
Abstract
A workshop entitled “Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis” was held at the 2018 Genetic Toxicology Association annual meeting. The objectives of the workshop were to provide an educational forum and use case studies and live multiple-choice polling to establish the degree of similarity/diversity in approach/opinion of the industry experts and other delegates present for some of the more challenging decision points that need to be considered when developing a compound-specific exposure limit (ie, acceptable intake or permissible or permitted daily exposure). Herein we summarize the relevant background and case study information for each decision point topic presented as well as highlight significant polling responses and discussion points. A common observation throughout was the requirement for expert judgment to be applied at each of the decision points presented which often results in different reasoning being applied by the risk assessor when deriving a compound-specific exposure limit. This supports the value of precompetitive cross-industry collaborations to develop compound-specific limits and harmonize the methodology applied, thus reducing the associated uncertainty inherent in the application of isolated expert judgment in this context. An overview of relevant precompetitive cross-industry collaborations working to achieve this goal is described.
Keywords
Introduction
The synthesis of an active pharmaceutical ingredient (API) involves the use of solvents, reagents, and synthetic intermediates, many of which carry through into the final API as low-level impurities. There are a number of International Council for Harmonisation (ICH) guidance documents which provide a framework for the identification, qualification, and control of impurities. A large majority of impurities related to the synthesis of an API and degradants associated with the stability of the final drug product are addressed within ICH Q3A 1 and Q3B, 2 respectively. More specific recommendations have been published for impurities that arise from the use of solvents (ICH Q3C), 3 elementals (ICH Q3D), 4 and impurities with mutagenic potential (ICH M7 [R1]). 5 In addition to establishing a common set of expectations for the identification and control of such impurities, the ICH Q3C, Q3D, and M7 guidelines provide compound-specific exposure limits for numerous chemicals that are commonly used in pharmaceutical synthesis. For example, the ICH M7 addendum (Appendix 3) provides compound-specific limits for 16 known mutagens and carcinogens. These compound-specific limits, referred to as permissible or permitted daily exposures (PDEs) and acceptable intakes (AIs, also a daily exposure), represent a dose of chemical that would present negligible risk to patients assuming lifetime daily exposure.
Publishing PDEs and AIs for chemicals that are relevant to pharmaceutical synthesis (and potentially other industries) is of high value, especially in the context of regulatory guidance. A major benefit is that it establishes a common and consistent view for both regulators and pharmaceutical sponsors of what represents a safe daily dose to patients. For chemicals that are not addressed in regulatory guidance, one can still consider establishing PDEs/AIs. The ICH M7 addendum, as well as ICH Q3C and ICH Q3D, provide guidance on the derivation of compound-specific limits for impurities. Subsequent to the ICH M7 addendum, further elaboration on methods to derive compound-specific limits for mutagenic carcinogens, nonmutagenic carcinogens, and nonmutagenic and noncarcinogenic drug substance impurities was provided in a publication that also included PDEs/AIs for 20 pharmaceutical reagents and byproducts. 6 Due to the need to apply expert judgment and the inherent subjectivity of some of the decisions made in the process of arriving at a value, it is reasonable to expect differences in approach and the stated PDE/AI value when a published limit is not available.
Materials and Methods
In 2018, a workshop entitled “Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis” was held at the Genetic Toxicology Association Annual Meeting at the University of Delaware. The main objectives of the workshop were to provide an educational forum as well as gauge the diversity in approach/opinion from the industry experts and other delegates present in the workshop for key decision points that need to be made in the development of compound-specific limits. The topics of the workshop included: (1) identification of the key toxicology study and the critical effect in that study to be used as the point of departure (POD); (2) selection of adjustment (ie, safety) factors; (3) determination of PDEs/AIs for different routes of exposure; and (4) approaches to consider when there are no toxicology data available to establish a PDE or AI. These were selected as they represent some of the more challenging aspects of deriving a PDE or AI. For each topic, case studies were presented, and the workshop delegates were asked to respond to several multiple-choice questions using the interactive Poll Everywhere electronic polling system. 7 The responses to the questions were tallied in real time which allowed attendees to see the degree of similarity/diversity of opinion of those present in the workshop. It is important to note that the experience/expertise of the workshop participants ranged from novice to expert. Subsequently, delegates engaged in a discussion to elaborate on the basis for their responses.
Herein relevant background information for each workshop topic and case study are summarized and significant delegate polling responses and discussion points highlighted. A common observation throughout was the requirement for expert judgment at some key decision points often resulting in different reasoning to arrive at a compound-specific PDE. As such, the following workshop report supports the value of precompetitive cross-industry collaborations to develop compound-specific limits and, as much as possible, harmonize the methodology applied to PDE limit derivation, which reduces the associated variability inherent in the application of isolated expert judgment in this context.
Results
Identification of the Key Toxicology Study and the Critical Effect in That Study to Be Used As the POD
The ICH M7 guideline outlines approaches for calculating AIs and PDEs for pharmaceutical impurities that are mutagenic carcinogens based on compound-specific risk assessments. The PDE approach as defined by ICH M7 is appropriate for mutagenic impurities with evidence for a nonlinear dose–response relationship, such as those compounds that interact with DNA but must overwhelm DNA-repair mechanisms to induce an adverse effect. Permissible or permitted daily exposure can be calculated based on carcinogenicity data, in vivo genetic toxicology studies when available, surrogate toxicity end points such as reproductive and developmental toxicology end points, or any toxicity finding in a chronic dosing study that is relevant to carcinogenicity (eg, preneoplastic lesion). The assumption is that exposure below the threshold dose does not pose a cancer risk. For mutagenic carcinogens with no evidence for a threshold, an AI is calculated based on carcinogenic potency and linear extrapolation to a theoretical excess cancer risk of less than 1 in 100,000 for a 50 kg individual. The TD50 is a numerical description of carcinogenic potency and represents the daily lifetime dose that induces tumors in half of the test animals over background. Equation 1 is used for AI calculation using linear extrapolation.
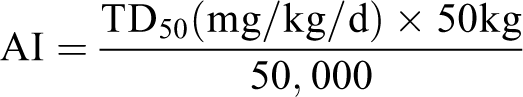
Equation 1: The equation used for AI calculation. An extrapolation factor of 50,000 is used to extrapolate from a 1 in 2 cancer risk to 1 in 100,000.
An alternate approach for linear extrapolation is the use of benchmark dose (BMD) analysis. The BMD is a statistical measure of the dose at a predefined response derived from experimental data. A BMDL10 is an estimated lower dose which will cause no more than a 10% cancer incidence in rodents at the lower bound 95% CI; BMDL10 may be a more accurate index of carcinogenic potency than TD50 when the dose–response data are sufficiently robust to provide insight into the shape of the dose–response curve for carcinogenesis, especially at the lower doses. To calculate an AI, BMDL10 would be divided by an extrapolation factor of 10,000.
In addition to choosing the most appropriate risk assessment method, multiple toxicology/carcinogenicity studies might be available with multiple organ-specific adverse effects observed, and the assessor must select one upon which to base the derivation of a limit. In the case of mutagenic carcinogens, it is recommended in ICH M7 that the most sensitive species, tumor site, and sex be selected to derive the AI. However, it is recognized that the derivation of the AI should also be based on high-quality studies (if possible), relevant to the exposure of interest, and tumors that are relevant to humans.
Workshop case study
During the workshop, ethylene oxide was used as an example mutagenic carcinogen, and the goal of the discussion was to identify the most appropriate carcinogenicity study for the AI calculation. Ethylene oxide is a DNA alkylating agent that is mutagenic in the bacterial reverse mutation assay and induces a variety of genotoxic effects in mammalian cells in vitro. 8 In vivo, ethylene oxide causes translocations, micronuclei, and chromosomal aberrations in rats and humans. Since ethylene oxide is a mutagen that directly interacts with DNA, the most appropriate approach for calculating an AI is linear extrapolation from a carcinogenicity study.
