
Abstract
Permitted daily exposure (PDE) values are used by some toxicologists to support the safety qualification of various types of impurities found in a drug substance (DS) or drug product (DP). Permitted daily exposure values are important tools for the toxicologist, but one must be aware of their limitations to ensure that they are used appropriately and effectively in the risk assessment process. First, a toxicologist must always perform a comprehensive analysis of all available animal and human safety data for an impurity, including identifying any data gaps that may exist. Second, if adequate data are available and there are no genotoxicity concerns, an appropriate well-designed repeat-dose toxicity study in animals should be chosen to calculate the PDE. It is important to note that PDE values qualify general systemic toxicity and not necessarily local toleration end points such as irritation and sensitization that are more concentration than dose dependent. In addition, a PDE value calculated from a general toxicity study in animals may not necessarily qualify for reproductive toxicology end points. Lastly, PDE values should never be thought of as analytical limits for or acceptable levels of an impurity in a DS or DP, as this ignores quality considerations. Using safety information from several chemicals as proxy impurities, this article serves as an educational primer to facilitate a better understanding of the development and use of PDE values in the risk assessment process.
Introduction
Toxicologists are responsible for the risk assessment and safety qualification of chemical impurities in a drug substance (DS) or drug product (DP). Although the presence of such impurities is inevitable, they deserve critical attention since they offer no therapeutic benefit to patients. Thus, from a safety perspective, a toxicologist must determine the potential risk to patients resulting from the unintentional exposure to chemical impurities in a pharmaceutical formulation when they are encountered. Some of the more common types of impurities found in a DS or DP include (but are not limited to) organic process-related impurities and degradants, solvents, elementals, and leachables. 1
Once an impurity is identified in a DS or DP, it must be qualified by a toxicologist. This process confirms the safety of the impurity a human will receive at the maximum daily dose of the drug containing it. The International Council for Harmonisation (ICH) provides guidance on analytical and toxicological considerations for organic process-related impurities and degradants assuming no genotoxicity concerns. 2,3 For these impurities, qualification is typically achieved through their presence in bulk material used to conduct repeat-dose toxicity studies during nonclinical development, ensuring that the doses the animals receive are adequate to support that which humans will at each impurity’s specified level. The qualification of solvents, elementals, and leachables in a DS or DP poses additional challenges for the toxicologist; since unlike organic process-related impurities and degradants, there is a marked structural distinction among them and to the drug itself, thereby increasing the likelihood of multiple and/or differing toxicological profiles. As evidence of this, the ICH guidance documents outlining analytical and toxicological considerations for residual solvents and elementals in pharmaceuticals lists profiles for these potential impurities ranging from those deemed relatively innocuous, to those having concerning enough toxicities that their presence in drugs should be strictly limited if not completely avoided. 4,5 Corresponding proposals on analytical and toxicological considerations for leachables derived from the manufacturing equipment and/or the container closure system are also available to help toxicologists address the presence and safety of these types of impurities present in pharmaceutical formulations given by inhalation and other parenteral routes of administration. 6 -8
To aid toxicologists in qualifying residual solvents and elemental impurities, permitted daily exposure (PDE) values were developed by the ICH, which provide safety thresholds for many of these types of impurities that are commonly encountered in a DS or DP. The process that the ICH uses to develop a PDE value has been clearly defined, being based on the results of repeat-dose animal toxicity data and employing several modifying factors to support extrapolation to humans. 4,5,9 The ICH procedure for developing a PDE is rigorous and methodical, including a thorough vetting process of the proposed values not only within the ICH Expert Working Group but also globally by external constituents. 10 The thoroughness of the research, development, and vetting process the ICH uses to establish their PDE values helps to ensure their worldwide regulatory acceptance.
There are many approaches toxicologists can use to leverage safety data to risk assess impurities in a DS or DP, including employing ICH-based PDE values for residual solvents and elemental impurities when available. Where an ICH-based PDE value does not exist for any given impurity, some toxicologists calculate their own using the ICH paradigm. Given such values are being developed outside the formal ICH process and subject to worldwide regulatory scrutiny, there are various matters toxicologists need to consider when taking this approach. Importantly, like the approach used by the ICH, a comprehensive evaluation of all available animal and human safety data for the impurity should be performed to fully understand its toxicological profile. The US Food and Drug Administration (FDA) has provided clarification on the types of data and information needed to establish acceptance criteria for impurities in pharmaceutical formulations, including some challenges and limitations that need to be considered to appropriately accomplish this task. 11 Similarly, this article outlines several important considerations and limitations concerning PDE values, especially those self-derived, to ensure that they are used appropriately and effectively in the context of the drug development process.
