
Abstract
Caffeine is a popular psychostimulant, which is frequently consumed with ethanol. However, the effects of caffeine on neuronal cells constantly exposed to ethanol have not been investigated. Apoptosis and oxidative stress occurring in ethanol-induced neurotoxicity were previously associated with decreased phosphorylation of the mTOR/p70S6K/4E-BP1 signaling proteins. Evidence also suggested that caffeine inhibits the mTOR pathway. In this study, human SH-SY5Y neuroblastoma cells were exposed to caffeine after pretreatment for 24 hours with ethanol. Results indicated that both ethanol and caffeine caused neuronal cell death in a dose- and time-dependent manner. Exposure to 20-mM caffeine for 24 hours magnified reduced cell viability and enhanced apoptotic cell death induced by 200 mM of ethanol pretreatment. The phosphorylation of mTOR, p70S6K, and 4E-BP1 markedly decreased in cells exposed to caffeine after ethanol pretreatment, associated with a decrease of the mitochondrial membrane potential (ΔΨm). These findings suggested that caffeine treatment after neuronal cells were exposed to ethanol resulted in marked cell damages, mediated through enhanced inhibition of mTOR/p70S6K/4E-BP1 signaling leading to impaired ΔΨm and, eventually, apoptotic cell death.
Introduction
Generalized toxic effects of ethanol on the brain have been widely demonstrated as impairing neurogenesis and promoting neuronal cell death. 1 -4 Mechanisms of ethanol-induced neurotoxicity are associated with the induction of oxidative stress, inflammation, and apoptosis. 5 -8 Apoptosis and oxidative stress occurring in the neurons have been shown to relate with the activity of the PI3K/Akt/mTOR pathway. 9 -11 The mTOR (mechanistic target of rapamycin) is a serine/threonine kinase that has been demonstrated to regulate a multitude of cellular processes such as survival, growth, protein synthesis, metabolism, cell cycle progression, cytoskeletal organization, and autophagy. 12 Inhibitory effects of ethanol on mTOR signaling have been shown in various cell types such as skeletal muscle, trophoblast cells, gastric epithelial cells, and B-cell lymphoma 13 -16 as well as brain cells. 17 -20 Data from an investigation of mouse cerebral cortical cells showed that chronic ethanol intake enhanced mTOR expression but reduced the expression of its downstream proteins p70S6K and 4E-BP1, with little effect on Akt signalling. 19 Although this evidence indicated that ethanol moderated mTOR signaling, the mechanisms related to its downstream consequences remain unclear and require further detailed investigation.
Caffeine (1,3,7-trimethylxanthine) is a popular psychostimulant, which is frequently consumed in conjunction with ethanol, especially among the younger generation. 21 -23 Caffeine and ethanol act on the adenosinergic system in distinct ways resulting in opposing physiological effects. 24 Some studies suggested that caffeine antagonized the cognitive and behavioral effects caused by ethanol intoxication, 25,26 while neuroprotective effects of caffeine on ethanol-induced neuronal cell death have also been reported. Previous studies in rats showed that low doses of ethanol, which singularly aggravates cerebral ischemia, reduced ischemic damage when applied with caffeine, 27,28 while another study in a rat model of alcoholism showed that the simultaneous ingestion of ethanol and caffeine reversed ethanol-induced neuronal and glial cell death in the cerebellar tissue. 29,30 However, other studies failed to demonstrate any neuroprotective effects of caffeine on ethanol-induced toxicity. A mouse study investigating the plus-maze discriminative avoidance task suggested that caffeine did not reverse ethanol-induced learning deficits, 31 and a study in rats showed that caffeine potentiated the apoptotic effect induced by ethanol in astrocytes. 32
Scientific knowledge and understanding concerning the effects of caffeine on alcohol are still lacking. SK-N-MC human neuroblastoma cells treated with caffeine at a concentration as high as10 mM for 24 hours exhibited several characteristics of apoptosis, including an increased caspase-3 activity. 33 Similarly, SH-SY5Y human neuroblastoma cells exposed to 10 mM of caffeine for 2 to 4 hours showed mild signs of membrane blebbing during apoptosis. 34 Evidence has shown that caffeine inhibited the PI3K/Akt/mTOR/p70S6K pathway in osteosarcoma cells, hematopoietic myeloid cells, and SH-SY5Y cells. 35 -37 In SH-SY5Y cells, exposure of cells to 10 or 25 mM of caffeine for 24 or 48 hours enhanced autophagic cell death, inhibited PI3K/Akt/mTOR/p70S6 signaling, and eventually resulted in apoptosis. 35
Here, we evaluated the effects of high concentration of caffeine on cell death and apoptosis, protein expression of the mTOR/p70S6K/4E-BP1 pathway, and the mitochondrial membrane potential in ethanol-treated SHSY5Y cells. Our findings may be useful in clarifying the effects of caffeine on ethanol-mediated neuronal death.
Materials and Methods
Materials
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide), dimethyl sulfoxide, and caffeine powder were purchased from Sigma-Aldrich (St Louis, Missouri). Ethanol absolute (ACS grade) was obtained from Merck (Darmstadt, Germany). Modified Eagle medium (MEM), Nutrient Mixture Ham’s F12 medium, fetal bovine serum (FBS), sodium pyruvate, nonessential amino acid, sodium bicarbonate, penicillin, and streptomycin were purchased from Gibco (Gaithersburg, Maryland). Rabbit anti-mouse primary antibodies against mTOR, p-mTOR, p70S6K, p-p70S6K, 4E-BP1, and p-4E-BP1 and goat anti-rabbit secondary antibodies conjugated with horseradish peroxidase (HRP) were purchased from Cell Signaling Technology (Danvers, Massachusetts). Horseradish peroxidase–conjugated horse anti-mouse secondary antibody against β-actin was purchased from Invitrogen (Eugene, Oregon). JC-10 mitochondrial membrane potential assay kit was purchased from Abcam (Cambridge, United Kingdom). Bradford protein assay reagent was purchased from Bio-Rad Laboratories (Hercules, California). Enhanced chemiluminescence (ECL) Western blotting substrate was purchased from Pierce Biotechnology (Rockford, Illinois).
SH-SY5Y Cell Culture
Human SH-SY5Y neuroblastoma cells were grown in a 1:1 mixture of MEM and Nutrient Mixture Ham’s F12 medium, supplemented with 10% heat-inactivated FBS, 1 mM sodium pyruvate, 0.1 mM nonessential amino acid, 1.5 g/L sodium bicarbonate, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 incubator. In the experiment, cells were subcultured and plated into appropriate culture plates. The number of cells to be subcultured was assessed under a phase-contrast microscope based on the exclusion of the trypan blue dye. The culture was maintained for 24 hours to allow for adhering to the plates. Thereafter, cells were treated with ethanol, caffeine, or ethanol followed by caffeine according to the experiment design.