The Carcinogenicity Potency Database (CPDB) is an internationally recognized resource for standardized carcinogenic potency data from diverse cancer bioassays where TD50 values are provided per study, per tissue, and per tumor type for each compound. 9 For those compounds where more than one carcinogenicity study was performed, the CPDB calculates a harmonic mean TD50, however the ICH M7 addendum states that using the lowest TD50 instead of the mean is a more conservative estimate and the harmonic mean should not be used. Thus, for compounds such as ethylene oxide, where more than one study is available, the risk assessor must choose the most appropriate TD50 value for calculating the AI. The CPDB, however, is no longer being updated. As a result, Lhasa Limited is now providing TD50 values through the Lhasa Carcinogenicity Database using methodology described in Thresher et al. 10 The main difference between the Lhasa Carcinogenicity Database and CPDB is that Lhasa excludes the calculation of a TD50 in certain cases where the carcinogenicity data were deemed not robust, such as when a single dose group is used in the study.
ICH M7 provides guidance for the selection of the most robust carcinogenicity study with additional considerations for selection of tumor and tumor site, route of administration, and human relevance (Table 1).
Guidance Provided Within ICH M7 5 for the Selection of the Most Robust Carcinogenicity Study.

Abbreviations: AI, acceptable intake; ICH, International Council for Harmonisation; TD50, a numerical description of carcinogenic potency which represents the daily lifetime dose that induces tumors in half of the test animals over background.
Table 2 summarizes the carcinogenicity data for ethylene oxide in the CPDB (date accessed December 2018) and for each study lists the TD50 value from the most sensitive tumor site, species, and sex. Some study details are not available in CPDB and specifics of pooled controls and forestomach tumors were obtained from the original publications. From Table 2, the workshop delegates were asked to choose which study is most appropriate for calculating the AI or whether to choose the harmonic mean of 21.3 mg/kg/d. The resulting AI which would result from selecting each of these TD50 options would range from 7.4 to 70.7 µg/d.
Summary of the Carcinogenicity Study Data for Ethylene Oxide From the CPDB (Date Accessed December 2018).
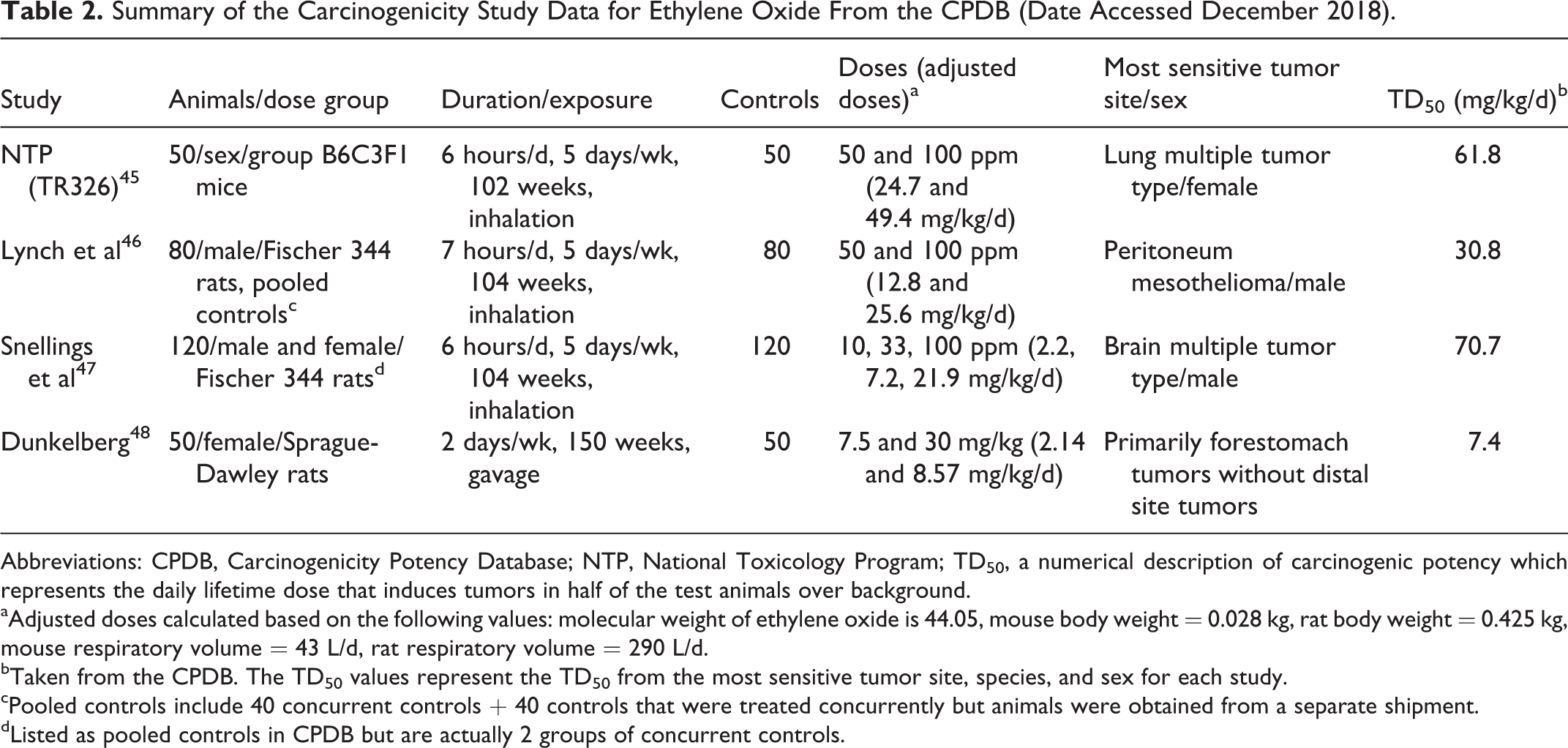
Abbreviations: CPDB, Carcinogenicity Potency Database; NTP, National Toxicology Program; TD50, a numerical description of carcinogenic potency which represents the daily lifetime dose that induces tumors in half of the test animals over background.
aAdjusted doses calculated based on the following values: molecular weight of ethylene oxide is 44.05, mouse body weight = 0.028 kg, rat body weight = 0.425 kg, mouse respiratory volume = 43 L/d, rat respiratory volume = 290 L/d.
bTaken from the CPDB. The TD50 values represent the TD50 from the most sensitive tumor site, species, and sex for each study.
cPooled controls include 40 concurrent controls + 40 controls that were treated concurrently but animals were obtained from a separate shipment.
dListed as pooled controls in CPDB but are actually 2 groups of concurrent controls.
The results of the delegate poll are presented in Supplemental Figure 1. These showed that 30.8 mg/kg/d was selected most often (50% of votes cast where the total votes cast in the poll was 16), followed by 70.7 mg/kg/d (25% of votes cast) and 7.4 mg/kg/d (13% of votes cast). The discussion that followed delegate polling revealed that most participants tended to choose the lowest TD50 value when studies had similar robustness taking into consideration the number of dose groups, number of animals per dose group/control group, and whether the controls were pooled or not. The study with a TD50 value of 30.8 mg/kg/d remained the most popular choice although the control animals were pooled and there were only 2 dose groups, because participants preferred to take a conservative approach and the robustness of the study was considered sufficient when compared with the other options. One participant chose 61.8 mg/kg/d because the study did not use pooled controls and thus in their opinion represented the most robust study. The study with a TD50 value of 7.4 mg/kg/d was excluded by most participants because forestomach tumors are not considered relevant for calculating human risk, and using the harmonic mean of 21.3 mg/kg was excluded by most delegates because ICH M7 does not recommend using the harmonic mean but rather the most sensitive site from the most robust study.