Methods
Following a review of the literature, several chemicals were identified with well-characterized toxicological profiles to serve as proxy impurities for this investigation. It is important to note that the proxy compounds chosen may or may not be entities that would typically present as impurities in a pharmaceutical product. As such, the purpose of this article is not to confirm their safety in a pharmaceutical formulation, but rather to use their particular data set to highlight important points to consider when developing and/or leveraging a PDE value for risk assessment purposes. For each chemical, if an ICH-derived PDE value was not listed, a range of potential choices was developed based on repeat-dose animal toxicity data as described by the ICH. 4,5,9 Regarding the latter, a general synopsis of the basic modifying factors developed by the ICH that must be considered when developing a PDE for a nongenotoxic impurity is presented in Table 1. F1 allows the surface area/body weight ratios of various nonclinical animal species to be compared to humans, F2 addresses individual human sensitivity differences to adverse effects, F3 enables extrapolation from the length of a particular animal study to support a chronic human safety threshold, F4 accounts for the seriousness of the toxicity reported in the animal study, while F5 takes into account if a no observed adverse effect level (NOAEL) was identified and if not were there any concerns about the toxicity being reported including dose-response considerations. Once the data from the animal studies were reviewed, the modifying factors were used to develop the PDE using the following general formula:
Standard Modifying Factors to Calculate a PDE Value From Repeat-Dose General Toxicity Studies Conducted in Animals.

Abbreviations: NOAEL, no observed adverse effect level; PDE, permitted daily exposure.
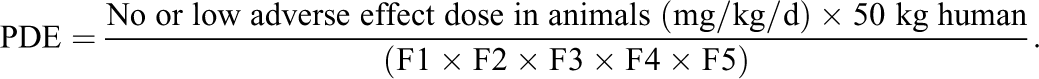
Once available, the ICH or self-derived PDE values were subsequently placed in the context of several scenarios that a toxicologist could encounter when developing a safety position for an impurity in a pharmaceutical product, with the goal being to enable a retrospective examination of their applicability and potential limitations in the risk assessment process. Although the self-derived PDE values are based on the standard ICH procedure, they are for instructional purposes only and should not be viewed as endorsed by any scientific or regulatory body. This article focuses on the derivation of PDE values and not any other safety-related thresholds for chemicals that may exist.
Results
Special Population Considerations
Benzyl alcohol (BA) is a solvent and antimicrobial preservative used in cosmetics, foods, and a wide range of pharmaceutical formulations. 12 Although BA has an overall favorable safety profile, a fatal toxic reaction known as “gasping baby syndrome” can occur in low birth weight neonates following its use as a preservative in solutions used to flush umbilical catheters. 13 Based on clinical data, the estimated threshold dose for BA to elicit this syndrome is 99 mg/kg/d from 2 to 28 days, although there is one report of a dose range of 32 to 105 mg/kg/d for 7 days causing breathing difficulty. 14 As a result of this syndrome, the US FDA has recommended that BA not be used in such umbilical flushing procedures and that the use of pharmaceutical formulations containing preservatives be avoided in newborns. 12
Given these data, a potential scenario of risk assessing the short-term intravenous administration of a DP containing a ≥16 mg/d dose of BA would be concerning for a preterm neonate weighing 0.5 kg (32 mg/kg/d × 0.5 kg). The longest oral and parenteral (inhalation) repeat-dose toxicity studies conducted in animals with BA reported in the literature were of 2 years and 28 days in duration, respectively. 15,16 In these studies, no adverse effects were reported in rats up to the highest oral (400 mg/kg/d) and inhalation (182 mg/kg/d) doses tested. Using the NOAELs for these studies in rats, a range of PDE calculations is provided in Table 2. Using the ICH paradigm that includes a standard human body weight of 50 kg, the PDE values calculated from both studies surpass the reported safety threshold for BA in preterm neonates (16 mg/d), with the one derived from the oral study (400 mg/d) clearly falling within the lethal range. Alternatively, in the context of the current risk assessment scenario, simply adjusting the calculation to focus on a body weight that is more reflective of the actual patient population brings both PDE calculations to below a level of known concern, with the value from the parenteral study (more consistent with the actual route of patient exposure) providing the most conservative margin. The importance of this example is not to qualify BA as an impurity in any particular DP, but to use data derived from it to stress the importance of considering body weight differences that may exist within a special population when developing or using a PDE to support safety.
Example PDE Calculations From Repeat-Dose Animal Toxicity Studies With Benzyl Alcohol.

Abbreviations: NOAEL, no observed adverse effect level; PDE, permitted daily exposure.
a NOAEL × 50 kg ÷ (5 × 10 × 1 × 1 × 1).
b NOAEL × 50 kg ÷ (5 × 10 × 10 × 1 × 1).
c NOAEL × 0.5 kg ÷ (5 × 10 × 1 × 1 × 1).
d NOAEL × 0.5 kg ÷ (5 × 10 × 10 × 1 × 1).