Measurement of Cell Viability
After exposure to ethanol, caffeine, or ethanol followed by caffeine, cell viability was measured by using the MTT colorimetric assay. Basically, in active mitochondria, the tetrazolium ring in MTT was reduced by mitochondrial dehydrogenases with NADH to purple formazan, which can be measured by a spectrophotometer. SH-SY5Y cells were seeded into a 96-well culture plate and incubated at 37°C under 5% CO2 in a humidified incubator for 2 days. Cells were then added with 100 µL of MTT (5 mg/mL), followed by 4-hour incubation at 37°C in a humidified atmosphere containing 5% CO2. Then, the media were removed out and replaced with MTT solubilization solution to allow dissolution of formazan crystals. The absorbance of the samples was measured at 570 nm and normalized by a wavelength of 690 nm, using a BioTek microplate reader with KC4 Synergy HT Software (BioTek, Shanghai, China).
Detection of Apoptotic Cells by Flow Cytometry
SH-SY5Y cells were seeded into 6-well plate at a density of 5 × 105 cells/well in 2 mL of medium and then incubated at 37°C under 5% CO2 in a humidified incubator for 24 hours. After exposure to ethanol, caffeine, or ethanol followed by caffeine, cells were trypsinized and centrifuged at 2,500 rpm, 4°C for 5 minutes. Apoptotic cells were detected using a fluorescein isothiocyanate (FITC) Annexin V/dead cell apoptosis kit purchased from Molecular Probes (Eugene). The pellet was gently washed twice with cold phosphate-buffered saline (PBS) and resuspended in 100 µL of Annexin V-binding buffer at a density of 2.5 × 105 cells/tube. Cells were then incubated in 5 µL of Annexin V-FITC at room temperature for 15 minutes in dark. Thereafter, Annexin V-binding buffer was added again and cells were incubated in 5 µL of propidium iodide (PI) at room temperature for 15 minutes in dark. The apoptotic cells were analyzed with an FACSCalibur flow cytometer (BD Biosciences, San Jose, California).
Western Blot Analysis for mTOR, p70S6K, and 4E-BP1 Expression
Following treatment, SH-SY5Y cells were washed 3 times with cold PBS, then the cells were lysed by RIPA lysis buffer containing protease inhibitor cocktail for 10 minutes at 4°C or on ice, then cells were scraped off, and the lysates were collected. The extracted lysates were centrifuged at 14,000 rpm for 15 minutes at 4°C, and the supernatants were collected and stored at −80°C, or immediately, the protein concentration were determined by the Bradford method. An equal amount of 30 μg proteins from each experimental group was separated by 10% to 15% sodium dodecyl–polyacrylamide gel electrophoresis. After that, the proteins within gel were transferred onto nitrocellulose membrane. The nonspecific binding was blocked by 5% skimmed milk, washed 3 times for 10 minutes each with Tris-Buffered Saline with 0.1% Tween 20 buffer and sequentially incubated with 1:1,000 dilution of mTOR, phospho-mTOR, p70S6K, phospho-p70S6K, 4E-BP1, and phospho-4E-BP1 primary antibodies or 1:5,000 dilution of monoclonal β-actin primary antibodies overnight at 4°C. After washing, the membrane was then incubated with 1:5,000 dilution of HRP-conjugated goat anti-rabbit IgG as the secondary antibody for targeted proteins and 1:10,000 HRP-conjugated horse anti-mouse IgG against β-actin at room temperature for 1 hour. The bands of protein were visualized using ECL Prime Western Blotting Substrate. Finally, the blots were scanned and a densitometry analysis was performed on the scanned images using ImageJ software (National Institutes of Health, Bethesda, Maryland).
Measurement of Mitochondrial Membrane Potential by JC-10 Flow Cytometry
After treatment depends on experimental designs, SH-SY5Y cells were trypsinized and centrifuged at 1,500 rpm for 5 minutes. The pellet was gently washed twice with cold PBS, and cells were resuspended in 500-µL JC-10 dye solution at the density 5 × 105 cells/tube. Cells were incubated in dark at 37°C under 5% CO2 for 30 minutes. JC-10 is capable of selectively entering into the mitochondria and reversibly changes its color from green to orange as the membrane potentials increase. Cells were analyzed with a flow cytometer measuring the fluorescence excitation at 488 nm, with 530 and 585 nm emission filters.
Statistical Analysis
All experiments were performed in triplicate. Statistical analyses were performed with one-way analysis of variance test followed by a post hoc analysis (Turkey’s multiple comparison tests) using Graph Pad Prism 5 Software for Windows (GraphPad Software, Inc, San Diego, California). All values were presented as the mean ± standard error of the mean for each group. P < 0.05 was considered statistically significant.
Results
Ethanol Decreased Cell Viability of SH-SY5Y Cells in Both a Dose- and Time-Dependent Manner
To determine the dose- and time-dependent responses for ethanol treatment, we assessed the effects of ethanol on cell viability by using the MTT assay. The cells were treated with various doses of 50, 100, 200, and 400 mM of ethanol for 0, 16, 24, 48, and 72 hours. At each time point, cell viability progressively decreased with increasing dose of ethanol, and at each particular dose, longer duration of exposure increased cell death of SH-SY5Y cells (Figure 1). For 400 mM of ethanol, cell viability significantly decreased to less than 25% at 16 hours of exposure (P < 0.001) compared to the control. However, at 200 mM of ethanol, cell viability showed a significant decrease to about 75% of the control at 24 hours of exposure. Therefore, we selected the dose of 200 mM ethanol and the exposure time of 24 hours for use in the later experiment.
