
Abstract
The liver is primarily thought of as a metabolic organ; however, the liver is also an important mediator of immunological functions. Key perspectives on this emerging topic were presented in a symposium at the 2018 annual meeting of the American College of Toxicology entitled “Beyond metabolism: Role of the immune system in hepatic toxicity.” Viral hepatitis is an important disease of the liver for which insufficient preventive vaccines exist. Host immune responses inadequately clear these viruses and often potentiate immunological inflammation that damages the liver. In addition, the liver is a key innate immune organ against bacterial infection. Hepatocytes and immune cells cooperatively control systemic and local bacterial infections. Conversely, bacterial infection can activate multiple types of immune cells and pathways to cause hepatocyte damage and liver injury. Finally, the immune system and specifically cytokines and drugs can interact in idiosyncratic drug-induced liver injury. This rare disease can result in a disease spectrum that ranges from mild to acute liver failure. The immune system plays a role in this disease spectrum.
Keywords
The Liver as an Immune System Organ
Of the numerous physiological functions associated with the mammalian liver, the pivotal role in immunity is often overlooked. The fetal liver is the primary site of hematopoiesis before birth, and the mature liver is the source of 80% to 90% of nonimmunoglobulin immune system proteins (eg, complement, haptoglobin, and cytokines), the site of first contact with products of the gut microbiome (distal from the intestines), important for control of inflammation, site of interaction between the immune system and various metabolic functions, and an important organ for destruction of pathogens. 1 There are several anatomic aspects of the liver that are associated with immune function. First, the liver is highly vascularized: In the adult human, 30% of total blood volume passes through the liver every minute. It is estimated that 108 lymphocytes interact with liver cells every day. The portal vein transports 80% of antigen-rich blood from the gut to the liver. The hepatic vasculature is lined with unique liver sinusoidal endothelial cells (LSECs) that are fenestrated—lacking a basement membrane—which allows for direct lymphocyte contact with hepatocytes. 2 About a third of the cells in the liver have immune function, and the adult human liver contains about 1010 lymphocytes. 3
The types and proportions of immune cells in the liver are also unique. For example, the majority of hepatic T cells demonstrate suppressor (CD8) versus helper (CD4) phenotype. 4 The liver also contains enriched populations of cells associated with immune tolerance. 5 Especially predominant are CD4+CD25+Foxp3+ regulatory T cells (TREG). 4 B cells are relatively sparse in the liver. The liver also contains enriched populations of natural killer (NK) and NK T cells (NKT) and special antigen-processing and -presenting cells (Kupffer cells [KCs], myeloid dendritic, and plasmacytoid dendritic cells [pDC]). 6 Hepatic dendritic cells produce interleukin (IL)-10 and transforming growth factor β (TGF-β), associated with immune tolerance rather than acquired immunity. 3 Hepatic dendritic cells are also associated with phenomena such as cross-priming and cross-presentation of antigens.These functions allow for greater control of immune responses. 7
The liver is important in innate immunity, recognition of foreign antigens without previous exposure, and acquired immunity. For example, most liver cells (including hepatocytes) express all of the Toll-like receptors (TLR) and respond to pathogen-associated molecular patterns (PAMPs) by producing proinflammatory molecules such as tumor necrosis factor α (TNFα), IL-6, and interferon (IFN). 8 The LSECs also demonstrate control of innate immunity, for example, mediating a phenomenon called “LPS tolerance” after repeated exposure to endotoxin. 2 Hepatic CCR9+ pDC respond to TLR ligands by producing anti-inflammatory molecules such as IL-10 and TGFβ. 4
Kupffer cells are a type of macrophage uniquely expressed in the liver, accounting for 30% of nonparenchymal hepatocytes. About 80% of total body fixed macrophages are KCs. 9 As with other macrophages, primary functions of KCs are phagocytosis and antigen presentation. In addition, KCs express TLR4 and respond to LPS by producing proinflammatory molecules such as TNF-α, IL-1β, IL-6, IL-12, and IL-18. However, unlike other macrophages, KCs may respond to PAMPs, damage-associated molecular patterns (DAMPs), and TLR ligands with anti-inflammatory cytokines such as IL-10. 4
As with other immune cell types, hepatic NK cells demonstrate unique characteristics. 10 For example, 30% of total hepatic leukocytes are NK cells and 50% of these express the CD56BRIGHT phenotype (vs 10% in peripheral blood). Hepatic NK cells express most TLRs but may respond to TLR3 ligands by inhibiting liver regeneration. Hepatic NK cells demonstrate other unique activities. For example, hepatic NK cells respond to TNF-related apoptosis-inducing ligand by inhibiting tumor initiation, growth, and metastasis. In addition, hepatic NK cells possess unique stress detection mechanisms such as the NKG2D system. 11 CD49a+DX5-NK cells may possess hapten-specific memory. 12,13
Hepatic T cells are predominately of the suppressor type. 14 In addition, the liver may have the most γδ T cells in the body (25%). These mucosal γδ T cells appear to be relatively invariant and recognize a limited number of stress proteins and nonprotein antigens. Hepatic B-cells makeup only about 8% of total lymphocytes in the liver and are enriched for CD5. The NKT cells, 30% of total hepatic lymphocytes, demonstrate very restricted αβ chain types (MHC 1 CD1d restricted), recognize lipid antigens, and have been described as 2 distinct populations. 15 Type I NKT cells are activated by IL-12, IL-18, TLRs, or IFN; express Th1, Th2, or Th17 cytokines, and can kill hepatocytes directly. Type II NKT cells recognize glycolipids and phospholipids and suppress proinflammatory type I NKT cells. Hepatic myeloid and pDC are considered “immature” but can be potent cytokine producers and T-cell stimulators (eg, CD141+ cells).