Reproductive Toxicology Considerations
Thalidomide was a sedative hypnotic widely used around the world until it was discovered to be a human teratogen in the early 1960s. 17 Even though thalidomide was banned from use in the United States during this time frame, it has become available for investigational use to treat several diseases. 18 -21 The product is available as an oral capsule formulation (50, 100, 150, and 200 mg), with the label having a “black box” warning that even a single dose (1 capsule, regardless of strength) can cause severe birth defects if taken by a pregnant woman. 22 Data from repeat-dose oral toxicity studies have been published characterizing the systemic adverse effects of thalidomide in dogs and rodents. 23,24 In a 53-week oral toxicity study in dogs (0, 43, 200, or 1,000 mg/kg/d), thalidomide did not induce any major systemic toxicity concerns. Findings included mammary duct dilatation and/or glandular hyperplasia in some of the treated females and the presence of bile pigment in the livers of high-dose males, with the authors designating the NOAEL as 200 mg/kg/d. Similarly, no seriously concerning toxicities occurred in 13-week oral toxicity studies conducted in mice and rats with thalidomide (0, 30, 300, or 3,000 mg/kg/d). Dose-related hepatic centrilobular hypertrophy was reported in male mice, an effect also observed in the high-dose females. In rats, the most prominent finding reported was a decrease in body weight in the treated animals, with a dose-related effect being more evident in the males. Collectively, the authors reported NOAELs of 30 mg/kg/d (male rats) and 3,000 mg/kg/d (mice and female rats) for these studies. Using the NOAELs from these studies, a range of PDE calculations is provided in Table 3. In 3 of the 4 scenarios presented, basing the derivation of the PDE value on repeat-dose toxicology studies in animals that assessed general systemic toxicity end points met or exceeded the teratogenic dose in humans, with the one exception being derived from male (not female) rats. Conversely, by employing modifying factors recommended by the ICH when using reproductive toxicology data, 4,5,9 basing the PDE on the lowest observed adverse effect level (LOAEL; 10 mg/kg/d) reported in an early embryonic development study conducted in female rabbits with thalidomide (0, 10, 50, or 100 mg/kg/d) where adverse embryonic effects occurred at all dose levels yielded a value (0.4 mg/d) below the dose of concern. 25 This example with thalidomide as a proxy impurity reinforces that reproductive toxicology data should always be considered separately from those derived from a general systemic toxicity study, as the resulting calculated PDE values derived from them can markedly differ from one another.
Example PDE Calculations From Repeat-Dose Animal Toxicity Studies With Thalidomide.

Abbreviations: NOAEL, no observed adverse effect level; PDE, permitted daily exposure; LOAEL, lowest observed adverse effect level.
a NOAEL × 50 kg ÷ (2 × 10 × 10 × 1 × 1).
b NOAEL × 50 kg ÷ (12 × 10 × 5 × 1 × 1).
c NOAEL × 50 kg ÷ (5 × 10 × 5 × 1 × 1).
d NOAEL × 50 kg ÷ (5 × 10 × 5 × 1 × 1).
e LOAEL × 50 kg ÷ (2.5 × 10 × 1 × 5 × 10).
Irritation Considerations
Ethanol, acetic acid, and formic acid are all ICH Q3C listed class 3 solvents with calculated PDE values of at least 50 mg/d (Table 4). 4,9 While class 3 solvents have an overall low degree of toxic potential, all 3 of those listed in Table 4 are irritants. The assessment of irritation is very often assessed in ocular or dermal studies conducted in animals using standard protocols with results typically reported as the administered dose. In ocular irritation studies conducted in rabbits with ethanol, formic acid, and acetic acid, doses of 79, 122, and 10.5 mg, respectively, were severely irritating to the eye. 26 -28 Thus, in all 3 of these examples, the calculated PDE values exceeded doses that are severely irritating to the eye, especially in the case of acetic acid.
ICH Q3C Listed Solvents and Irritation Potential.
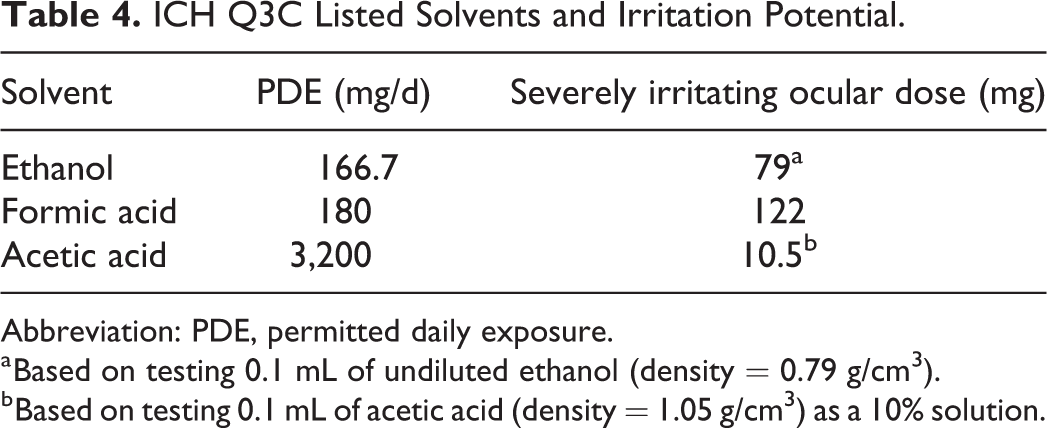
Abbreviation: PDE, permitted daily exposure.
a Based on testing 0.1 mL of undiluted ethanol (density = 0.79 g/cm3).
b Based on testing 0.1 mL of acetic acid (density = 1.05 g/cm3) as a 10% solution.