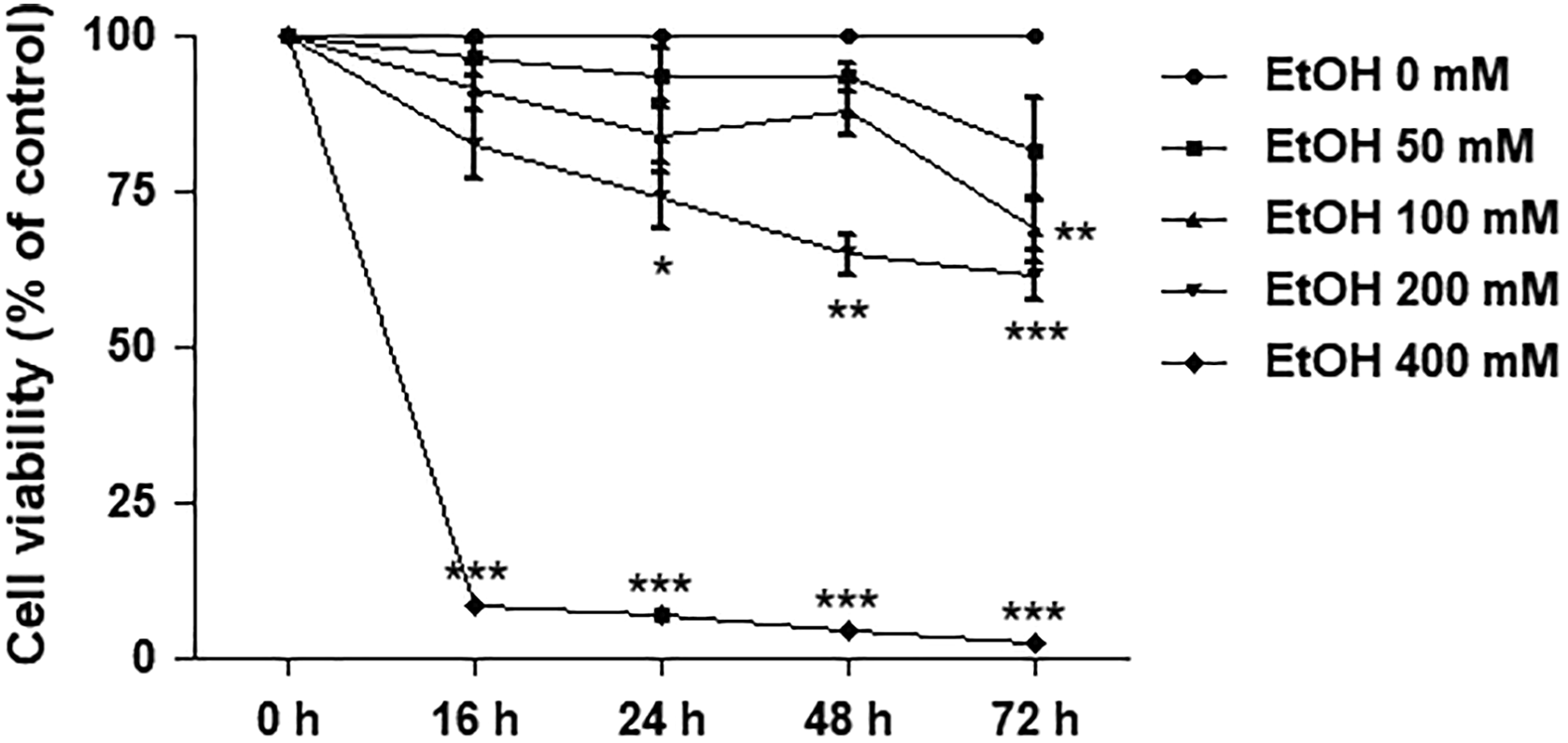
Effects of ethanol on cell viability in SH-SY5Y cells. Cells were treated with 50, 100, 200, and 400 mM of ethanol for 16, 24, 48, and 72 hours. Line graph represents a separate time-dependent experiment. Data are expressed as the means ± standard error of the mean obtained from 3 dependent experiments, each of which included 3 replicates. *P < 0.05; **P < 0.01; ***P < 0.001 compared with the control group.
Caffeine Decreased Cell Viability of SH-SY5Y Cells in Both a Dose- and Time-Dependent Manner
Cell viability of SH-SY5Y cells was investigated by using the MTT assay. The cells were treated with various doses of 5, 10, and 20 mM of caffeine for 0, 16, 24, 48, and 72 hours. Similar to that of ethanol, caffeine affected cell viability in both a dose- and time-dependent manner (Figure 2). For 20 mM of caffeine, cell viability significantly decreased to about 34% at 24 hours of exposure (P < 0.001) compared to the control. Therefore, this dose and exposure time of caffeine were selected for use in the later experiment as the intervening agent.
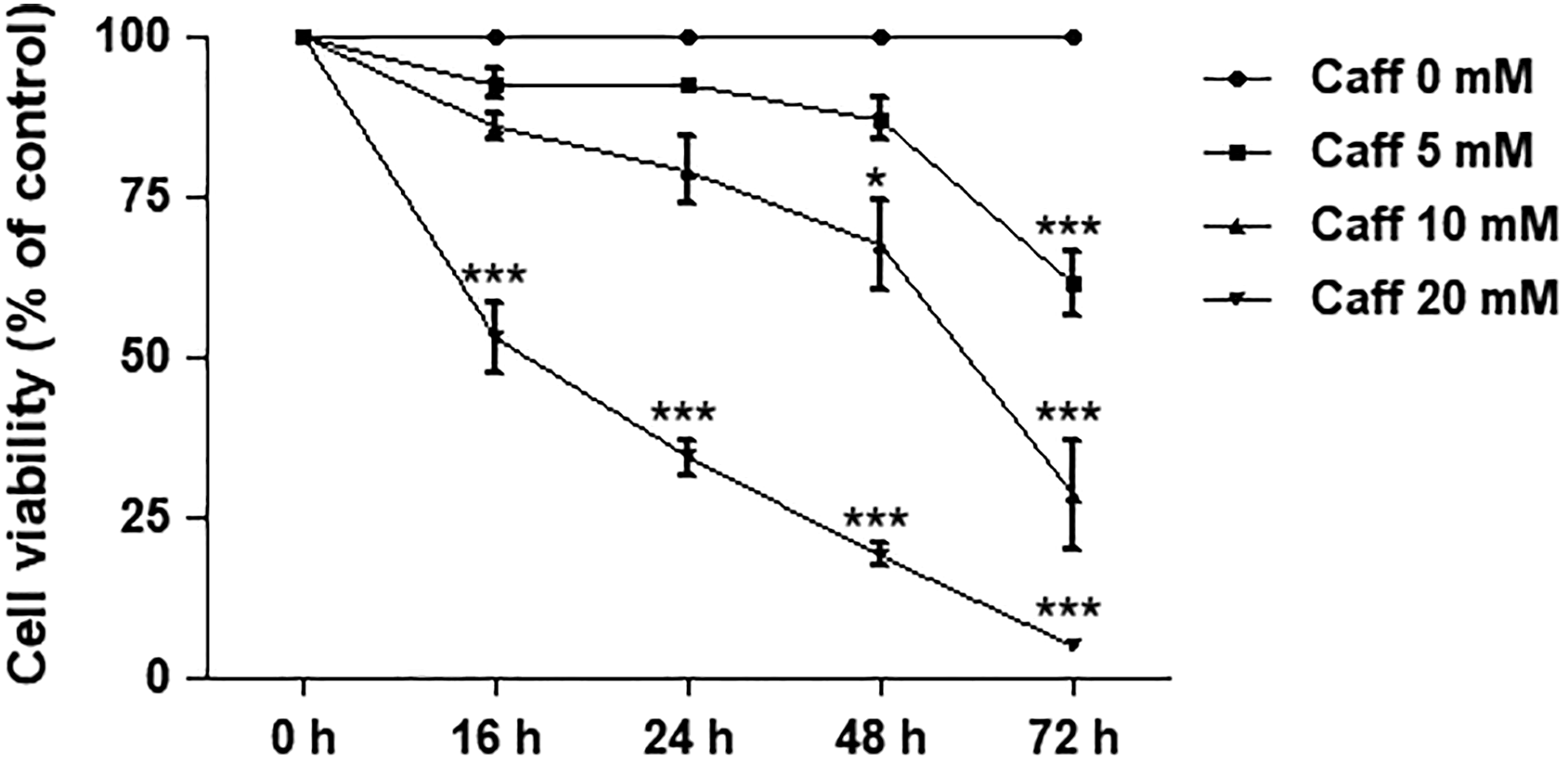
Effects of caffeine on cell viability in SH-SY5Y cells. Cells were treated with 5, 10, and 20 mM of caffeine for 16, 24, 48, and 72 hours. Line graph represents a separate time-dependent experiment. Data are expressed as the means ± standard error of the mean obtained from 3 dependent experiments, each of which included 3 replicates. *P< 0.05; **P< 0.01; ***P< 0.001 compared with the control group.