The liver plays an important role in immune tolerance. Not only is the fetal liver the primary site of hematopoiesis, but it is also pivotal in maintenance of central tolerance, which prevents activation of the maternal immune system. 16 Peripheral immune tolerance is an active process controlled by antigen-specific TREG cells. The KCs, hepatic myeloid-derived suppressor cells (MDSC), and even hepatic neutrophils can respond to immunostimulation by producing IL-10, TGF-β, or other immunosuppressive molecules, resulting in downregulation of co-stimulatory cell surface markers, impaired T-cell activation, and upregulation of antigen-specific TREG cells.
The healthy liver appears to be “tolerogenic,” that is, it is maintained in a “controlled inflammatory state.” 17 Both immune and nonimmune cells (hepatocytes, hepatic stellate cells, and LSECs) express pattern recognition receptors (PRR) for PAMPs, DAMPS, and other ligands that, when activated, result in expression of either pro- or anti-inflammatory cytokines. Whether PRR activation results in inflammation or not depends on both the nature of the ligand and the status of the liver. Constituent expression of proinflammatory cytokines (eg, IL-2, IL-7, IL-12, IL-15, and IFNγ) is characteristic of many hepatic nonimmune cells, but these also demonstrate constituent expression of anti-inflammatory cytokines (eg, IL-10, IL-13, and TGFβ). Which pattern predominates depends on factors that are not clearly understood, although some appear to be important. For example, KCs and hepatic dendritic cells when activated by antigen or PRR ligands undergo a change in metabolism from oxidative phosphorylation to aerobic glycolysis, a process known as the Warburg effect. 18 This is a proinflammatory activation of immune cells associated with upregulation of hypoxia-inducible factor 1-α and IL-1β. The liver is also the source of acute-phase proteins (APPs; such as complement, haptoglobin, fibrinogen, prothrombin, IL-6, C-reactive protein) produced in response to proinflammatory signals from extrahepatic sites, a phenomenon called the acute-phase response (APR). Hepatic inflammasomes are multiprotein complexes (essentially “virtual organs”) that can be activated by PRR ligands such as PAMPs and DAMPs, reactive oxygen species, cholesterol, triglycerides, and products of metabolism such as succinate, resulting in proinflammatory activation of caspase-1 and release of IL-1β. 19 This process may help to explain the relationship between chronic hyperlipidemia and increased susceptibility to chronic inflammatory diseases. Hepatic parenchymal cells express liver X receptors (LXRs), which regulate lipid homeostasis but also, when activated, produce anti-inflammatory effects. 20 For example, activated LXRs inhibit TLR (2, 4, and 9) signaling to nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK), their downstream effectors. Some APR proteins have anti-inflammatory activity (eg, C-reactive protein inhibits TNFα production, α2-macroglobulin inhibits neutrophil function, and serum amyloid A activates MDSC). The pro- and anti-inflammatory mechanisms in the healthy liver are thus balanced to respond appropriately when challenged by foreign antigens and products of metabolism.
Hepatic immune response to foreign antigens (microbial, viral, or food) depends on numerous factors. 1 Depending on the nature of the antigen, the initial response may be activation of innate immunity. The KC response to PRR activation can be either pro- or anti-inflammatory. If pro-inflammatory, KCs increase production of IL-1, IL-6, TNFα, MIP-1α, RANTES, and other mediators of inflammation. Hepatic T cells can be activated without CD4+ T cells (T-independent activation). The mechanisms of acquired immunity (T-helper cells, specific B-cell activation, and antibody production) play a role, but the liver does not appear to be an important site for this aspect of the immune response. If the immune system eliminates or otherwise neutralizes the foreign antigen, mechanisms to control inflammation become important. Hepatic NK cell-mediated hepatic stellate cell (Ito cells) cytotoxicity controls inflammation and inhibits fibrosis and formation of scar tissue. In addition, activated hepatic NK cells increase the expression of matrix metalloproteinases (MM) and decrease the expression of MM inhibitors, which is an anti-inflammatory response. Regeneration of damaged hepatic tissue can be mediated by proinflammatory cytokines (IL-1α, IL-6, and TNFα), growth factors, and KCs.
Chronic liver inflammation can result from several factors: inability to eliminate a pathogen, failure to neutralize a foreign antigen, chronic exposure to PRR ligands or cytotoxic xenobiotics, and/or failure to repair damaged tissue. In addition, some pathogens escape immune system elimination by, in some way, manipulating hepatic immune tolerance mechanisms. Liver pathology due to chronic inflammation can include fibrosis, autoimmunity, cancer, and liver failure. Thus, it is important to remember that liver inflammation is a Janus-headed phenomenon: protective when acute and likely to be pathologic if chronic.