Skin Sensitization Considerations
Methylisothiazolinone (MI) is a preservative used in several personal care products and cosmetics. When used in this capacity either alone or in conjunction with the biocide methylchloroisothiazolinone, allergic contact dermatitis (ACD) can occur. 29 -31 Based on human patch test data, doses of MI ranging from 14.3 to 60 µg (expressed per square centimeter) have elicited ACD. 32 Data from repeat-dose toxicity studies have been reported characterizing the systemic adverse effects of MI in rats and dogs. 31 In 3-month oral toxicity studies conducted in rats (0, 6.5, 19, or 66 mg/kg/d) and dogs (0, 3, 10, or 41 mg/kg/d) with MI, no adverse effects were reported up to the highest doses tested. Using the NOAELs from these studies, PDE calculations are provided in Table 5. In both scenarios, basing the derivation of the PDE value on repeat-dose toxicology studies in animals that assessed general systemic toxicity end points exceeded doses causing ACD in patch tests conducted in humans. Likewise, using modifying factors recommended when using reproductive toxicology data, 4,5,9 basing the PDE for MI on the NOAELs reported in early embryonic development (rats and rabbits) and 2-generation studies (rats) where no adverse effects occurred up to the highest doses tested also exceeded that reported to cause ACD. 31
Example PDE Calculations From Repeat-Dose Animal Toxicity Studies With Methylisothiazolinone.
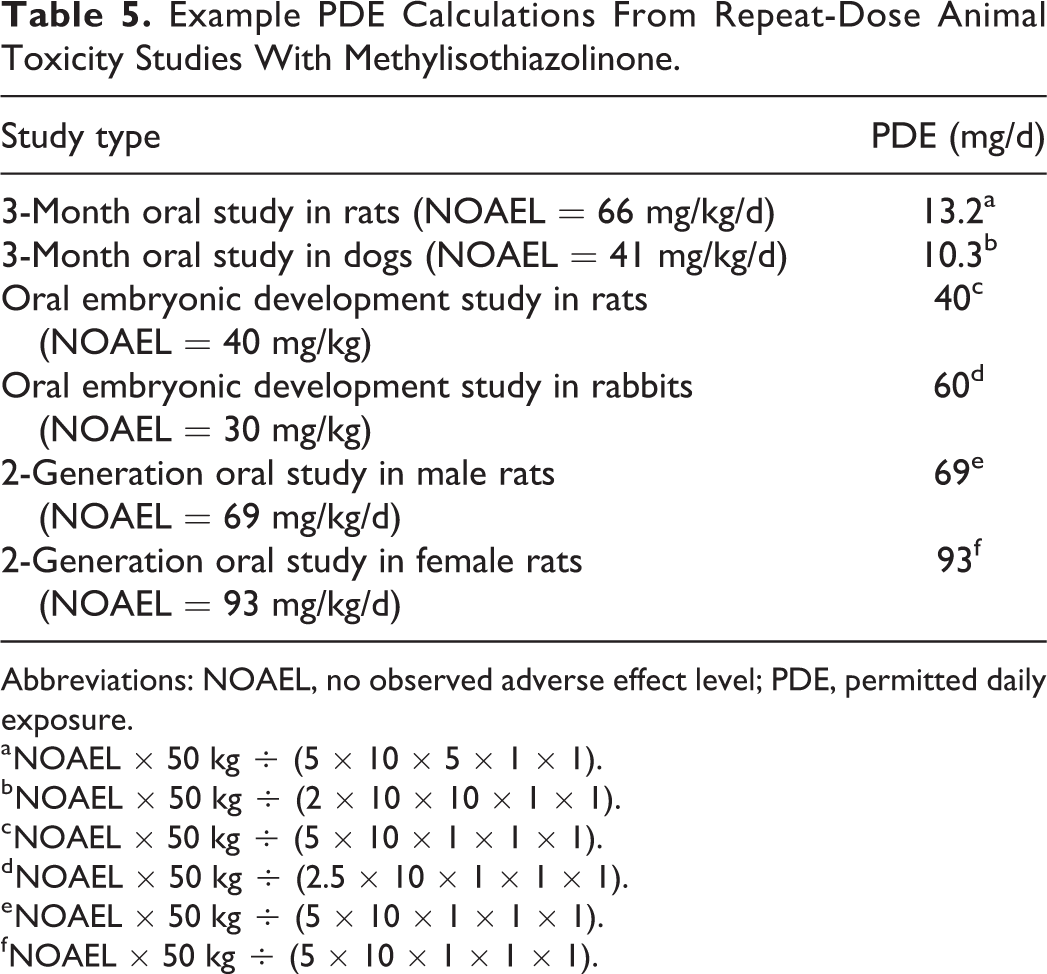
Abbreviations: NOAEL, no observed adverse effect level; PDE, permitted daily exposure.
a NOAEL × 50 kg ÷ (5 × 10 × 5 × 1 × 1).
b NOAEL × 50 kg ÷ (2 × 10 × 10 × 1 × 1).
c NOAEL × 50 kg ÷ (5 × 10 × 1 × 1 × 1).
d NOAEL × 50 kg ÷ (2.5 × 10 × 1 × 1 × 1).
e NOAEL × 50 kg ÷ (5 × 10 × 1 × 1 × 1).
f NOAEL × 50 kg ÷ (5 × 10 × 1 × 1 × 1).