Exposure to ethylene oxide in humans can also occur from endogenous sources such as lipid peroxidation, oxidation of methionine and hemin, and metabolism by intestinal bacteria. 11 In another poll, 94% of participants (total votes cast = 18) agreed that endogenous exposure should be factored into the risk assessment, but the approach should be case-by-case depending on the available data. Several ideas were expressed and included both calculating the AI based on carcinogenicity data and considering endogenous exposure as a reference point while others suggested forgoing AI calculation if data for calculating endogenous levels were robust enough.
The delegate polling exercise revealed that even with the extensive guidance provided in ICH M7, the assessor must apply their expert judgment to make multiple decisions on a case-by-case basis and those decisions may differ between individual assessors. Nonetheless, by having multiple experts contributing to the discussion, a consensus can be reached that is acceptable to most and therefore gives greater confidence in the derivation of a safe exposure limit.
Selection of Adjustment Factors
Adjustment factors (AFs) for impurities have been referred to as safety, modifying, or uncertainty factors. 12 The intent of the AFs is to lower the POD, that is, the starting dose for the derivation of a PDE, to a value that would not result in harm to the large population of patients who consume the drug. Although the AF approach may differ depending on the organization, this section describes the general principles used to determine the PDE using Equation 2, as described by ICH Q3C and ICH M7. It also describes challenges and thoughts when applying the AF, as exemplified by the results of the workshop delegate polling exercise.
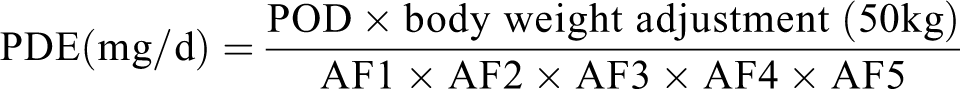
Equation 2: The equation used for PDE calculation, where POD is the point of departure, AF1 is for interspecies variability, AF2 for intraspecies or human variability, AF3 for duration of exposure, AF4 for severity of effect, and AF5 for lowest observed adverse effect level (LOAEL) to no-observed (adverse) effect level (NO(A)EL) extrapolation.
Adjustment factors are not intended to be a “checkbox” approach. They should be used at the discretion of the toxicologist. Although some AFs may be obvious to apply, others may be unclear and require expert judgment from the toxicologist. The rationale for each AF should be well documented in a summary monograph document facilitating transparency and peer-review. If the total AF is too large (ie, >5,000), then the degree of uncertainty may be too great to derive a PDE and other approaches may be appropriate. 12
Interspecies variability (AF 1)
In the ICH Q3C, 3 Q3D, 4 and M7 guidelines, 5 interspecies variability has been defined by the allometric differences between animals and humans. Surface area factors are used to estimate the metabolic differences between animal species and humans. If the toxicity data are derived from humans, the AF would be 1. The default factors for interspecies extrapolation that are recommended in the ICH impurity guidelines are presented in Table 3.
Summary Tables for Interspecies Extrapolation (AF 1), Intraspecies or Human Variability (AF 2), Different Exposure Duration (AF 3), and Severity of Effect (AF 4) Adjustment Factors.
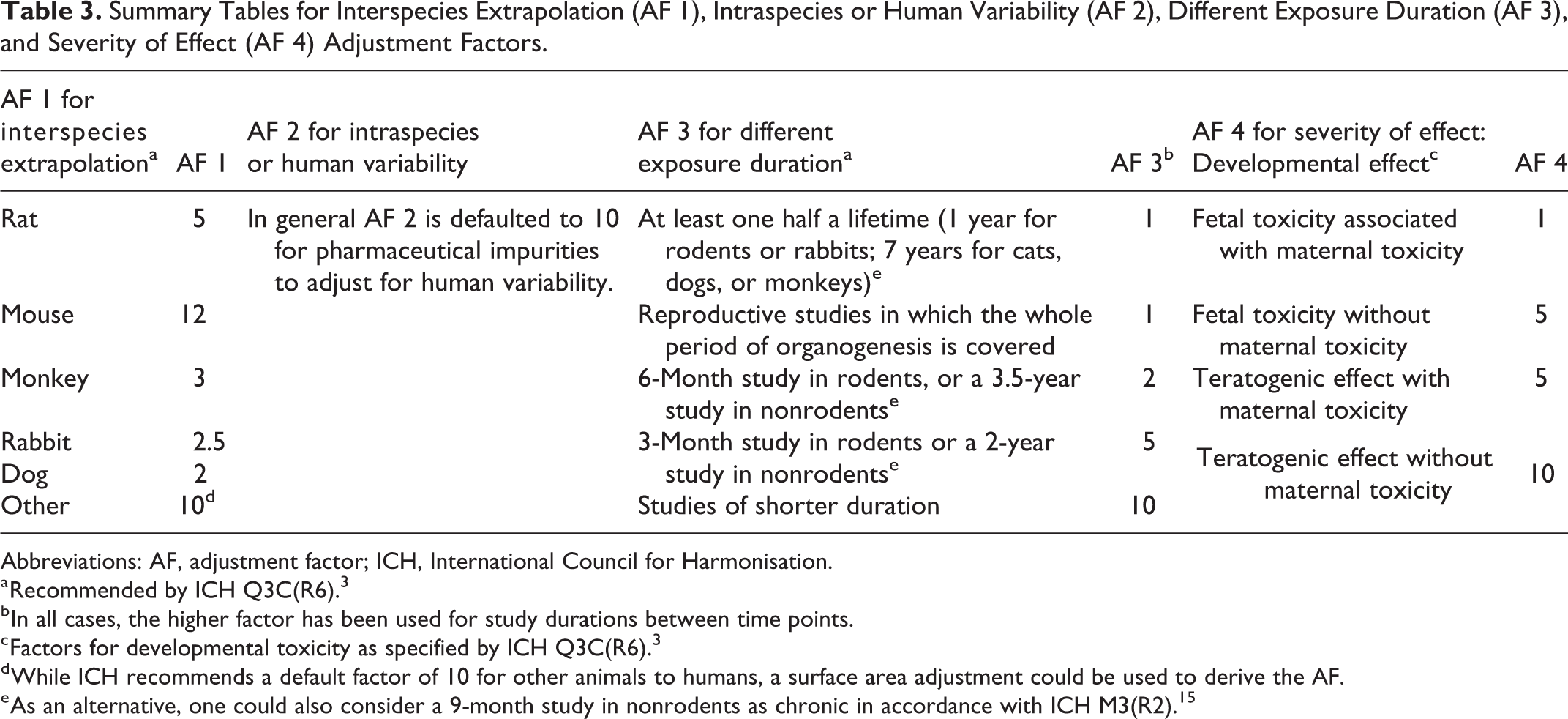
Abbreviations: AF, adjustment factor; ICH, International Council for Harmonisation.
a Recommended by ICH Q3C(R6). 3
b In all cases, the higher factor has been used for study durations between time points.
c Factors for developmental toxicity as specified by ICH Q3C(R6). 3
d While ICH recommends a default factor of 10 for other animals to humans, a surface area adjustment could be used to derive the AF.
e As an alternative, one could also consider a 9-month study in nonrodents as chronic in accordance with ICH M3(R2). 15
Although not included in the ICH impurities guidelines, it is also acceptable, or even preferable if data are available, to derive a chemical-specific AF (CSAF) based on the toxicokinetic and toxicodynamic differences between animals and humans. 13,14 Although it is not often that pharmaceutical impurities will have enough data to derive an AF in this manner, this should not exclude its use, if appropriate.
Intraspecies or human variability (AF 2)
In general, the intraspecies or human variability AF 2 is defaulted to 10 for pharmaceutical impurities to adjust for human variability. An AF of 10 takes into consideration that individual responses to the same chemical exposure will vary across a large population of patients. For example, some people may metabolize a chemical more or less efficiently than others, or individuals may respond differently from a toxicodynamic perspective. Therefore, AF 2 conservatively reflects the uncertainty in this inherent variation.