Viral Pathogens of the Liver: Viral Hepatitis, Pathology, and Immunity
Viral hepatitis causes significant morbidity and mortality worldwide. While viral hepatitis can be self-limiting such as in the case of hepatitis A virus (HAV), in other instances such as Hepatitis B virus (HBV) and hepatitis C virus (HCV), the infection can lead to a variety of problems such as chronic infection, cirrhosis, and hepatocellular carcinoma. 21 Worldwide about 50% of the cases of HCV and 30% of HBV infections are not diagnosed, and only 33% of HCV and only 10% of HBV infections are treated. Infection with HBV is a particularly important health concern in the absence of infant vaccination because mother-to-child transmission is the most common means of transmitting HBV and results in a great number of chronic infections of HBV. 22
A prominent group of viruses that infect the liver are the hepatotropic viruses, which specifically target the liver for infection and have developed a specialized means to gain entry into liver cells. Additionally, HBV and HCV have developed strategies that allow them to evade and suppress the response of the immune system and results in a pathological process that is extremely harmful to the host. 23,24
The immune response to liver infection is multifaceted, and clearance of viral infections typically results from the induction of cytotoxic T-lymphocytes (CTLs) that are directed at the viruses and are subsequently followed by the development of neutralizing antibodies. 25 Development and expression of CTLs are the primary mechanisms involved in the clearance of these infections, and neutralizing antibodies play a variable role in the clearance of the infection. Additionally, these antibodies provide some protection from subsequent infections. 26 -28
The CTL response is directed by the expression of various cytokines and chemokines that are released during the process of infection and prominent among these cytokines are IFNγ and TNFα, which possess various antiviral and macrophage-directing activities.
The response of the immune system to the acute infections of HBV and HCV are similar but different in terms of the outcomes that result in a chronic phase of infection. In acutely ill patients who successfully clear HBV, the T-cell response is robust and multiclonal with broad specificity, whereas in chronically infected individuals, it is insubstantial and narrowly focused. 29 The story with HCV is slightly different. As with an acute infection with HBV, the T-cell response during the acute infection with HCV is strong and multispecific, but during the chronic phase of HCV infection, unlike HBV infection, the immune response remains vigorous. In this case, a high rate of HCV mutation, due to a faulty RNA polymerase, creates various resistant forms of virus, and these new quasi-species of virus have a survival due to alterations in immune recognition sites. 30 -36
The hepatotropic viruses are named straight forwardly A, B, C, D, and E.
Hepatitis A virus: It is a nonenveloped picornavirus, containing a single-stranded RNA packaged in a protein shell. Hepatitis A virus is unlike other picornaviruses in that it requires an intact host eukaryote initiating factor 4G to make viral protein. 37 Infections with HAV typically are self-limited and acute because the immune response is almost always robust and effective in eliminating the virus. The HAV rarely leads to chronic hepatitis because of the vigorous cell-mediated immune response. 38 Hepatitis A virus enters liver cells through HAV cellular receptor 1 also known as T-cell immunoglobulin and mucin domain 1 which 39 also is used by Ebola 40 and Dengue viruses. 41 The HAV viral particles replicate not only in hepatocytes and KCs but also in gastrointestinal epithelial cells. After replication, viral particles are released into blood and bile. To exit the liver, HAV particles are secreted across the cellular membrane into the biliary canaliculus and pass into the bile and small intestine.
Hepatitis B virus: The HBV virus may cause acute or chronic infections. Hepatitis B virus is a small DNA virus and has some features similar to those of retroviruses because the viral DNA integrates into the host genome by forming a covalently closed circular form of DNA. 42 Thus, the viral genome persists in the infected host. Hepatitis B virus gains entry into hepatocytes through binding to the human sodium taurocholate cotransporting polypeptide heparin sulfate proteoglycans. Upon exiting a liver cell, DNA-containing capsids acquire an envelope consisting of cellular membrane lipids and are released through the constitutive secretory pathway via the Golgi apparatus. 43 Although much of the pathology associated with hepatitis is directly due to the actions of the immune system, some aspects of hepatic pathophysiology may be due to the virus. In case of HBV, proteins coded by one of the genes, gene X, may stimulate the development of liver cancer. 44 This viral action and a few others are exceptions to the general rule that the virus does not directly contribute to pathogenicity.