Caffeine Increased Cell Death in Ethanol-Treated SH-SY5Y Cells
To determine the effect of caffeine on ethanol-treated neurons, SH-SY5Y cells were pretreated with 200 mM of ethanol for 24 hours prior to exposure to 20 mM of caffeine for the subsequent 24 hours. Cell viability was assessed by using the MTT assay. Exposure to caffeine alone showed a significant reduction in the cell viability (47.04% ± 6.8%; P < 0.001) compared to the control. Treatment with 200 mM of ethanol alone decreased cell viability to 82.79% ± 2.7% (P < 0.01) as shown in Figure 3. Pretreatment with 200 mM of ethanol for 24 hours, followed by 24 hours of exposure to 20 mM of caffeine, significantly decreased the viability of the SH-SY5Y cells (P < 0.001) compared to ethanol alone and the control.
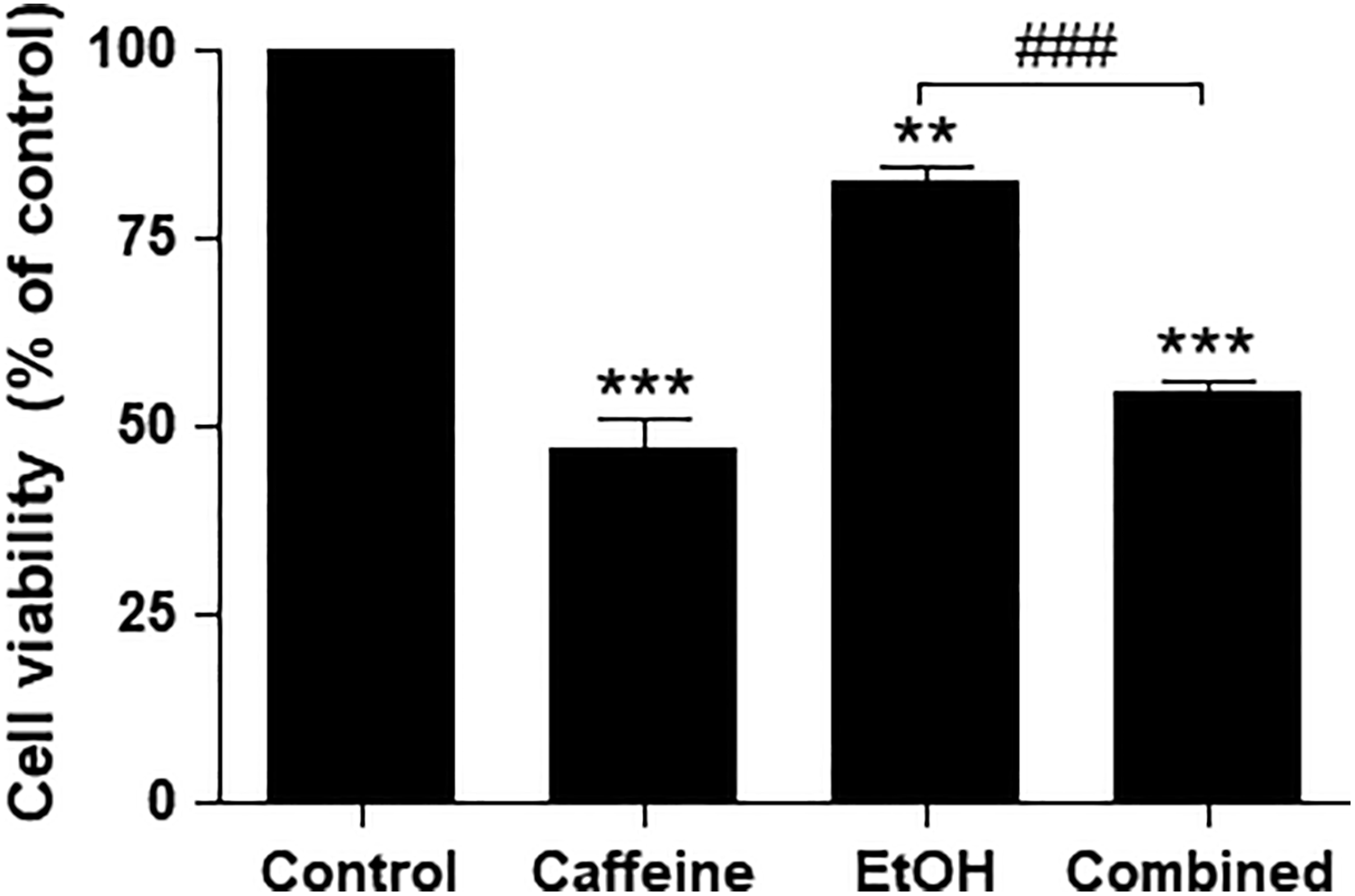
Effects of caffeine on cell viability in ethanol-treated SH-SY5Y cells. Cells were pretreated with ethanol (200 mM) for 24 hours before exposure to caffeine (20 mM) for 24 hours. Data are expressed as the means ± standard error of the mean obtained from 3 dependent experiments, each of which included 3 replicates. **P < 0.01; ***P < 0.001 compared with the control group; ###P < 0.001 compared with the ethanol-alone group.
Caffeine Increased Early Apoptosis in Ethanol-Treated SH-SY5Y Cells
Morphology of the ethanol-treated cells affected by caffeine was assessed by phase-contrast microscopy (Figure 4A). In cells treated with caffeine alone and ethanol alone, there were increasing numbers of small, round cells suggesting cell shrinkage and cytoplasm condensation, indicators of early apoptosis. 38 These characteristic morphological features of early apoptotic cells were markedly observed in cells treated with ethanol followed by caffeine.
