U.S.C., §105 provides that copyright protection under this title is not available for any work of the US Government. Title 17, U.S.C., §1010 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person’s official duties.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.