Hepatitis C virus: The HCV is a small, enveloped virus that is single stranded with a positive-sense RNA. The virus was formerly known as nonA, nonB virus. Infection with HCV can result in acute and chronic states. There are 6 major HCV genotypes and a large number of subtypes. Hepatocytes are the major target of HCV, but the other targets of virus may include nonhepatic cells. 45 -47 Hepatitis C virus enters through interactions with cell surface molecules such as CD81 as well as several other molecules, including the LDL receptor among others (Scavenger Receptor Class B, type 1, Dendritic Cell-specific Intercellular adhesion molecule-3-Grabbing Non-integrin, Claudin-1, and Occludin). 48 The release of HCV from the hepatocyte involves the very low-density lipoprotein secretory pathway. 49 Hepatitis C virus presents a major challenge to the host immune system due to the creation of escape or resistant mutants that are formed during the process of infection. As noted, this results from a defective viral replication process that creates quasi-species due to a viral RNA-dependent, RNA polymerase (NS5b) that lacks proof-reading capability. Despite these challenges to the immune system, approximately 20% of acute HCV infections can be spontaneously cleared giving hope that a vaccine can someday be devised. 50
Hepatitis D virus (HDV): The genome of HDV consists of a single-stranded RNA, and the virus can only propagate in the presence of the HBV. 51 Of note, coinfection with HBV and HDV results in the highest fatality rate of all the acute hepatitis infections (20%); additionally, progression to cirrhosis is also faster in chronic patients with combined infections, when compared to HBV monoinfection, thus suggesting an interaction between the viruses regarding disease progression. Transmission of HDV can occur either as a simultaneous infection with HBV (coinfection) or as superimposed on individuals with chronic hepatitis B or hepatitis B carrier state (superinfection). The fate of HDV is determined by the host response to HBV, and HDV is cleared if HBV is cleared. 52
Hepatitis E virus (HEV): HEV is a small, nonenveloped virus with a single-stranded RNA genome and was discovered in 1983. It typically causes a self-limiting short-lived disease, and the majority of infections remain asymptomatic. Hepatitis E virus has one important distinction and that it causes a high mortality rate in pregnant women. 53
The fact that liver injury is largely due to the immunologic response rather than the direct action of the virus is supported by a large body of scientific literature. The significant delay between the onset of HBV and HCV infections and the appearance of signs and symptoms of the disease underscores the lack of direct toxicity of these viruses and ability of the viruses to limit an immune response. During the process of infection, virus-specific, cytotoxic T lymphocytes (CTLs), generated by the immune system in the liver, cause apoptosis and necrosis due to the responses of various immune cells acting directly and through the expression of various cytokines and chemokines. 23,54 Furthermore, antigen-nonspecific inflammatory cells enhance CTL-induced immunopathology. 55 This may be augmented by platelets that are activated at the site of infection by the inflammatory response and cause an accumulation of CTLs in the liver mediating damage to the host liver. 56
For example, after infection with HCV, a strong and sustained level of CD8+ T-cell activity as well as CD4+ T-cell helper activity is observed and promotes clearance of the infection. Patients who spontaneously recover from HCV infection demonstrate vigorous multiepitope-specific CD4+ and CD8+ T-cell responses. By contrast, patients who develop chronic hepatitis C tend to have delayed, transient, or more narrowly focused T-cell responses. 57 In comparison to cell-mediated immune responses, the role of an antibody response to HCV is less clear. The HCV-specific antibodies are much more variable in infected patients, and early after HCV infection no antibodies appear, and in some cases, do not appear at all. Often strain-specific, neutralizing antibodies are present at low levels during the first 6 months of HCV infection and may take as long as 6 to 12 months for antibodies to reach levels that approximate neutralization titers.
Both the cell-mediated and antibody responses are regulated by the innate and adaptive arms of the immune system with the innate inflammatory response mediating the initial immune response to viral infection. This critical response depends on the recognition of nonself-molecules. In the case of HCV, it is the intracellular presence of viral RNA as a nonself-molecules that triggers an immune response. The innate response depends on 2 complementary groups: the PAMPs which include viral RNA and the PRRs such as TLRs. While some PRRs are located at the cell surface, others are located at intercellular sites. In response to HCV infection, important cytoplasmic PRRs include the 2 TLR viral RNA receptors: melanoma differentiation-associated protein 5 (MDA-5) and retinoic acid-inducible gene I like receptor (RIG-I). Both MDA-5 and RIG-I use a common signaling adaptor molecule, mitochondrial antiviral-signaling protein, which results in the expression of IFNβ, among other cytokines. Besides cytokine responses, immune cellular responses are important. The main function of NK and NKT cells are to limit viral replication via their expression of cytokines and that function may become compromised during chronic infection and contribute to viral persistence.
The initial innate response is critical to the outcome of the infection in terms of the subsequent adaptive response. The current literature suggests that a strong innate response leads to an effective, adaptive immune activation and control of viral replication. Inversely, an inefficient activation of the innate immune pathways gives rise to a lower and less rigorous adaptive response that cannot clear the infection and has deleterious effects on the liver.
The adaptive immune system is needed to control and eradicate viral infections via the processing of viral antigens, and in the case of HAV, the viral infection is eliminated by the production of antibodies. However, in the cases of HBV and HCV, the viruses are often successful at suppressing and evading the immune system. Thus, setting up the situation in which a continuing immune response takes hold and results in significant pathology as part of the immune response to clear the infection, infected hepatocytes activate CD4+ T cells that subsequently interact with virus-specific CD8+ T cells and clears the virus by noncytolytic and cytolytic effector mechanisms. This T cell-mediated protection depends on a constant supply of activated effector CD8+ T cells to maintain the immune response and to prevent the emergence of resistant virus. 21,25
If the T cells and subsequent humoral responses fail to control and eradicate the infection, a persistent infection is established. 58 Unfortunately, the exact mechanisms are poorly understood but are thought to involve failure of virus-specific CD8+ T cell. But most likely, other mechanisms contribute to T-cell failure, such as emergence of viral escape mutants, T-cell repression that is mediated by inhibition, lack of sufficient CD4+ T-helper cells, or development of Tregs.