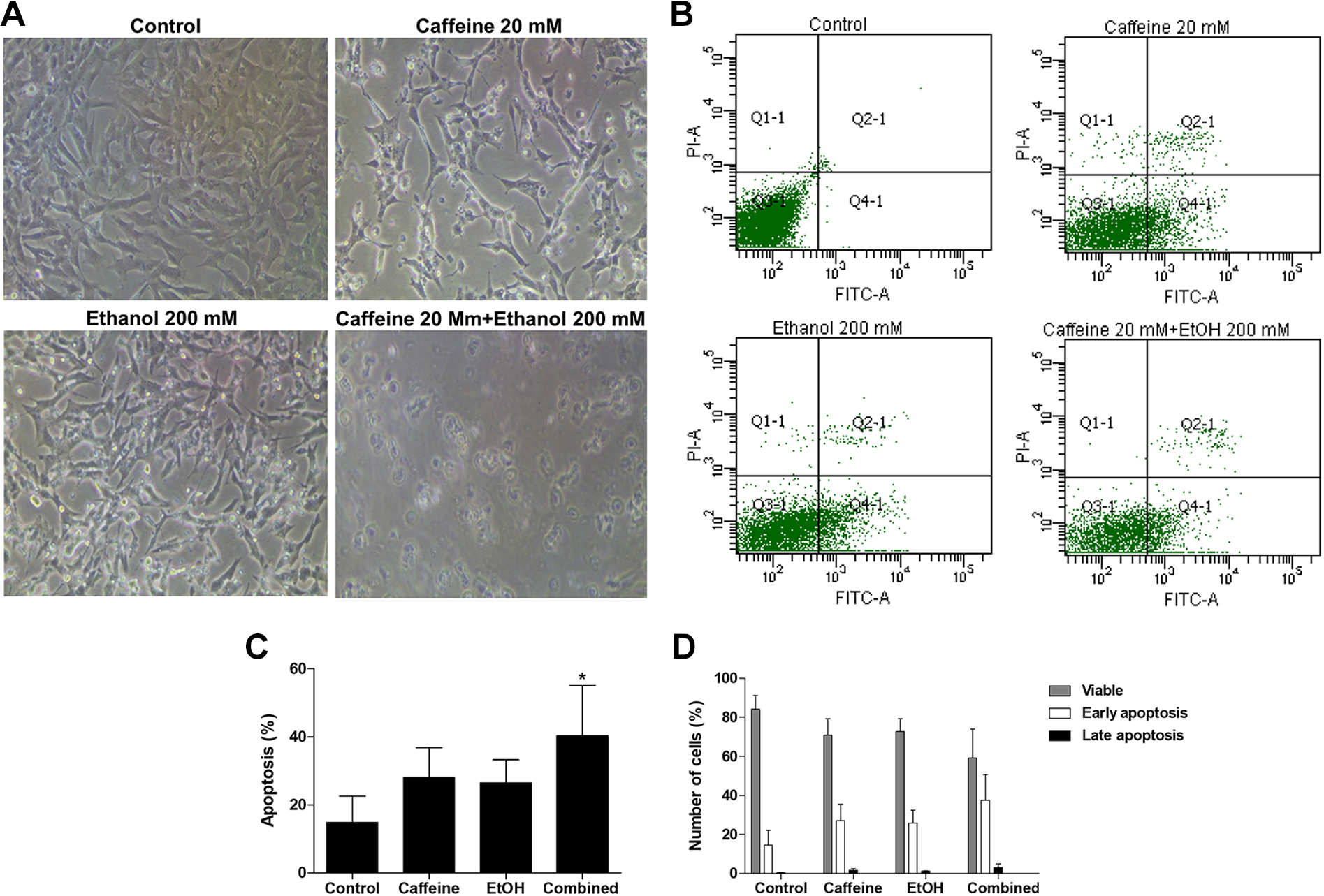
Caffeine increases apoptosis in ethanol-treated SH-SY5Y cells. Cells were pretreated with ethanol (200 mM) for 24 hours before exposure to caffeine (20 mM) for 24 hours. A, Representative of phase-contrast microscopic images. B, Flow cytometry profile represents Annexin-V-FITC staining in the x-axis and PI in the y-axis. The right upper quadrant represents Annexin V+/PI+ cells, and the right lower quadrant represents Annexin-V+/PI− cells. C, Quantification of total apoptotic cells from flow cytometry data. D, Percentage of early and late apoptotic cells quantified from flow cytometry data. Data are expressed as the means ± standard error of the mean obtained from 3 dependent experiments. *P < 0.05 compared with the control group. FITC indicates fluorescein isothiocyanate; PI, propidium iodide.
To confirm that the cell death was a result of apoptosis, flow cytometry was performed using Annexin V-FITC and PI to quantitate apoptotic cells. With Annexin V-PI flow cytometry, viable cells remained unstained (Annexin V-FITC−/PI−). Early apoptotic cells showed Annexin V-FITC+/PI− staining patterns, whereas late apoptotic cells exhibited Annexin V-FITC+/PI+ staining patterns due to a loss of cell membrane integrity (Figure 4B). The percentage of Annexin-V-positive cells was calculated and shown in Figure 4C. After exposure to 20-mM caffeine alone or 200-mM ethanol alone, the percentage of apoptotic cells increased but was not significant when compared to the untreated control. Exposure to caffeine after pretreatment with ethanol significantly increased the apoptosis of the SH-SY5Y cells (P < 0.05) compared to the control and the proportion of early apoptotic cells was markedly higher than that of late apoptotic cells (Figure 4D).
Caffeine Inhibited the Activity of mTOR/p70S6K/4E-BP1 in Ethanol-Treated SH-SY5Y Cells
To examine the effects of caffeine on the mTOR/p70S6K/4E-BP1 pathway in ethanol-treated SH-SY5Y cells, Western blotting was performed. Treatment with 20 mM caffeine to SH-SY5Y cells for 24 hours did not significantly decrease the expression of p-mTOR/mTOR and p-p70S6K/p70S6K but significantly decreased p-4E-BP1/4E-BP1 expression (P < 0.05) compared to the control (Figure 5A-D). Treatment with 200-mM ethanol for 24 hours significantly decreased the expression of p-mTOR/mTOR (P < 0.05) and p-p70S6K/p70S6K (P < 0.05) but did not significantly decrease the p-4E-BP1/4E-BP1 expression. Exposure to caffeine after pretreatment with ethanol significantly decreased the expression of p-mTOR/mTOR, p-p70S6K/p70S6K, and p-4E-BP1/4E-BP1 (P < 0.01 for all) compared to the control and significantly decreased the expression of p-p70S6K/p70S6K (P < 0.05) and p-4E-BP1/4E-BP1 (P < 0.01) when compared to ethanol alone.