As with interspecies variation, it is possible to develop a CSAF, 13,14 however, this is less frequently applied due to a scarcity of information regarding toxicokinetic and toxicodynamic variability data in humans.
Duration of exposure (AF 3)
The duration of exposure AF 3 is applied to extrapolate from shorter exposures to longer exposures. Typically for a PDE to be applied broadly across pharmaceuticals, it is assumed that exposure can be chronic. Studies in animals may not necessarily be conducted over a chronic duration as defined for that species. ICH Q3C 3 and Q3D 4 recommend certain factors based on the duration of exposure in toxicology studies which are listed in Table 3. However, these guidelines recommend that a chronic study in nonrodents would be for durations up to 7 years. It is extremely rare, impractical, and unnecessary for a nonrodent to be tested for up to 7 years. Alternatively, one may consider guidance from ICH M3(R2), 15 which considers a 9-month study chronic for nonrodents; however, the regulatory acceptance of this approach is unknown.
Other considerations may be warranted when selecting an AF for duration of exposure. Examples include the ability of the chemical to accumulate over time, the observation of a lowered threshold for toxicity (eg, lower NOAEL or LOAEL) when dosing over a longer duration, or knowledge of the mechanism of action. In addition, the available study data may not fit the subcategorization of AFs presented in Table 3. An example is a human study, where it is not possible to test a compound over 70 years. In such cases, knowledge of the chemical’s toxicity and pharmacokinetics in humans can be used to determine the AF 3 value needed to adjust for short-term to chronic exposures.
Severity of effect (AF 4)
The ICH impurities guidelines indicate that the severity of effect AF 4 is a variable factor up to 10 that can be used to account for the observation of severe effects in the toxicology study. In the ICH Q3C guidelines, it is recommended that this AF be used for toxicities such as neurotoxicity, developmental toxicity, and nongenotoxic carcinogenicity. 3 The factors in Table 3 are recommended in the ICH Q3C guidance for developmental toxicity. However, making an adjustment to account for the observation of severe toxicity, especially when the effect is only observed at the highest dose tested in a toxicology study, may be overly conservative. It is important to keep in mind that toxicity studies are often designed with the intent to observe severe toxicity. Depending on the incidence and type of adverse effect observed at the lower doses tested, a reduced AF 4 value for severe toxicity may be considered. The AF 4 value selected by the expert risk assessor can be subjective and requires a good supporting description of the effects observed and why the value was chosen.
Lowest observed adverse effect level to NO(A)EL extrapolation (AF 5)
In a toxicity study, the no-observed effect level (NOEL) or NOAEL may be defined. The difference between the 2 is for an NOAEL an effect may be observed but it is not considered adverse. Many animal studies focus on defining the NOAEL as it is more relevant in the context of human risk. 16 Ideally, an NO(A)EL (adverse is in parenthesis to reflect both an NOAEL or NOEL) would be identified in the key toxicology study being selected as the POD to calculate the PDE. However, in some cases the NO(A)EL may not be identified. In these cases, the risk assessor should extrapolate to an NO(A)EL using a variable AF of up to 10. A reduced factor can be applied when a dose–response is observed, and the threshold is being approached.
Alternatively, a BMD analysis can be calculated from toxicity dose–response data 17,18 and utilized as an NOAEL for noncarcinogenic effects. The benchmark response is dependent on the type of data (continuous vs quantal) and toxicity end point (eg, repeat-dose, developmental, genotoxicity, etc). Calculating the BMD can be a better way of extrapolating the NOAEL as it considers the full dose–response curve (not just doses selected in the study) and variability of response at each dose. If a BMD is derived and applied, then the AF 5 value is 1. ICH Q3D also mentions an F5 range of 1 to 5 for an NOAEL (as opposed to a 1 for an NOEL), but for most elements the value was set to 1. 4
Workshop case studies
Two case studies were presented in the workshop for discussion; triphenylphosphine and hydroxylamine. Details of the toxicology data are described in Bercu et al. 6 The workshop delegates were polled on which AFs they would have chosen in the study, based on the toxicity study data presented to the delegates. Due to workshop time limitations, not all AFs were able to be polled for both compounds, however the delegates were relatively harmonized on the selection of AFs when polled. The major exception was in the selection of AFs related to severity of effect (AF 4) where there was the most diversity in responses across the workshop attendees.
The critical effect for triphenylphosphine toxicity was neurotoxicity in a 5-week study in dogs. Although ICH Q3C considers neurotoxicity as “severe toxicity”, the data indicated the neuropathic changes were of questionable human relevance because this effect occurs spontaneously in dogs, a limited number of dogs in the study were used, and it was not confirmed in a 3-month study. When polling the workshop delegates (Supplemental Figure 2), there was a variety of proposals for the value of AF 4, with AF 4 = 5 being the most commonly proposed (43% of votes cast where the total votes cast in the poll was 14), followed by AF 4 = 3 (29% of votes cast), AF 4 = 1 (14% of votes cast), and finally AF 4 = 2 and 10 (7% of votes cast, respectively). Notably, this delegate poll demonstrated a marginally reduced response rate relative to the other polls conducted during the workshop (14 delegate votes cast vs a maximum response rate of 18 delegate votes). This may reflect greater uncertainty in the expert judgment of the voting workshop delegates.
Group discussion demonstrated a large variety of opinions with some expressing there was uncertainty about the neurotoxicity and that an AF 4 greater than 1 may not be needed, but recommended a higher factor to be conservative, while others also expressed that an AF 4 of 10 was not needed given the lack of human relevance, and a reduced factor could therefore be applied. In the publication of Bercu et al, 6 the authors applied an AF 4 of 1 due to the questionable relevance in humans.
The critical effect for hydroxylamine is tumors in male rat spleens induced by hemosiderosis, a nongenotoxic mode of action. Based on a BMD analysis, a BMDL10 of 0.23 mg/kg/d was derived. The delegates also presented a heterogeneity of responses when polled over the most appropriate AF 4 to apply within this case study (Supplemental Figure 3), with AF 4 = 10 being the most commonly proposed (46% of votes cast where the total votes cast in the poll was 13), followed by AF 4 = 1 (31% of votes cast) and AF 4 = 5 (23% of votes cast). Again, a reduced response rate relative to other workshop delegate polls may indicate reduced conviction in these selections across the delegates (13 delegate votes cast vs a maximum response rate of 18 delegate votes).
After discussion, it emerged that the respondents who chose an AF 4 = 5 or 10 did so because they considered nongenotoxic carcinogenicity a “severe toxicity” as described in ICH Q3C. Others who chose an AF 4 = 1 did so because hydroxylamine has been reported to be a product of normal cellular metabolism. In the publication of Bercu et al, 6 the authors had selected an AF 4 = 10 for severe toxicity because the mode of action was nongenotoxic carcinogenicity.
The delegate polling indicated that there was a degree of harmonization in the audience on many of the AFs polled. Where there was the most variation and discussion was on the basis of severe toxicity AF 4. This is not surprising given that severe toxicity is more subjective and based on the experience and expertise of the individual risk assessor. Therefore, to account for this potential variability, it is important to integrate a suitably impartial peer-review process and wider collaborative discussion among scientific experts to ensure that the AFs selected are based on good scientific rigor and consistent interpretation of the relevant toxicity data identified.
Determination of PDEs/AIs for Different Routes of Exposure
When establishing PDEs for pharmaceutical impurities, ideally the POD should be selected from a robust study administered via the route of exposure to the risk assessment situation, that is, the route of administration for the drug. This may not always be available and therefore approaches for route-to-route extrapolation need to be considered to ensure patient safety.