In the case of HBV, the virus has evolved various mechanisms that contribute to establishing a chronic condition 59 that results in a continued immune-mediated pathology. 29 In this instance, both viral and host factors influence the development of a chronic infection. These factors include host genetic susceptibility in terms of the MHC class II locis 60 -62 and cytokine receptors, high levels of replication of the virus, and expression of viral proteins like HbsAg, 63 HBeAg, 64 or HBx. 65 The latter is thought to suppress innate responses by inhibiting TLR signaling. 66 Many of these factors are related to virus mutation and the subsequent evasion of T and B cell-mediated responses. In the case of HBV, other factors contribute to weakening the host response or help the virus evade detection and in particular through the formation of a covalent closed circular form of viral DNA in the nucleus. 67 Other suppressive factors include production of IL-10 which suppresses of CD8+ T-cell activity. 68
The ability of HCV to persist within a host is believed to be related to the numerous mechanisms. As noted previously, one important escape mechanism results from the formation of quasi-species due to the high rate of mutation that takes place during HCV replication and is the result of lack of a proofreading capacity of its error-prone RNA-dependent, RNA polymerase. This results in a rapid diversification of the viral population. In some cases, this delays and hinders the emergence of effective adaptive cellular and humeral immune responses due to the lag between the point in time that one set of immune responses has evolved, which may be appropriate for the initial virus and the generation of escape mutants or quasi-species. Rapid genetic mutation may also impair effective responses involving MHC–peptide complexes, since genetically modified viral peptides can come about and alter the interaction with CD4+ T cells in a manner effecting the expression of the TH1 and TH2 responses. Genetic diversification results in viral peptide modification, and its MHC interaction may also weaken T-cell recognition, alter binding of viral peptide to MHC molecules, or impair antigen processing, thus decreasing immune effectiveness. 69 HCV infections are reported to impair the functionality of antiviral T cells by disrupting their maturation and altering their effector function. 70 Analysis of HCV-specific CD8+ T cells in the liver and peripheral blood has revealed various defects in the effector cell populations. 71 These include an altered maturation phenotype with a diminished capacity for expansion, poor peptide-specific cell killing ability, and decreased secretion of IFN-γ and TNFa. 72
Both HBV and HCV are associated with an increase in autoimmune disease that takes on various manifestations. 73,74 This may be the result of actions on Tregs also known as suppressor T cells. These cells are believed to be important components in the ability of the system to maintain tolerance to self-antigens and prevent autoimmune disease due to their ability to produce IL-10 and TGF-β. During chronic infection with HBV, Treg cells were observed to increase in their number when compared to acute infection or healthy patients, and the increase correlated with greater viral loads 75 suggesting an association with viral replication. Additionally, an increased number of Tregs were found in the peripheral blood of patients with HBV liver failure, 76 suggesting that they play an active role in downregulating effector immune responses. HBcAg was also able to induce significantly higher levels of IL-10 in peripheral blood mononuclear cells from patients with chronic HBV infection. 77
Extrahepatic disease is observed with HBV or HBC and only occasionally in patients with hepatitis A. The increase associated with autoimmune disease in the case of HBV may involve several causes, including molecular mimicry between HBV antigens and self-proteins and the generation of immune complexes with resultant extrahepatic disease. 74 By far among various aliments the most common extrahepatic condition following HCV infection is that of mixed cryoglobulinemia, and almost half of all patients with chronic HCV exhibit this condition sometime during the course of their illness. Type I cryoglobulinemia predominantly affects the skin, kidney, and bone marrow, whereas types II and III predominantly involve the skin, peripheral nervous system, and kidney. The exact mechanism by which HCV leads to essential mixed cryoglobulinemia is not clear. One possibility lies in the ability of HCV to bind B lymphocytes via the CD 81 cell surface receptors and thereby lower the activation threshold of B lymphocytes, leading to B-cell clonal expansion, overproduction of immunoglobulins, and ultimately formation of cryoglobulins that affect brain, joints, blood vessels, bones, and kidneys. 78
Regarding HBV infections, immunecomplexes may be formed between antibodies and HBsAg or HBeAg. These complexes may be responsible for local activation of the complement 79 at various sites such as the medium- or small-sized arteries of the kidney and the skin. This is the mechanism implicated in the occurrence of polyarteritis nodosa systemic vasculitis and membranous glomerulonephritis. 80
Besides hepatotropic viruses, there are other viruses that cause damage to the liver but without specifically infecting the liver itself. 81 These viruses target other organs such as the lung but can indirectly affect the liver as part of a generalized infection of the host. Viruses that demonstrate this phenomenon include the herpes viruses (Epstein-Barr virus, cytomegalovirus [CMV], and herpes simplex virus), parvovirus, and adenovirus. Liver involvement in these nonhepatotropic, viral infections can range from mild elevations in liver enzymes to fulminant liver failure. In case of infection with Epstein-Barr virus, hepatitis is thought to be a consequence of viral-specific CD8+ T cells, generated in response to the viral infection occurring outside the liver. 82
Vaccination or previous exposure to hepatitis A gives long-term immunity, and since the 1990s, several vaccines against HAV have been commercially available, which include both an inactivated and live attenuated vaccine. The first inactivated HAV vaccine was produced from a strain of the virus that was propagated in cell culture. This was then purified and inactivated using formalin, and the purified HAV strain was then grown in cell culture. Besides the monovalent forms of the vaccine, combination forms are also available which include antigens to either hepatitis B or typhoid virus. These include Twinrix, which is a vaccine against both hepatitis A and hepatitis B, and Vivaxim, which is a vaccine against hepatitis A and typhoid fever.