Route/Target Organ Considerations
Maleic anhydride (MA) is a chemical used in the manufacture of various products including resins, dyes, pharmaceuticals, and pesticides. 33 Data from repeat-dose oral toxicity studies have been reported characterizing the systemic adverse effects of MA in rats and dogs. 34,35 In a 2-year oral toxicology study in rats (0, 10, 32, or 100 mg/kg/d), a dose-related decrease in body weight occurred, and the NOAEL was reported as 10 mg/kg/d. Similarly, no seriously concerning toxicities occurred in a 3-month oral toxicology study conducted in dogs with MA up to the highest dose tested of 60 mg/kg/d. Using the NOAELs from these studies, PDE calculations are provided in Table 6. Regarding parenteral exposure to MA, in a 1-month inhalation toxicity study conducted in rats, various adverse effects were reported including prominent damage to the upper respiratory tract and lungs down to the lowest dose tested of 1.7 mg/kg/d. 36 The PDE for MA calculated from the inhalation study in rats (0.0017 mg/d) is markedly lower than those developed following oral administration to animals (10-15 mg/d), with the pulmonary system not being adversely affected in the latter studies. This example with MA as a proxy impurity highlights that marked differences in toxicity (in terms of target organ effects and potency) can occur with the same chemical depending on route of exposure.
Example PDE Calculations From Repeat-Dose Animal Toxicology Studies With Maleic Anhydride.
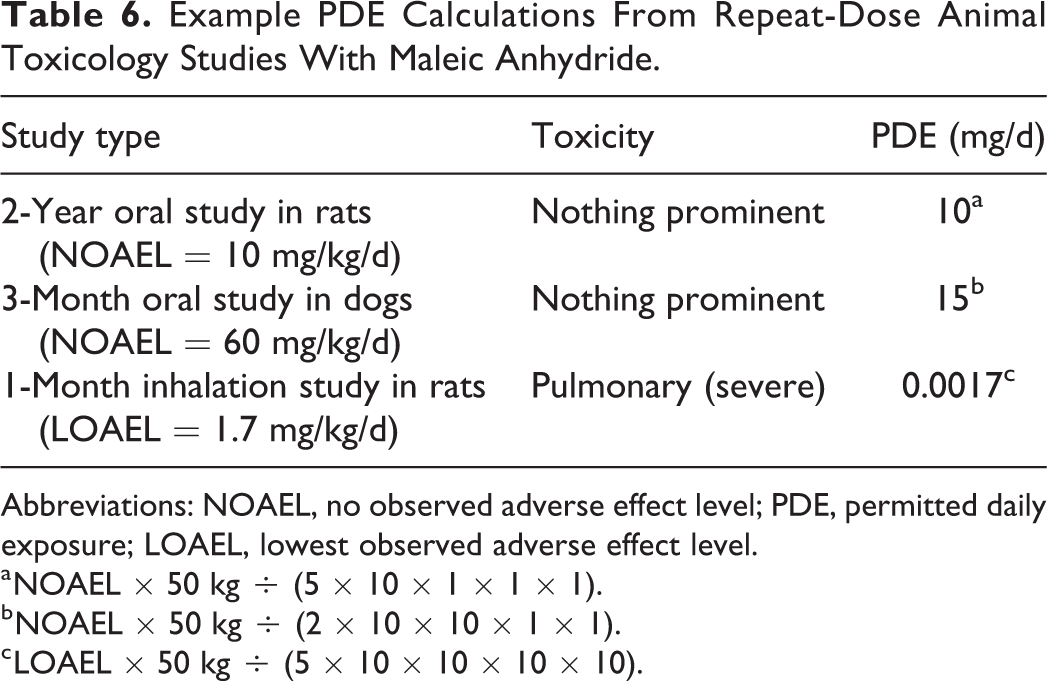
Abbreviations: NOAEL, no observed adverse effect level; PDE, permitted daily exposure; LOAEL, lowest observed adverse effect level.
a NOAEL × 50 kg ÷ (5 × 10 × 1 × 1 × 1).
b NOAEL × 50 kg ÷ (2 × 10 × 10 × 1 × 1).
c LOAEL × 50 kg ÷ (5 × 10 × 10 × 10 × 10).