The ICH Q3C 3 methodology for deriving PDEs is generally applicable to all routes of administration, as solvents are assumed to be well-absorbed systemically. Likewise, in ICH M7 addendum 5 where robust data were available from carcinogenicity studies for more than 1 route and the tumor sites did not appear to be route-specific, the route with the lowest TD50 value was selected for the AI calculation. As such, the AI is considered suitable for all routes. However, careful review of available toxicology data may identify route-specific safety concerns, such as site-of-contact carcinogenicity. In which case, if tumors are only observed at the site of contact, for example, inhalation exposure resulting in respiratory tract tumors with no tumors at distal sites and the inhalation TD50 is lower than for other routes, then a separate route-specific AI should be developed to safeguard patients. Similarly, a route-specific PDE can be derived utilizing ICH Q3C methodology if the data indicate route-specific toxicity. The following examples highlight approaches that may be used to establish safe limits when extrapolating from different routes of administration.
Equivalent bioavailability
For volatile chemicals such as solvents, toxicity studies are often conducted via an inhalation route of administration. Therefore, the POD used to derive a PDE for an impurity that is administered orally may be limited to critical studies from a different route of administration. Bioavailability following inhalation may be higher than oral due to the absence of first-pass metabolism, therefore a conservative approach could assume equivalent bioavailability to oral administration. This means an exposure limit based on inhalation data could be applicable to all routes of administration. The derivation of an AI for bis(chloromethyl)ether as published in ICH M7 addendum 5 is a good example of using this approach.
Site of contact toxicity
If local or site of contact effects are observed for a particular route of administration, then route-specific exposure limits may need to be calculated. As toxicity is driven by high local concentrations at the site of contact rather than the systemic dose, it may be overly conservative to extrapolate between routes of administration. 16,19,20 ICH M7 addendum 5 monographs for dimethylcarbamyl chloride and hydrazine illustrate this approach, where careful review of carcinogenicity data reveals more potent carcinogenicity following inhalation administration. Site of contact nasal tumors are observed resulting in a lower TD50 value compared to oral administration, therefore route-specific PDEs were derived.
Route-specific and species-specific effects
When reviewing carcinogenicity and toxicology studies conducted by different routes of administration, species-specific effects should also be evaluated. Poorly soluble particles (PSPs), such as pigment-size titanium dioxide (ie, non-nanosized), is a site of contact carcinogen in rats following inhalation exposure, however, is not carcinogenic in rodents following dietary administration. 21 Rodent sensitivity has been well described, as they have limited capacity to clear PSPs. Consequently, the lungs become overloaded resulting in adenomas and carcinomas. 22 This observation does not translate to humans with epidemiological studies in titanium dioxide or carbon black exposed workers clearly demonstrating that long-term occupational exposures to these particle-types do not cause lung cancer or noncancerous diseases of the respiratory tract. 22 Therefore, when deriving a route-specific PDE for pigment-size titanium dioxide from the inhalation carcinogenicity studies, AF 1 for species-specific allometric should be modified to account for rat sensitivity, thus providing a more realistic route-specific exposure limit for human safety assessment. 23
Bioavailability adjustment
To address differences in bioavailability between routes of exposure, additional toxicokinetic information can be utilized to establish route-specific PDEs. This may be necessary when toxicology studies for intravenous (iv) or subcutaneous administration are lacking and standards only exist for daily intake of substances in food, water (oral exposure), air and/or occupational exposure (inhalation/dermal exposure). Methods for adjusting for bioavailability have been well described previously, 19 and the ICH guideline for elemental impurity safety assessment in pharmaceuticals, ICH Q3D, 4 specifically addresses oral-to-parenteral extrapolation (Table 4). In the absence of relevant inhalation and parenteral data, including potential local tissue toxicity, one could apply an AF of 100 (assuming <1% bioavailability). Physical–chemical properties and/or indications of local toxicity may allow for a better estimate such as ≥1% to <50% bioavailability (ie, apply an AF of 10) but this should be evaluated case by case. 4
Oral-to-Parenteral Bioavailability Adjustment Factors for Elemental Impurities Including Metals.a
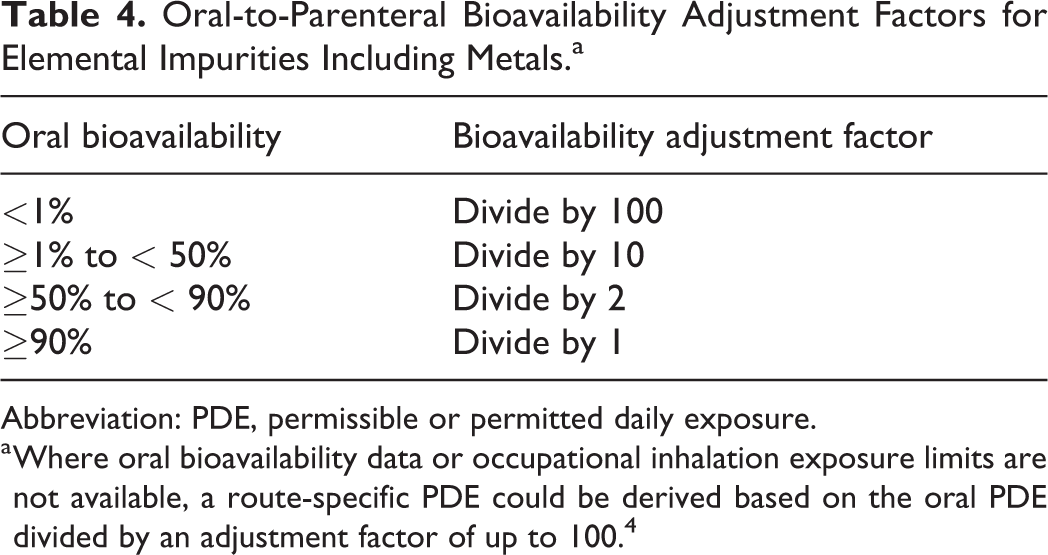
Abbreviation: PDE, permissible or permitted daily exposure.
a Where oral bioavailability data or occupational inhalation exposure limits are not available, a route-specific PDE could be derived based on the oral PDE divided by an adjustment factor of up to 100. 4
Workshop case studies
Two case studies were presented for discussion with workshop attendees provided with an accompanying handout of relevant summary toxicological data for acrolein and methyl bromide. Both were recently published in the study of Bercu et al 6 as examples of mutagenic noncarcinogens with route-specific PDEs derived for oral and inhalation routes of exposure. Delegates demonstrated a clear understanding of the application of different methodologies to calculate compound-specific exposure limits, with all respondents agreeing that the ICH Q3C methodology is appropriate for mutagenic noncarcinogens. As observed with the other case studies, poll answers were varied for which data and AFs to use in the derivation of a route-specific PDE.
Acrolein is an irritant, therefore site of contact toxicity is responsible for the critical effect in the oral and inhalation studies. A dose-related decrease in survival in rats is observed and attributed to gastric irritation following oral gavage administration for 2 years with an NOAEL of 0.05 mg/kg/d determined. Similarly, mortality and stomach lesions are the predominant effect in long-term oral studies with dogs as well as 13-week studies in rat and mice (note: for the purposes of the workshop and polling question, these studies were termed “chronic oral study data”). The 2 inhalation carcinogenicity studies are not considered robust enough to derive a PDE (1 year duration with a single exposure concentration in each study, and a limited number of animals per group), therefore the 13-week inhalation studies in rats, hamsters, and rabbits were also reviewed. Concentration-related lesions of the nasal cavity were observed in all species, and rat was the most sensitive with an LOAEL of 0.4 ppm (0.9 mg/m3) or 0.1 mg/kg/d. Minimal histopathological changes in the nasal cavity were observed at this level. As an NOAEL was not determined, we cannot compare this dose/concentration with the chronic oral studies so a route-specific PDE should be derived.