The first hepatitis B vaccine was approved in the United States in 1981. A recombinant monovalent version was marketed in 1986. First HBV vaccine developed was a subviral particle of hepatitis B surface antigen and was purified from the inactivated plasma of asymptomatic carriers of HBV. Subsequently, a vaccine was developed from cloned HBV DNA fragments encoding the S protein. Monovalent forms of the vaccine are believed to provide indefinite protection. The monovalent form of hepatitis B vaccine is also available in combination with vaccines to prevent other diseases such as diphtheria and polio.
There is no available vaccine for HCV although efforts are underway to develop both preventative and therapeutic vaccines. The major obstacle to development of both is the high rate of replication and expression of related but different immunological versions of the virus. This causes the emergence of resistant mutations within the virus population which come to dominate the viral population.
For preventative vaccines, the envelope epitopes E1 and E2 have been targets of clinical development. Additionally, the HCV core protein has been considered a candidate target for a therapeutic T-cell vaccine, since this is the most highly conserved region. However, studies suggest that the immunology may be more complex than originally thought as the core protein itself may interfere with development of innate and adaptive anti-HCV immune responses.
In summary, HAV, HBV, and HCV are important health concerns, despite the fact that an effective vaccine for HAV and HBV exists. Although infection with HAV typically produces a self-limiting disease without significant sequelae and an effective vaccine exists, HAV remains a health concern. Both HBV and HCV are able to evade and suppress the immune system through various mechanisms, and lacking an effective control of the infection may lead to significant morbidity and mortality. To combat these infections, scientific efforts are underway which may lead to an effective vaccine to prevent HCV and therapeutic vaccines for HBV and HCV.
Bacterial Pathogens of the Liver: Liver Immune Response to Bacterial Infection
The liver is the largest gland in the body and has many important functions, including detoxification, protein synthesis, and the production of bile acids that help digest food. In addition, the liver is also considered an innate immune organ, playing a key role in host defense against bacterial infection. 6 The first evidence for a critical role that the liver plays in innate immunity is the enrichment of KCs in the liver, which accounts for 80% to 90% of the total population of fixed tissue macrophages in the body. In addition, KCs together with LSECs form the hepatic reticuloendothelial system, which is the major site for removing circulating macromolecules and microorganisms including bacteria from the systemic circulation. Hepatocytes constitute approximately 70% to 85% of the liver volume and play an essential role in metabolism, detoxification, and protein synthesis. Accumulating evidence suggests that hepatocyte is also a key cell type for innate immunity against bacterial infection. 8 Here we discuss how hepatocytes fight bacterial infection by secreting a large amount of innate immunity proteins and how the expression of these innate immunity proteins is upregulated after bacterial infection.
Hepatocytes constitutively express and secrete a variety of proteins that play important roles in innate immunity against bacterial infection. 6,8 Most of these proteins are further upregulated in response to bacterial infection. These proteins either directly kill pathogens or orchestrate the immune system for efficient pathogen clearance. As many of these proteins are quickly produced after stimulation, they are also called APPs. For example, hepatocytes constitutively produce most of the complement proteins and maintain sufficiently high levels in the circulation, eliminating bacteria. 6,8,83,84 Many of these complement proteins are further upregulated after bacterial infection or inflammatory stimulation. For instance, the most abundant complement in the blood is C3 complement protein (∼130 mg/dL), which is mainly produced by hepatocytes. 85 These high basal levels of serum C3 are further increased by 50% during the APR and bacterial infection. In addition, hepatocytes produce many other plasma complement components and their soluble regulators, including the classical (C1 r/s, C2, C4, C4 bp), alternative (C3, factor B), lectin (MBL, MASP1-3, Map19), and terminal (C5, C6, C8, C9) pathways of the complement system as well as soluble regulators (factors I, H, and C1 inhibitor). 6,8,83,84 The bactericidal functions of complements are mediated via the activation of chemical cascades to create pores in the membranes of invading bacteria or pathogenic host cells and lyse the targets. Three pathways activate the complement system: the classical pathway, the alternative pathway, and the lectin pathway. 86 In addition to complements, many other opsonins such as C-reactive protein, serum amyloid a component, and serum amyloid p component are predominately produced by hepatocytes. 6,8 These opsonin proteins assist in the phagocytosis of foreign pathogens and complement activation.