Mutagenicity/Carcinogenicity Considerations
2-Nitrotoluene (o-nitrotoluene) is a chemical intermediate that is a DNA-reactive mutagenic carcinogen. 37 In 2-year oral (in-feed) carcinogenicity studies conducted in rats (0, 25-30, 50-60, or 90-100 mg/kg/d) and mice (0, 150-165, 320-360, or 700-710 mg/kg/d) with 2-nitrotoluene, a dose-related increase in tumor incidence at multiple sites occurred in both species. 38,39 The PDE values of 0.25 mg/d (250 µg/d) and 0.63 mg/d (630 µg/d) are calculated for 2-nitrotoluene based on the LOAELs of these studies in rats and mice, respectively (Table 7). In both instances, the PDE values exceeded the threshold of toxicological concern (TTC) of 1.5 µg/d, the chronic daily dose developed for the control of DNA-reactive mutagenic impurities in pharmaceuticals to limit potential carcinogenic risk. 40
Example PDE Calculations From Repeat-Dose (Carcinogenicity) Studies With O-Nitrotoluene.
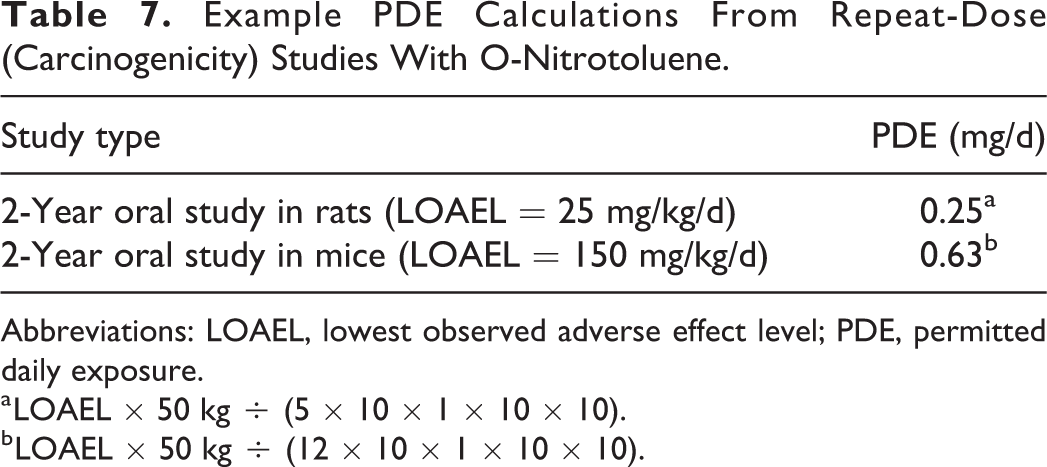
Abbreviations: LOAEL, lowest observed adverse effect level; PDE, permitted daily exposure.
a LOAEL × 50 kg ÷ (5 × 10 × 1 × 10 × 10).
b LOAEL × 50 kg ÷ (12 × 10 × 1 × 10 × 10).
Discussion
The process of risk assessing impurities in pharmaceutical products is both a science and an art. As any toxicologist regularly performing this function can attest to, extensive research and interpretation of the available safety information for an impurity is critical, but subsequently leveraging it to develop a comprehensive, compelling position supporting acceptable human exposure to it can be complex and challenging. The process must always begin with a review of the available in silico, in vitro, animal, and possibly human safety data for the impurity to fully characterize its toxicological profile. Subsequently, developing a safety qualification position for the impurity must consider critical matters of dose, duration of exposure, route of administration (including potential bioavailability differences), patient population (such as juvenile patients or women of childbearing potential), the disease being treated, and any other pertinent health matters that may be commonly encountered in the target population. 41 -43 Even if a toxicologist encounters an impurity they may have risk assessed previously, the aforementioned considerations demand continued due diligence, since what was acceptable in one situation may not be in another. In addition, the toxicologist must always be cognizant that scientific data and regulatory perceptions for any given chemical impurity can change over time. For example, the ICH-derived PDE value for the solvent tetrahydrofuran was lowered from 121 to 7.2 mg/d due to nonclinical safety data being generated confirming it was a nongenotoxic animal carcinogen. 4 In addition, beginning some years ago, human exposure to phthalate plasticizers (especially in children) came under scrutiny in many regulatory circles, 44 -46 making risk assessing them as leachable impurities from a container closure system more challenging, given the availability of replacement materials that could fulfill their function in products. Thus, the toxicologist must take into account a wide range of matters when risk assessing an impurity, always carefully considering the most current available information (scientific and regulatory) and using it to develop a fit for purpose safety position addressing the specific clinical situation where exposure is anticipated. Following these principles when making a risk assessment of an impurity is critical, especially in situations where a PDE value may be used to support a safety qualification. Since PDE values (ICH or self-derived) are used by many individuals in risk assessment processes, the examples provided in this article demonstrate the importance of fully understanding their scope to ensure they are leveraged appropriately.