Delegates were asked which data should be used to calculate a route-specific PDE for acrolein. Polling showed (Supplemental Figure 4) that the options for using the “Carcinogenicity study data” (oral and inhalation available) and “Dermal, nasal and ocular irritation study data” were equally supported (43% of votes cast, respectively, where the total votes cast in the poll was 7—note that the reduced sample size is due to the formation of 2 cohorts within the workshop delegates each contributing to a single case study), followed by using “Chronic oral study data” (14% of votes cast). The dermal, nasal, and ocular irritation data indicate a hazard that should be considered when interpreting the oral and inhalation repeat dose toxicology studies. Due to the acute duration and limited systemic toxicological assessment in such studies, they are not routinely used to derive exposure limits, however they provide important information on potential local effects. In this case, the oral carcinogenicity studies and 13-week inhalation studies are most appropriate to derive route-specific exposure limits.
The second case study focused on methyl bromide which is mutagenic but not a rodent carcinogen via oral or inhalation routes. Chronic inhalation studies were more sensitive than the oral studies, and rats were the most sensitive species. The critical effect following inhalation at low doses is primarily in the nasal region. As rats are obligate nose breathers, this is consistent with a site-of-contact toxicant following inhalation administration. Since the main adverse effects of methyl bromide at low doses are localized at the site of contact, a route-specific PDE was generated. The inhalation carcinogenicity study by Reuzel et al 24 was considered the most sensitive and was also utilized by US Environmental Protection Agency (USEPA) for the derivation of the reference concentration. 25
Although robust dietary carcinogenicity studies exist, these were not available at the time the USEPA derived the oral reference dose (RfD). 25 Instead, the RfD was based on the results from a subchronic (13-week) rat study with the critical site-of-contact effect of epithelial hyperplasia of the forestomach. This highlights the need to conduct a thorough literature review; even with the availability of regulatory limits, the toxicologist should consider the most current toxicity data to derive exposure limits. Given 2 robust dietary chronic studies are now available, the lowest NOAEL of 2.2 mg/kg/d based on bodyweight changes at the highest doses was used to derive an oral PDE. When polled, the delegate cohort who contributed to this case study responded collectively that they would utilize the rat inhalation carcinogenicity study data to calculate the route-specific exposure limit for methyl bromide.
The route of administration impacts the absorption, distribution, metabolism, and excretion of a pharmaceutical and the associated impurities. Therefore, where the data set allows, route-specific pharmacokinetics and pharmacodynamics should be accounted for when deriving impurity exposure limits. Several approaches are outlined providing the toxicologist with opportunities to conduct a route-specific assessment if deemed necessary following expert review. Selection of the critical studies again highlights potential variability during the process and emphasizes the need for discussion, collaboration, and peer-review. In the case of acrolein, delegate polling showed different responses strengthening the point that clear documentation of the critical study and AF rationale is important to clearly communicate the derivation of safe impurity-exposure limits.
Approaches to Consider When There Is No Toxicology Data Available to Establish a PDE or AI
Although the previous sections discussed the methodologies for selecting studies or AFs to determine PDEs, often there is interest to develop a PDE where no or very little toxicology data exist for the compound in question. There are some guideline-driven options as well as alternative approaches that can be considered to set safe exposure levels for patients, as summarized below.
ICH M7-related limits
The ICH M7 guideline 5 describes methodology for evaluating process-related impurities for the potential to increase carcinogenic risk as direct DNA-reactive substances and provides exposure limits for mutagenic impurities with unknown carcinogenic potential for varying durations of exposure that are considered acceptable for clinical trial subjects and patients exposed to the impurity in a marketed pharmaceutical. The impurity limits are based on the premise that there is a linear relationship between cumulative exposure to a mutagenic impurity and carcinogenic risk. Limits for mutagenic impurities without carcinogenicity default to the acceptable intake of 1.5 µg/d (the threshold of toxicological concern for mutagenic carcinogenicity or TTC) for lifetime dosing, which is associated with less than a 1 in 100,000 excess risk of cancer for a majority of carcinogens, as well as acceptable less than lifetime intakes. For example, controlling an impurity to the TTC for a drug to be taken daily for chronic therapy at 100 mg/d requires analytical controls to 15 ppm. Higher limits are allowable for less-than-lifetime exposures. Impurities requiring low level control are identified by using 2 complementary computational methods (also called in silico assessment), one expert rule-based and the other statistically based. A negative prediction in the in silico evaluation or negative result in the Ames test allows for higher daily exposures (see section below regarding ICH Q3A/Q3B thresholds). Expert review of in silico results can also be applied to support the classification of the impurity as a potentially mutagenic impurity or not.
In some cases, AIs can be developed for a class of mutagenic carcinogens. This is because the TTC was developed to be conservative, assuming that any mutagenic compound is also a potent carcinogen. Class-specific AIs are based on an analysis of carcinogenic potencies of compounds with a particular structural alert. There needs to be a sufficient number of compounds in the class and a common mechanism for mutagenicity or carcinogenicity and secondary alerts (alerting structures for mutagenicity not related to the class of compounds) should be identified to avoid confounding alerts. Class-specific AIs may be higher or lower than the TTC. Examples of class-specific AIs are the monofunctional alkyl chloride and bromide compounds. 3,6,26
ICH Q3A/Q3B thresholds
Depending on the source of the impurity (process impurity or storage degradant), if nonmutagenic, application of the default ICH Q3A or Q3B qualification thresholds is appropriate. These guidelines provide a framework for the identification and qualification of impurities/degradants based on daily dosage of the drug. The qualification thresholds provide a daily exposure below which is assumed safe for chronic (lifetime) administrations and above which requires a toxicological justification of safety. Harvey et al 27 described an approach which derived higher qualification thresholds in the drug substance and drug product for early clinical development. In addition to the qualification threshold expressed as a percentage in the drug substance, ICH Q3A also indicates an exposure of 1 mg/d for drug substance impurity for a lifetime could be considered safe. This value has been assessed by Harvey et al who scientifically justified this 1 mg/d exposure based on evaluation of the distribution of NOAELs in hundreds of animal toxicology studies. This work also reasons that for shorter duration exposures such as early clinical trials, exposure to higher levels than suggested by ICH Q3A/Q3B could be justified. 27
Threshold of toxicological concern for noncarcinogenic effects and non-API related impurities
There are different limits used for noncarcinogenic impurities generated separately from the synthesis of an API. This is because synthetic impurities are typically part of the API, tested nonclinically with the API, and also typically close in structure to the API. In these cases, the ICH Q3A/Q3B thresholds have been used to demonstrate when additional safety data are needed for an impurity. However, other types of impurities are not governed by ICH Q3A/Q3B since they are not generated during API synthesis. Such examples can include leachables and extractables, which are chemicals that migrate into the drug substance or drug product during contact with equipment or container/closure systems. The use of published classification systems that base expected toxicity on structure and similarity to known compounds could be considered when evaluating control of an impurity with no toxicity data. An example presented during the workshop was the use of the Cramer decision tree to categorize chemicals into several classes. Cramer classified compounds based on structure and analogy to known toxicants into categories of low, intermediate, and significant toxicity potential. 28 Munro et al 29 provided quantifiable levels for these categories (Table 5). These were then correlated with TTCs based on noncarcinogenic effects.