Immune cells can recognize bacteria via binding to PAMPs of bacteria and subsequently become activated to fight bacterial infection. The most well-characterized PAMP is lipopolysaccharide (LPS), which is the major molecular component of the outer membrane of gram-negative bacteria. The LPS is recognized by TLR4 on immune cells, followed by activation of NF-κB activation, and downstream inflammatory responses. 87 Interestingly, LPS does not directly bind TLR4 and only interacts with TLR4 after forming a receptor complex with several proteins that are mainly produced by hepatocytes. 87 These proteins include LPS-binding protein (LBP), soluble CD14 (sCD14), and soluble MD-2 (sMD-2). 87 Therefore, hepatocytes play a key role in regulating LPS signaling via the production of LBP, sCD14, and sMD-2 and subsequently activating innate immunity against bacterial infection. 6,8
Hepatocytes are known to play an important role in controlling iron metabolism via the production of several iron metabolism-related proteins: lipocalin-2, transferrin, hemopexin, and hepcidin. 6,8 Many of these proteins also play a critical role in inhibiting bacterial growth by preventing bacteria from obtaining iron. Among them, lipocalin-2 is probably the most extensively studied iron metabolism-related proteins for its role in inhibiting bacterial infection. Lipocalin-2 is highly elevated in many types of liver diseases and is a predicator for mortality and bacterial infection in patients with liver cirrhosis. 88 Lipocalin-2 was first identified in neutrophils and was also called neutrophil gelatinase-associated lipocalin. Lipocalin-2 binds to the siderophore enterobactin and subsequently prohibits iron uptake by bacteria. Many types of pathogenic bacteria acquire iron with enterobactin-like siderophores, including Escherichia coli, Salmonella spp., Brucella abortus, Bacillus anthracis, Burkholderia cepacia, Corynebacterium diphtheriae, and Paracoccus spp. 89 It has been shown that lipocalin-2 protects mice from E coli in peritoneal infection and pneumonia models as well as the Klebsiella pneumoniae infection model. 89 -91 Despite its initial discovery in neutrophils, it is now known that hepatocytes are mainly responsible for the production of lipocalin-2 in the circulation after bacterial infection, and neutrophils mainly contribute to local lipocalin-2 elevation. 92,93 Basal serum levels of lipocalin-2 in mice are ∼62 ng/mL, which can be elevated approximately 100-fold, reaching to ∼ 6000 ng/mL after bacterial infection. Genetic deletion of the lipocalin-2 gene specifically in hepatocytes markedly increased the susceptibility of mice to systemic bacterial infection (via the intraperitoneal injection of bacteria) with significant reduction in serum lipocalin-2 elevation. 92 Hepatocyte-specific lipocalin-2-knockout mice and global lipocalin-2-knockout mice are equally susceptible to systemic bacterial infection, indicating the dominance of hepatocytes in protecting against systemic bacterial infections by producing lipocalin-2. 92 Interestingly, both hepatocytes and neutrophils express high levels of lipocalin-2 after bacterial infection, and a recent study suggests that hepatocytes and neutrophils cooperatively suppress bacterial infection by differentially regulating lipocalin-2. 93 This study has demonstrated that hepatocytes play an important role in fighting against systemic infection by secreting lipocalin-2 protein into the circulation postintraperitoneal injection of bacteria. However, for the local lung infection site postintratracheal intubation of bacteria, the circulating lipocalin-2 produced by hepatocytes cannot efficiently reach to the local infection area. Instead, neutrophils can carry lipocalin-2 protein in their specific granules and reach to the local infection area, playing a critical role in combating lung local infection. 93 In addition, lipocalin-2 from neutrophils but not from hepatocytes is a component of neutrophil extracellular traps (NETs) and inhibits bacterial infection via the NETs. 93
Normal hepatocytes constitutively express high basal levels of many innate immunity proteins. For example, approximately 150 to 350 mg/dL fibrinogen, 130 mg/dL complement C3 protein, and 10 mg/dL peptidoglycan recognition protein 2 are found in the blood from healthy individuals. 85,94,95 These high basal levels of hepatocyte-derived innate immunity proteins are controlled by liver-enriched transcription factors such as hepatocyte nuclear factors (HNFs) and CCAAT/enhancer-binding protein (C/EBP). 6,8 Hepatocyte nuclear factors are expressed mainly in the liver but are also expressed and play critical roles in regulating gene expression in other tissues. The C/EBP family members include C/EBP-α, C/EBP-β, C/EBP-δ, and C/EBP-Ζ, many of which are enriched in the liver. The promoter regions from the majority of hepatocyte-derived innate immunity proteins contain binding sites for these liver-enriched transcriptional factors, HNFs and C/EBP, which contribute to high basal levels of innate immunity proteins produced by hepatocytes. 6,8 Additionally, some of these liver-enriched transcription factors are also upregulated postbacterial infection and subsequently stimulate the inducible expression of innate immunity proteins in hepatocytes.
After bacteria enter the body, immune cells will first recognize them and become activated to produce a wide variety of cytokines. These elevated cytokines not only further activate adaptive and innate immunity but also stimulate hepatocytes to produce a large amount of innate immunity proteins that subsequently fight against bacterial infection. Based on their downstream signaling pathways, we can group these cytokines into 2 categories. The first category of cytokines which includes IL-6, IL-6 family cytokines, and IL-22, mainly activate STAT3 and subsequently upregulate the expression of innate immunity proteins in hepatocytes. 96 -98 The second group of cytokines which includes IL-1β and TNF-α predominately activate NF-κB and subsequently increase the expression of many innate immunity proteins in hepatocytes. Both STAT3 and NF-κB do not significantly contribute to the production of the basal levels of innate immunity proteins, but they can work together to promote maximal induction of many innate immunity proteins in hepatocytes, playing essential roles in combating bacterial infection. 99
In summary, hepatocytes play a key role in suppressing bacterial infection by producing large amounts of innate immunity proteins (Figure 1). Because hepatocyte function is decreased in diseased livers, especially in liver cirrhosis, the production of innate immunity proteins is blunted, which likely contributes to an increased risk of bacterial infection in chronic liver disease. Although many patients with cirrhosis have elevated circulating innate immunity proteins due to high basal levels of pro-inflammatory cytokines that upregulate these proteins, these patients respond poorly to the further elevation of innate immunity proteins post bacterial infection and are more susceptible to bacterial infection. 100,101 Future studies to further understand how the liver responds to bacterial infection may help in the design of better strategies for the treatment of bacterial infections.