The sensitivity of the human pediatric population to the toxicity of many substances differs from that of adults, with both increased and decreased responses reported depending on the chemical. 47 The BA example highlights the potential seriousness of these differences; since while mortality can occur in preterm infants following intravenous exposure to it during catheter flushing procedures, its widespread use in many marketed pharmaceutical formulations attests to an overall acceptable safety profile in the general population. 12 Other examples of sensitivity differences to excipients that exist between adult and pediatric patients are well recognized, with many being highlighted in the Safety and Toxicity of Excipients for Paediatrics (STEP) Database developed by the European Paediatric Formulation Initiative. 48 The standard formula the ICH uses to develop a PDE assumes a 50-kg individual, 4,5,9 a value intentionally chosen for risk assessment purposes to conservatively represent an all-encompassing worldwide weight for an adult. The use of BA as a proxy impurity to develop a PDE value using the standard ICH calculation paradigm would not have supported its safety (or any other chemical with a similar toxicological profile) in preterm infants, while adjusting the weight to more accurately reflect that of the actual patient population (from 50 to 0.5 kg) does. This highlights the importance of understanding the components of the standard ICH calculation paradigm and ensuring they accurately reflect the actual patients being exposed when special population circumstances exist in any given risk assessment scenario.
As described by the ICH, a PDE value can be calculated from repeat-dose animal toxicity studies assessing either general or reproductive toxicology end points. 4,5,9 As the example with thalidomide as a proxy impurity demonstrates, the range of PDE values calculated from studies assessing general systemic toxicity end points in mice, rats, and dogs (6-600 mg/d) exceeded that derived from a teratology study conducted in rabbits (0.4 mg/d), while only the latter study yielded a value below the teratogenic dose in humans. Thus, one must not assume that a PDE value calculated from a general toxicity study will always sufficiently support reproductive toxicology safety. In this example, using thalidomide as a proxy impurity represents a striking confirmation of this point, which could be theoretically applicable to any chemical impurity that may possess a similar liability, but not have a comprehensive enough reproductive toxicology safety data set to confirm it. In instances where both general systemic and reproductive toxicity data are available for an impurity such as with MI (Table 5), one should consider calculating PDE values based on both sets of data, using the more conservative one to support both end points. Where reproductive toxicity studies are not available for an impurity, the toxicologist may consider using corresponding data from similarly structured surrogate compounds to develop a safety position. In addition, several publications are available defining thresholds of toxicological concern for reproductive and developmental toxicology end points that could be used to reinforce a safety position. 49 -51 These are critical issues to consider for any impurity, especially when exposure of women of childbearing potential is anticipated. In addition, any potential reproductive toxicology hazards posed by the drug itself containing the impurity are appropriate to consider in the overall risk–benefit analysis.
Irritation and sensitization are important toxicological end points to consider when risk assessing impurities, especially when topical/parenteral exposure is anticipated. Regarding the examples presented characterizing the solvents ethanol, formic acid, and acetic acid as potentially irritating substances, ICH Q3C does not specifically address this end point, which is more concentration than dose dependent. As such, the ICH-derived PDE values (presented as dose) do not qualify this end point since they were not developed to do so. Intuitively, the less concentrated a potentially irritating solution is, the more likely it will be tolerated. Thus, there are various approaches toxicologists can take to adequately risk assess a potentially irritating impurity. No observed adverse effect concentration (NOAEC) values for a chemical tested in models to assess irritation (in vitro or in vivo) can be leveraged to support safety. Occupational workplace exposure level concentrations may be available for a potentially concerning agent (such as Permissible Exposure Limits by the US Occupational Safety and Health Administration) that are designed to protect workers from both local (including mucous membrane irritation) and systemic toxicity issues following repeated, prolonged daily exposure. The FDA Ophthalmic Division has presented a concentration-dependent acceptance criterion for impurities in ocular products, which would include those that may be irritants. 52 Regarding sensitization, specifically chemicals that cause the delayed T-cell–mediated mechanism resulting in ACD, reactions become less severe in human patch test studies when exposure to the same dose is spread over a greater surface area. 30 Thus, the concept of a threshold concentration exists for these types of chemicals. Like with the end point of irritation, PDE values were not developed to consider the toxicological end point of sensitization. There are examples of safety thresholds for certain chemicals that elicit ACD. Specifically, Danish authorities have regulated objects with nickel-coated surfaces coming into contact with the skin at 0.5 µg/cm2/wk. 53 In addition, based on a comprehensive review of available safety data for a number of these agents, the European Scientific Committee on Consumer Safety developed a concentration of 0.01% (in cosmetics) as a reasonable safety threshold for chemicals capable of eliciting this effect. 54 Of special mention are chemical impurities capable of eliciting an immediate (immunoglobulin E-mediated) hypersensitivity or anaphylactic reaction. Given the seriousness of the potential outcome of this reaction, it is difficult to envision many circumstances where the presence of an impurity that can cause this effect could be justified in a pharmaceutical formulation. Collectively, the qualification of the toxicological end points of irritation and sensitization is more appropriately performed by leveraging animal and/or human data specifically developed to assess these issues after analytical verification of the actual concentrations of the potentially offending agent in the final product are confirmed.