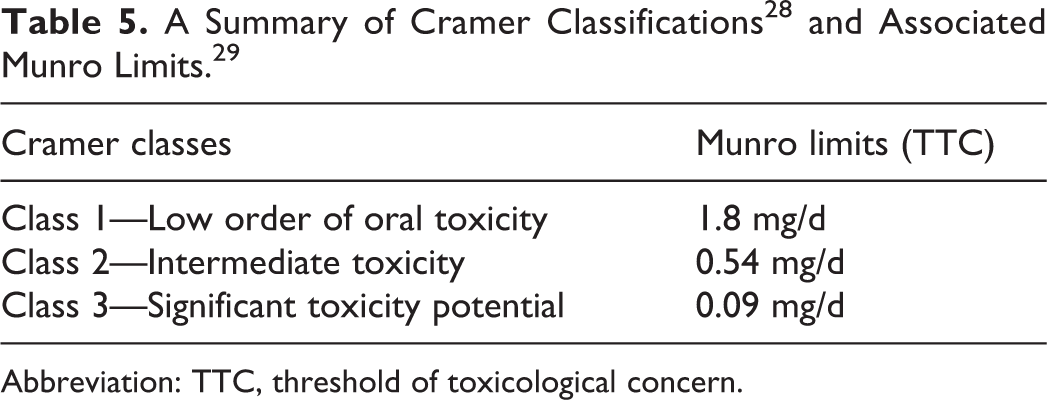
Abbreviation: TTC, threshold of toxicological concern.
Similarly, Dolan et al 30 developed TTCs for APIs in pharmaceutical manufacturing operations to limit cross-contamination of equipment. The categories were based on potential to be carcinogenic, highly toxic, or not likely to be potent, toxic, or carcinogenic. These values are, respectively 1, 10, and 100 µg/d.
Read-across
Another common approach for non-API related impurities that is embodied in the TTC classification systems is read-across of data from sufficiently similar chemicals and assuming they share the same toxic potential with the chemical of interest. Publications from authors and agencies provide detailed methodologies. 31 -33 Read-across can incorporate chemically similar structures, physical/chemical properties, and biological outcomes. Careful consideration is needed for the data set to be used. As previously described, the quality of the data can affect the outcome, and additionally, assuring that the comparator set of compounds is appropriately similar will lower uncertainties in the analysis.
Endogenous exposure
ICH M7 indicates that the conservative default limits for a mutagenic impurity can be exceeded if exposure to the impurity may be greater from natural sources such as food or endogenous formation. It would seem logical that endogenous exposures may present many fold-higher exposures relative to a low-level microgram dose from a pharmaceutical. The quantities of various chemicals found in foods, such as the amount of citric acid in orange juice, have been published. An example was provided for arginine, a structurally alerting (CASE Ultra v.1.6.0.3, MultiCASE Inc 34 ) and Ames positive naturally occurring amino acid. 35 Simple evaluation of the synthetic route and Ames test results may lead to unnecessarily controlling arginine to default TTC levels. However, the literature shows consumption or safe levels of exposure to arginine could be as high as 20 g/d, 36 and the US Food and Drug Administration accepts arginine as a food additive. Thus, higher limits could be acceptable for arginine, up to ICH Q3 A or B levels for nonmutagenic impurities. Another potential approach for establishing an acceptable daily exposure for an impurity is to consider the metabolic fate. For example, a compound could be metabolized readily to an endogenous product. An example of this approach is described in the study of Bercu et al 6 for vinyl acetate.
Workshop case studies
Two case studies were presented within the workshop where little or no in vivo toxicity data exist to illustrate these situations, and the audience was asked to describe their preferred approach. The responses illustrate the diversity of opinions on how to arrive at a limit for an impurity when there are no existing data.
The first case study was based on 4-butylaniline. This chemical has no structural alerts for mutagenicity (CASE Ultra v.1.6.0.3, MultiCASE Inc 34 ) and no repeated dose-toxicity or carcinogenicity data are available. It has a high degree of structural similarity to 4-methylaniline, which is carcinogenic in mice but Ames negative in the presence of rat S9 (CPDB and Japan Chemical Industry Ecology-Toxicology and Information Center 37 ). Despite the lack of alerts and because of the structural similarity to 4-methylaniline, the sponsor managed 4-butylaniline as a potentially mutagenic carcinogen, with a daily intake limit calculated from the carcinogenicity data on 4-methylaniline. This is a very conservative approach given the absence of structural alerts and that 4-methylaniline is not mutagenic in the Ames assay. The attendees were asked if they would control 4-butylaniline as an ordinary impurity (ICH Q3A), if they would test it in an Ames assay, generate a PDE based on read-across of 4-methylaniline, or conduct a more detailed review of similar substances to derive a PDE. The overwhelming response was to conduct an Ames test to determine whether the chemical is mutagenic (89% of votes cast where the total votes cast in the poll was 18), although there was still consideration by some for understanding, if 4-butylaniline was Ames negative, whether this class of chemical has a propensity for carcinogenicity.
The second case study was the tertiary amine, diisopropylethylamine (DIPEA), which was described as nonmutagenic with no other toxicity data available. Munro et al 38 suggest a 0.56 µg/kg/d exposure limit for DIPEA, based on read-across toxicity data from a similar compound, diisopropylamine. Read-across data from the European Chemicals Agency with trimethylamine from a repeated dose toxicity study with developmental/reproductive end points describe a 40 mg/kg/d NOAEL, 39 while the European Food Safety Authority describes tertiary amines as food additives with a Cramer classification of 1 (1.8 mg/d threshold of concern, low level of toxicity). 40 When the workshop delegates were asked what approach they would use to establish a safe exposure limit for DIPEA as a pharmaceutical impurity, the attendees were divided in opinion (Supplemental Figure 5), with the largest cohort of delegates suggesting they would use the read-across data from trimethylamine (a class 3 solvent per ICH Q3C) to establish a PDE using the principles described earlier (33% of votes cast where the total votes cast in the poll was 15). Other workshop participants indicated they would apply the Cramer TTC of 1.8 mg/d (27% of votes cast), while others thought it was appropriate to control to ICH Q3A qualification threshold, 0.15% or 1 mg/d whichever is lower (20% of votes cast). Regarding use of read-across with the data available for review, it was thought this was a more robust approach to setting a limit versus a general classification of “low toxicity” as in the Cramer scheme.
The diverse responses and arguments for or against any of these approaches highlight the potential for inconsistencies. A consensus derived by a panel of experts may be a reasonable approach to global acceptance of PDEs when no or limited data exist, although in most circumstances the need to derive a PDE for general use may not exist and such situations would have to be handled on a case-by-case basis by sponsors and regulators.
Discussion
The workshop summaries and case studies presented here have highlighted the flexibility available to industry risk assessment experts in the application of their personal expertise for interpreting toxicology data and applying this interpretation to the generation of a compound-specific AI or PDE limit. The inherent flexibility in this approach is a key driver for uncertainty to exist in the routine derivation and application of these limits across the pharmaceutical industry. Hence there is a need to establish greater cross-industry consistency in the methodology applied to AI or PDE derivation and to limit situations where comparable toxicologists with similar experience and knowledge in setting AIs or PDEs may generate different compound-specific safety limits for chemicals of concern and apply these safety limits in their work. In addition to establishing a common/consistent view for both regulators and pharmaceutical sponsors of what represents a safe daily dose to patients, collaborating to further harmonize the approach to compound-specific limit setting also benefits industry through affording greater efficiency through reducing duplicated effort and therefore provides significant time and cost savings.
Several precompetitive cross-industry collaborations have emerged which share a common goal of enhancing harmonization and reducing uncertainty in the methods for generating compound-specific AI or PDE limits. Some examples of these collaborations are discussed below.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Guidelines
The remit of ICH is to achieve greater harmonization across the pharmaceutical industries to ensure that safe, effective, and high-quality medicines are developed and registered in the most resource-efficient manner.
41
Harmonization is achieved through the development of ICH guidelines which involve a process of reaching scientific consensus between regulatory and industry experts working side-by-side in expert working groups. The derivation of some of these ICH guidelines has resulted in several publicly available and globally harmonized compound-specific exposure limits being published as exemplars within specific ICH guidance, including: the ICH M7 (R1) guidance for the “Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk,”
5
where the addendum section (Appendix 3) describes the application of a harmonized methodology for the derivation of 16 compound-specific exposure limits (AIs and PDEs) for chemicals which are considered to be mutagens and carcinogens; the ICH Q3C (R6) “Guideline for Residual Solvents”
3
which focuses upon the recommended acceptable amounts of residual solvents in pharmaceutical drug substances, excipients, and drug products. The ICH Q3C guideline describes the harmonized methodology for generating and applying PDE limits for 29 class 2 and several class 3 residual solvents; and the ICH Q3D (R1) “Guideline for Elemental Impurities”
4
which describe the potential for residual elemental impurities to be present in drug products. The ICH Q3D guideline considers the methodology for establishing compound-specific exposure limits for elemental impurities and provides harmonized PDE limits and monographs for 24 elemental impurities spanning elemental impurity classes 1, 2A, 2B, and 3.