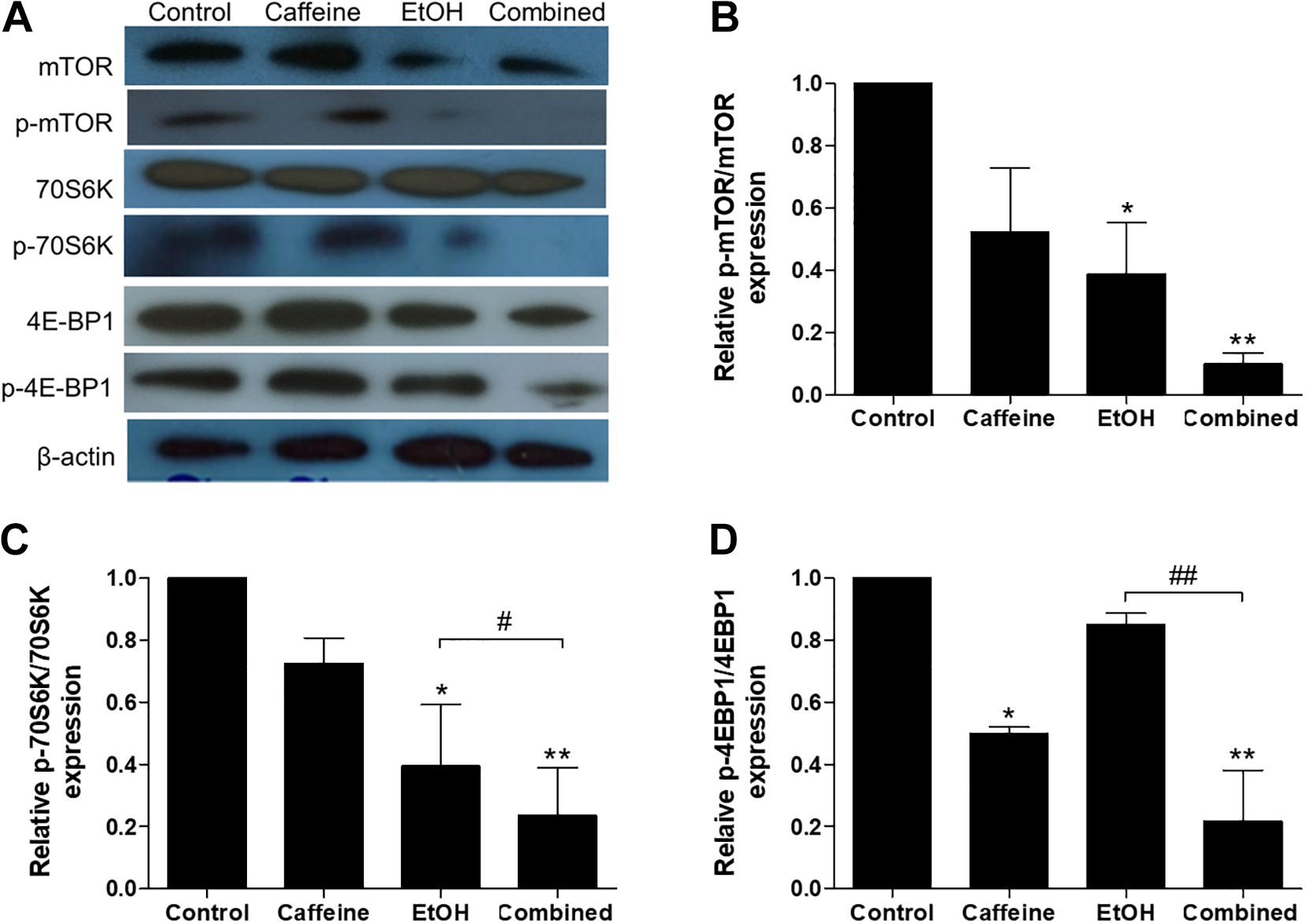
Caffeine inhibits phosphorylation of mTOR, p70S6K, and 4E-BP1proteins in ethanol-treated SH-SY5Y cells. Cells were pretreated with ethanol (200 mM) for 24 hours before exposure to caffeine (20 mM) for 24 hours. A, Representative images of protein expression examined by Western blot. β-actin was used as a normalization control. B-D, Bars graphs show quantifications for the indicated proteins. All data were determined in proportion between phosphorylated and unphosphorylated forms. The band intensity was normalized by β-actin and relative to untreated control. Data are expressed as the means ± standard error of the mean from 3 independent experiments. *P < 0.05; **P < 0.01 compared with the control group; #P < 0.05; ##P < 0.01 compared with the ethanol-alone group.
Caffeine Decreased the Mitochondrial Membrane Potential in Ethanol-Treated SH-SY5Y Cells
Previous evidence showed that the mTOR pathway could regulate mitochondrial membrane potential (ΔΨm) and survival. 35,39 We further assessed the ΔΨm by using fluorescent JC-10 flow cytometry. It is known that the JC-10 probe passes through the mitochondrial membrane and accumulates in the mitochondrial matrix, where accumulation is maintained by the membrane potential. In normal cells, JC-10 forms red fluorescent aggregates. However, in apoptotic cells, JC-10 exists in monomeric form and stains cells green. The ratio of green to red fluorescent emission was calculated and was shown in Figure 6A. Exposure to caffeine alone showed a significant reduction in the ΔΨm (P < 0.001) compared to the control, while treatment with 200 mM of ethanol alone significantly decreased the ΔΨm (P < 0.05) as shown in Figure 6B. Pretreatment with ethanol followed by exposure to caffeine significantly decreased the ΔΨm of the SH-SY5Y cells compared to ethanol alone (P < 0.05) and the control (P < 0.001).