Using MA as a proxy impurity highlights the critical importance of considering route of exposure and target organ toxicity when calculating a PDE, especially when they are interlinked. The PDE values calculated for MA from the repeat-dose oral toxicity studies in rats (10 mg/d) and dogs (15 mg/d) are almost 5,900-fold greater than when exposure to rodents was via inhalation (0.0017 mg/d). In addition, the pulmonary system was the target organ of toxicity following exposure to MA via inhalation, an effect not observed in the oral studies. It is difficult to envision that such a profound route of administration difference in the toxic response to MA is related to low oral bioavailability. Even using an additional modifying factor of 100 as described by ICH Q3D to convert the oral PDE value to a parenteral one would not have provided as conservative of a safety threshold for MA as the one developed from an actual inhalation toxicology study. 5 Regardless, the adverse effects on pulmonary tissue caused by MA in the inhalation study likely resulted from a direct local effect of a chemical characterized as strongly irritating, 33 an end point previously discussed as concentration dependent and therefore not supported by a PDE value. This example of MA as a proxy impurity underscores the importance of performing a comprehensive examination of all critical safety data for any given chemical before deciding on the most appropriate rationale to develop a safety position. If one were to envision a chemical with this toxicological profile presenting as a leachable in an inhalation product intended to treat patients with chronic obstructive pulmonary disease who already have compromised pulmonary function, its presence would be difficult to justify at any level. Indeed, in this scenario, if such a chemical presented as an extractable derived from the product’s container closure components, the toxicologist would be justified to recommend a material change rather than attempting to develop what would likely be a precarious risk assessment position.
The potential of a chemical impurity in a drug to elicit a carcinogenic response in humans is one of the most serious outcomes a toxicologist will encounter during the risk assessment process. As such, the ICH M7 guideline provides TTC-based acceptable daily intake values for a mutagenic impurity in a pharmaceutical formulation ranging from 1.5 (≥10 years of daily exposure) to 120 µg/d (≤1 month of daily exposure). 40 Using 2-nitrotoluene as a proxy impurity demonstrates that the PDE values calculated from the 2-year carcinogenicity studies conducted in rats (250 µg/d) and mice (630 µg/d) exceeded both the short-term (120 µg/d) and chronic (1.5 µg/d) TTC values for mutagens defined by ICH M7. In addition, the compound-specific TTC value calculated for 2-nitrotoluene based on the animal carcinogenicity data (4.66 µg/d) is also breached. 37 Collectively, as conservative as the ICH-based PDE calculation paradigm is, it is not sufficient to support a safety position for compounds with mutagenicity/carcinogenicity concerns that are more appropriately addressed by ICH M7. Unlike with 2-nitrotoluene, carcinogenicity data for impurities may not readily exist. In such cases, the toxicologist must consider appropriate in silico analyses and/or genetic toxicology studies in setting an acceptable safety threshold based on ICH M7 lifetime to less than lifetime limits developed for mutagenic impurities. 40
A PDE value only defines a safety limit for a chemical and should never be viewed as an acceptable level of an impurity in a pharmaceutical formulation. Quality considerations are paramount when dealing with impurities in drugs, which should always be controlled based on best manufacturing process capabilities. This ensures diminished exposure to a chemical with no therapeutic value to the patient that someone might still have an unanticipated adverse reaction to. One would only be highlighting to regulatory authorities a poorly controlled manufacturing process by suggesting the presence of a 100 mg/d dose of ethanol as a “residual solvent” in any DP is justified and reasonable since it is below its ICH PDE value of 167.7 mg and therefore safe. One should always be mindful of 3 basic principles concerning impurities in pharmaceutical formulations. First, there must be a justifiable and logical reason for its presence in the drug, otherwise the product may be viewed as adulterated by a regulatory authority. Second, once the impurity is analytically confirmed in the drug, the amount present should be deemed reasonable from a quality perspective. Third, the toxicologist needs to support safety, which may include the use of PDE values. Thus, a PDE value can never be used to justify either the presence or the amount of an impurity in a pharmaceutical formulation, only its safety. The PDE value was developed as a tool to risk assess impurities present in pharmaceutical formulations, and its use in any other circumstance necessitates input from a toxicologist to ensure it is being used appropriately.
In conclusion, the PDE value is an important tool that many toxicologists use to risk assess impurities in pharmaceutical formulations. Whether or not an ICH-derived PDE value already exists for a chemical, it is the responsibility of the toxicologist to fully understand the toxicological profile of each individual impurity encountered in any given drug. Subsequently, whatever method a toxicologist uses to risk assess an impurity, be it a PDE value or not, it must be tailored to address the specific clinical use of the drug containing it and the patients exposed to it. Following this process helps to ensure appropriate leveraging of the available toxicology data to successfully qualify the impurity and ensure patient safety.
Footnotes
Acknowledgments
The authors are grateful to Andrea Nilson for valuable discussions and critical review of the manuscript.
Author Contributions
Ball, D.J. contributed to conception and design; analysis and interpretation; drafted manuscript; and critically revised manuscript. Beierschmitt, W. contributed to conception and design; acquisition, analysis, and interpretation; drafted manuscript; and critically revised manuscript. Both authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy. Both authors contributed to the material in this manuscript based on their combined experiences risk assessing impurities in pharmaceutical products in a regulatory environment and their involvement in ICH-based initiatives to develop safety thresholds for chemicals. Both authors were involved throughout the process of developing the components of this manuscript and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.