The ICH working groups actively continue to maintain and extend the agreed scope of these guidance documents, as exemplified by the currently on-going revisions to the ICH M7 (R2), ICH Q3C (R8), and ICH Q3D (R2) guidance, and the formation of the ICH Q3E working group focused on developing guidance for extractables and leachables (E&Ls). 41
The Lhasa Limited AI/PDE Data Sharing Project
The AI/PDE Data Sharing Project consortium was established by Lhasa Limited in 2017 and currently involves industry experts from 9 global pharmaceutical organizations, who are collaborating with the shared aim of: sharing existing in-house or newly generated AI or PDE limit data to generate a series of AI and PDE limits for common impurities of mutual interest (eg, reagents, solvents, nonproprietary impurities); collaborating through intraproject monograph peer-review to harmonize the shared AI and PDE limits and to further standardize the methodology applied when conducting these safety assessments; gaining access to the harmonized shared compound-specific AI or PDE limit data and monographs in addition to identified publicly available PDE limit data through Lhasa Limited’s Vitic database platform; consistently applying the harmonized AI or PDE limit data in their work and in a regulatory context leading to greater certainty in the use of the compound-specific limits for the shared chemicals.
The pharmaceutical organizations involved in this project start each data sharing cycle by agreeing the number of compound-specific AI or PDE monographs to be shared within the project in each calendar year; to date this has been 2 perorganization per year. Lhasa Limited then facilitates the sharing and peer-review of the shared monograph limit data through Lhasa Limited’s Vitic database platform, such that each shared monograph document is independently peer-reviewed by at least 2 other organizations. Following collation of the peer-review comments raised, the organizations involved in this project then meet to openly discuss these peer-review comments in the context of each compound-specific monograph, prior to the original donor organization refining the shared monograph to reflect the agreed upon peer-review comments, consequently leading to a harmonized compound-specific AI or PDE limit. This refined monograph is then made available to the project consortium through Lhasa Limited’s Vitic database platform. Additionally, to aid with consistency in the approach to deriving and presenting supporting PDE limit data, a monograph template has been developed and refined within the project consortium. To date, a total of 51 compound-specific PDE limits and supporting monograph documents have been shared by the pharmaceutical companies involved, which when supplemented with identified public compound-specific limit data (eg, from Bercu et al, 6 ICH M7 addendum, 5 ICH Q3C/D 3,4 , etc) has produced a database of compound-specific exposure limits for 148 substances (Database Version 2020.1.0). It is the intention of this consortium to continue to share compound-specific limit data and expand this harmonized PDE limit database resource over the coming years.
This data sharing initiative therefore offers the participating pharmaceutical organizations with the forum to collaborate to drive a greater level of consistency and certainty in the generation and application of the shared compound-specific exposure limits through the cross-project harmonization and peer-review exercises. This, in addition to being able to access the Vitic database of harmonized compound-specific exposure limits, affords these organizations with the benefits of reducing cross-industry duplication of effort, which has significant direct time and cost efficiency savings, in addition to the benefit of realizing the consistent application of the harmonized limits within their organization and in a regulatory submission context.
Precompetitive Publications Co-ordinated Across the Pharmaceutical Industry
A recent precompetitive collaborative publication 6 which was co-ordinated across 11 pharmaceutical organizations resulted in further cross-industry standardization of the methodology for developing 20 harmonized compound-specific exposure limits for common synthetic reagents and byproducts which are potential impurities in drug substances. Additionally, this collaboration developed a class-specific limit for the monofunctional alkyl bromide impurity class. Publishing these 20 compound-specific limits and the harmonized methodology for their generation facilitates the consistent application and acceptance of these limits across the pharmaceutical industry both in routine corporate work and within a regulatory context.
The Extractables and Leachables Safety Information Exchange Consortium
The Extractables and Leachables Safety Information Exchange Consortium (ELSIE) was established in 2007 and is comprised of pharmaceutical, biotechnology, and medical device companies. These organizations collaborate precompetitively to collect, review, and compile within a database, publicly available toxicological data from peer-reviewed scientific journals, government reports, and other databases for E&Ls which originate from packaging materials and delivery systems. 42 The aim of this consortium is to reduce the duplication of effort burden for individual organizations through precompetitive collaboration and data sharing. The ELSIE consortium is considering ways to collaboratively generate PDEs for the E&L substances held within the database; to date this has been applied to 2 commonly occurring leachables where oral and iv limits were developed. 43 A further publication by Parris et al 44 derives PDEs for common E&Ls such as butylated hydroxytoluene, Irganox 1010, and Irgafos 168.
Supplemental Material
Supplemental Material, sj-tif-1-ijt-10.1177_1091581820982547 - Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop
Supplemental Material, sj-tif-1-ijt-10.1177_1091581820982547 for Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop by William C. Drewe, Krista L. Dobo, Zhanna Sobol, Joel P. Bercu, Patricia Parris and John Nicolette in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-tif-2-ijt-10.1177_1091581820982547 - Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop
Supplemental Material, sj-tif-2-ijt-10.1177_1091581820982547 for Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop by William C. Drewe, Krista L. Dobo, Zhanna Sobol, Joel P. Bercu, Patricia Parris and John Nicolette in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-tif-3-ijt-10.1177_1091581820982547 - Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop
Supplemental Material, sj-tif-3-ijt-10.1177_1091581820982547 for Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop by William C. Drewe, Krista L. Dobo, Zhanna Sobol, Joel P. Bercu, Patricia Parris and John Nicolette in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-tif-4-ijt-10.1177_1091581820982547 - Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop
Supplemental Material, sj-tif-4-ijt-10.1177_1091581820982547 for Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop by William C. Drewe, Krista L. Dobo, Zhanna Sobol, Joel P. Bercu, Patricia Parris and John Nicolette in International Journal of Toxicology
Supplemental Material
Supplemental Material, sj-tif-5-ijt-10.1177_1091581820982547 - Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop
Supplemental Material, sj-tif-5-ijt-10.1177_1091581820982547 for Deriving Compound-Specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis: Challenges in Expert Decision-Making Exemplified Through a Case Study-Based Workshop by William C. Drewe, Krista L. Dobo, Zhanna Sobol, Joel P. Bercu, Patricia Parris and John Nicolette in International Journal of Toxicology
Footnotes
Acknowledgments
The authors would like to acknowledge and thank those delegates who attended and actively participated in the “Deriving Compound-specific Exposure Limits for Chemicals Used in Pharmaceutical Synthesis” workshop held at the Genetic Toxicology Association meeting at the University of Delaware, May 2018.
Author Contributions
Drewe, W. contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript, and critically revised the manuscript. Dobo, K. contributed to conception and design; contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript. Sobol, Z. contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; and critically revised the manuscript. Bercu, J. contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; and critically revised the manuscript. Parris, P. contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; and critically revised the manuscript. Nicolette, J. contributed to conception and design; contributed to acquisition, analysis, and interpretation; drafted the manuscript; and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared receipt of the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employees of the organization to which they are affiliated. John Nicolette owns stock in AbbVie, Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work described in this manuscript was conducted in the course of the authors’ employment. The authors’ employers had no involvement in the study design; in the collection, analysis, and interpretation of data; in the writing of the manuscript, and in the decision to submit the article for publication. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.