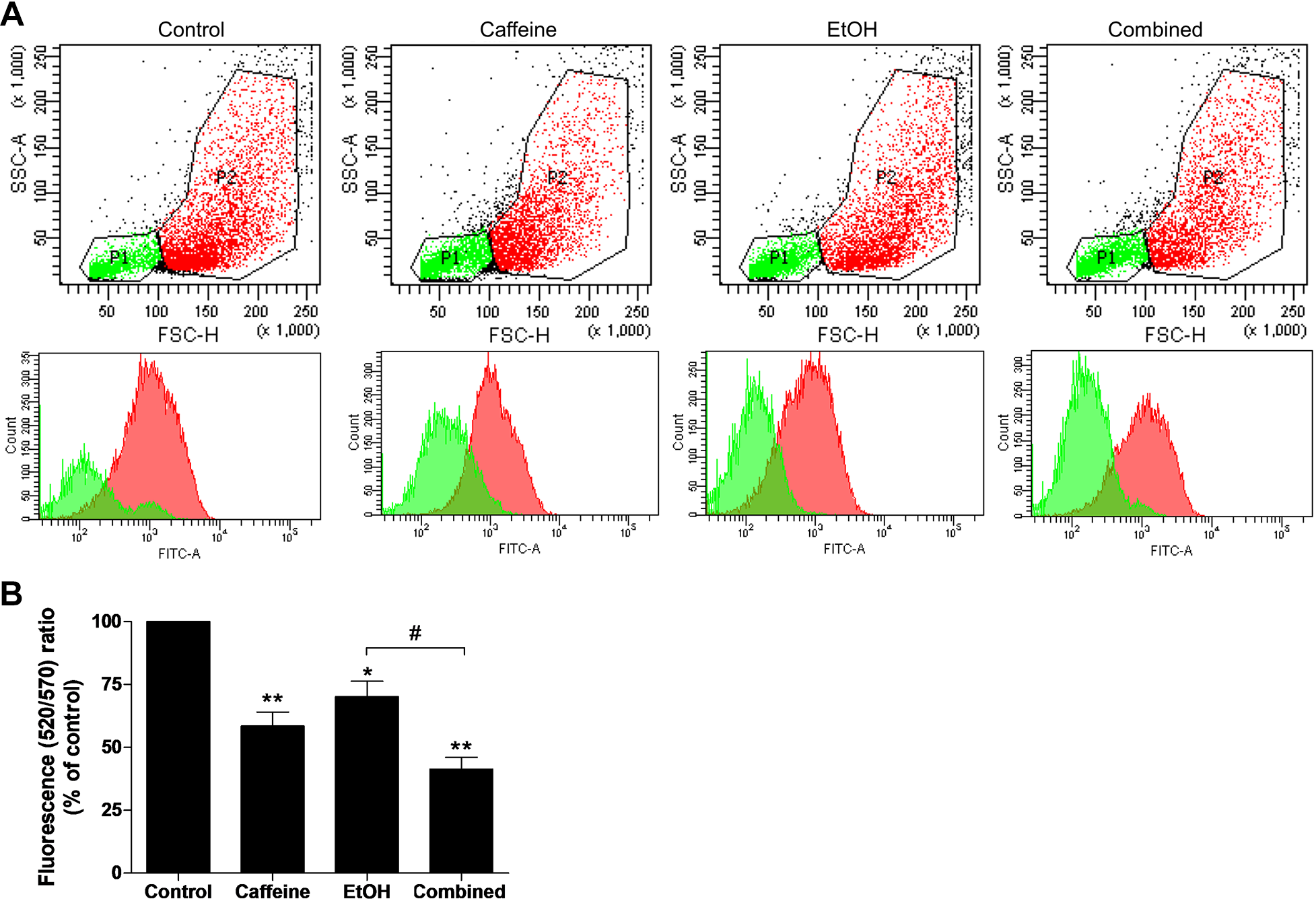
Caffeine reduces the mitochondrial membrane potential in ethanol-treated SH-SY5Y cells. Cells were pretreated with ethanol (200 mM) for 24 hours before exposure to caffeine (20 mM) for 24 hours. The mitochondrial membrane potential was examined using the fluorescent JC-10 probe and analyzed with a flow cytometer. A, The scatter plots (upper panel) and histograms (lower panel) show the quantity of cells emitting light of JC-10 dye. P1 area indicates apoptotic cells emitting green fluorescence, and P2 area indicates healthy cells emitting red fluorescence. Data are expressed as the means ± standard error of the mean from 3 independent experiments. *P < 0.05; **P < 0.01 compared with the control group; #P < 0.05 compared with the ethanol-alone group.
Discussion
People who consume alcoholic beverages may sometimes have a daily caffeine drink. 22 However, the effects of caffeine on neuronal cells constantly exposed to ethanol have not been investigated. Our results showed that caffeine exacerbated the effects of ethanol in human SH-SY5Y neuroblastoma cells. The SH-SY5Y cell line has been widely used as an in vitro cellular model for studying neurotoxicity. 40 Results demonstrated that (1) both ethanol and caffeine caused neuronal cell death in both a dose- and time-dependent manner, (2) exposure of cells with caffeine after ethanol treatment increased the magnitude of cell death, (3) the mechanism of ethanol- and caffeine-induced cell injury was related to early apoptosis, and (4) the superimposed effect of caffeine in ethanol-treated cells might be mediated through the mTOR/p70S6K/4E-BP1 pathway and further impaired the ΔΨm, which might result in mitochondrial dysfunction.
Cell viability results showed that exposure to 200 mM of ethanol for 24 hours led to significant cell death at about 25%. This allowed sufficient cells for further experiments. The length of 24 hours chosen here for ethanol exposure represented an in vitro model of acute ethanol abuse. 41 For caffeine, cell viability significantly decreased to about 34% at 24 hours of exposure to 20 mM. At this dose and length of caffeine exposure, a previous study in hepatic stellate cells showed that apoptosis induced by caffeine was mediated by increasing endoplasmic reticulum stress and could trigger autophagy. 42 Endoplasmic reticulum stress was also shown to inhibit neuronal death by promoting autophagy, 43 a degradation pathway by which the cell digests its own components. This process occurs in all cell types at a low basal level, serving a homeostatic function to recycle proteins and organelles. 44 Thus, this setting of caffeine treatment was considered as appropriate for the remainder of the experiment.
Since blood alcohol levels at higher than 100 mg/dL can suppress human brain activity 45 and caffeine is a brain stimulant, the popular opinion is that caffeine can antagonize or at least reduce the neuropsychiatric and motor deficits induced by alcohol intoxication. 25,26 However, only some animal experiments supported the benefits of caffeine on neuronal functions. In a rat model of ischemic stroke, a combination of low-dose ethanol and caffeine protected the brain from damage only when treated immediately before or for 2 hours after focal stroke 28 but not for chronic daily oral ethanol plus caffeine prior to ischemia. 27,28 In granular, Golgi and glia cells of rat cerebellum, simultaneous ingestion of ethanol and caffeine reversed the apoptotic process induced by ethanol in these cells, as shown by the reduction of caspase-3 and x-linked inhibitor of apoptosis. 30 Toxic effects of the co-administration of ethanol and caffeine were observed in mice, showing that co-administration failed to reverse learning deficits compared to ethanol treatment alone. 31 Similarly, but in rat astrocytes, treatment with ethanol plus caffeine potentiated cytosolic Ca2+ release leading to higher cell death than ethanol treatment alone. 32 It should be noted that these neuroprotective and toxic results were observed from a combination treatment of ethanol and caffeine on cells that were not preexposed to ethanol. Here, we showed that neuronal cells preexposed to ethanol could be more vulnerable to the toxic effect of caffeine.
Ethanol and caffeine are well known to cause apoptosis in neuronal cells, 5,7,30,33,34 and our results confirmed these traits. Our study also showed that the magnitude of early apoptosis markedly increased in cells pretreated with ethanol followed by caffeine. These findings concurred with the decreased activity of mTOR/p70S6K/4E-BP1 signaling. Previous studies have indicated that the inhibition of PI3K/Akt/mTOR signaling can induce apoptosis of neurons through the mitochondrial pathway. 9 -11 mTOR interacts with other proteins to form a complex, mTORC1. This, then, acts as a key downstream signal of the PI3K/Akt pathway to regulate cell survival. 46 The 2 well-characterized downstream signaling molecules of mTORC1 are p70S6K and 4E-BP1. Both 4E-BP1 inhibition and p70S6K activation by mTORC1 activation promote protein synthesis. 47 A previous study in SH-SY5Y cells showed that treatment of 10-mM caffeine for 24 hours inhibited phosphorylation of mTOR, p70S6K, and 4E-BP1. This inhibition was observed as early as 3 hours of the caffeine treatment. 36 In addition, caffeine was shown to inhibit the phosphorylation of Akt, the upstream regulator of mTOR. Our results demonstrated a similar trend of inhibition by caffeine, especially for 4E-BP1. Inhibition of mTOR/p70S6K/4E-BP1 phosphorylation by ethanol has also been demonstrated in the cerebral cortex from mice fed with a 4% alcohol diet for 16 weeks. 19 However, inhibition of Akt phosphorylation did not occur during this chronic ethanol consumption. In SH-SY5Y cells, treatment with 0.4% ethanol for 24 hours also decreased the phosphorylation of p70S6K, which could be observed at the beginning from 1 hour of treatment. 17
Our results clearly demonstrated that exposure of neuronal cells pretreated with ethanol to caffeine dramatically decreased the phosphorylation of mTOR, p70S6K, and 4E-BP1 compared to those without caffeine exposure. It was observed in our results that the combination of ethanol and caffeine did not produce a decrease in viability any greater than the caffeine group alone. This may be due to the effect of ethanol pretreatment on the mTOR pathway, particularly 4E-BP1 which is higher in ethanol alone than caffeine alone, which is supported by previous studies showing that the increased 4E-BP1 levels can be protective of dopaminergic neurons. 48,49 Therefore, cells that are pretreated with ethanol and exposed to caffeine may not show a reduction in viability relative to the group of caffeine alone.
It is known that the mTOR pathway can regulate mitochondrial membrane potential (ΔΨm) of neurons. 35 Both ethanol and caffeine impair the mitochondrial function of neurons by decreasing the ΔΨm. 35,50 Disruption of the ΔΨm is an important process in triggering the apoptotic cascade. 51 Mutual suppression of the mTOR pathway by a combination of caffeine and rapamycin, an mTOR inhibitor, 32 or of caffeine and perifosine, an Akt inhibitor, 52 induces synergistic effects on apoptotic cell death, suggesting that superimposed inhibition of the mTOR/p70S6K/4E-BP1 signaling by caffeine following ethanol exposure would be a rational explanation for augmented apoptotic neuronal death.
As shown by earlier research, apoptosis induced by caffeine depends on autophagy through selective lysosomal degradation (mitophagy). 35 Damaged mitochondria, as reflected by the loss of ΔΨm, is a trigger for mitophagy. 53 Therefore, we used JC-10, a potentiometric probe and a derivative of JC-1 useful for determining ΔΨm, which has been used to reflect mitophagy in various cells including SH-SY5Y cells. 54,55 Although the autophagic pathway may play an important role in neuronal cell survival, several findings suggest that imbalance of autophagic flux leads to neuronal cell death. 56 The present JC-10 results may imply that excessive activity of mitophagy may occur in cells exposed to caffeine following ethanol and lead to apoptotic cell death as indicated in the Annexin V-PI results. Direct measurement of markers of autophagy such as LC3, p62, Atg5, or Beclin-1, and in particular, colocalization of mitochondria and the autophagosomal marker MAP1LC3B for mitophagy would be essential to support this assumption. It was observed in our results that the combination of ethanol and caffeine produced a slight decrease in the ΔΨm, compared to the caffeine group alone. Although the decrease was not statistically significant, it may reflect that caffeine potentiates the mitophagic process initiated by ethanol, leading to the lower ΔΨm when compared to the caffeine group alone that induce mitophagy in cells without ethanol pretreatment.
Based upon published data, it is known that caffeine is absorbed rapidly and completely with 99% being absorbed within 45 minutes of ingestion. 57 It is also known that ethanol is distributed completely and uniformly throughout the body’s water, thus the brain is exposed to a similar alcohol concentration as the blood. 58 Such data indicate there are limitations to the present study. First, the effects of 20-mM caffeine, following ethanol exposure at relatively high concentrations, were far less than shown by the equivalent to toxic plasma level of 100 mg/L in humans. 59 Based on previously reviewed experimental outcomes, high doses of caffeine are likely to be harmful. Since the brain is exposed to similar concentrations as the blood, it is unlikely that the concentrations of ethanol and caffeine used in these experiments would ever be encountered in humans as these concentrations are well beyond known lethal concentrations. Although such high concentrations may help us to understand mechanisms, the results do not provide evidence of what will happen at physiologic levels of caffeine and ethanol after humans consume these substances. Thus, our results do not indicate that consuming ethanol and caffeine in normal physiologic quantities causes neurotoxicity. However, these data still provide some value in terms of potential mechanisms for toxicity. Second, the direct cytotoxic impact of ethanol on cells is one of the main concerns using in vitro ethanol treatment for levels above 100 mM, 42 while acute in vitro exposure to ethanol may be hampered by evaporation. Thus, the reported accuracies of the ethanol concentrations in our study might be inconsistent. However, the declining in vitro ethanol levels may be desired to imitate in vivo ethanol elimination. Lastly, there is a drawback due to the lack of accounting for in vitro metabolism of caffeine and ethanol that could influence toxicity.
In conclusion, caffeine treatment after neuronal cells were exposed to ethanol-produced marked cell damages, mediated through enhanced inhibition of mTOR/p70S6K/4E-BP1 signaling leading to impaired ΔΨm and, eventually, apoptotic cell death. Further detailed research is, however, required to understand the biology of caffeine on ethanol-induced neurotoxicity and to explore the clinical relevance of our findings for drinkers who consume caffeine to antagonize the depressant effect of alcohol.
Footnotes
Authors’ Note
This research is under the research framework of Mahidol University.
Author Contribution
Sangaunchom, P. contributed to acquisition, analysis, or interpretation of data and drafted the manuscript. Dharmasaroja, P. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, drafted the manuscript, critically revised the manuscript. Both authors gave final approval and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from Army Institute of Pathology, Royal Thai Army (to P.S.) and in part from Faculty of Science, Mahidol University (to P.D.).