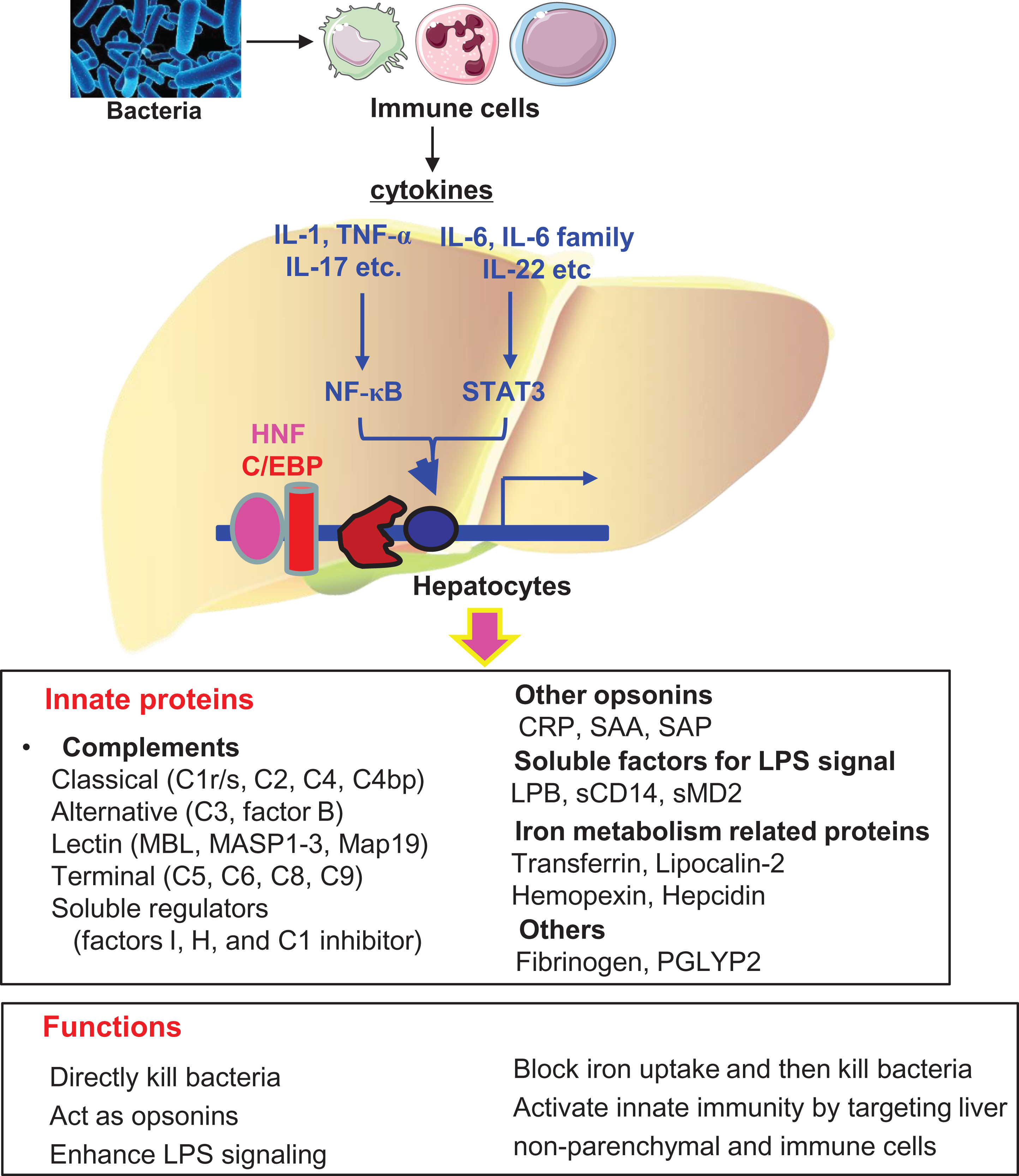
Liver immune response to bacterial infection: After bacteria enter the body, immune cells recognize them and become activated to produce many cytokines, which then stimulate hepatocytes to produce a wide variety of innate immunity proteins, including (1) bactericidal proteins that directly kill bacteria, (2) opsonins that assist in the phagocytosis of foreign bacteria, (3) several important components of the lipopolysaccharide (LPS) signaling pathways that enhance LPS signaling, (4) several key iron metabolism-related proteins that constrain bacterial growth via the inhibition of iron uptake by bacteria, and (5) the coagulation factor fibrinogen that indirectly kills bacteria by activating complements and by recruiting neutrophils to the local inflammatory site.
The Role of Cytokines in Drug-Induced Liver Injury
Idiosyncratic, drug-induced liver injury (IDILI) reactions are rare and occur in susceptible individuals at doses that are not toxic to most people. The mechanisms of IDILI are not understood, although evidence from human patients and from experimental animal models suggests that immune responses contribute to the pathogenesis. Polymorphisms in specific HLA alleles have been identified as risk factors for liver injury induced by some drugs. For example, HLA-B*57:01 has been associated with IDILI in patients taking abacavir and other antiretroviral drugs, antituberculosis drugs, and flucloxacillin. 102 -104 HLA-A*33:01 has been implicated in liver injury from terbinafine, fenofibrate, and ticlopidine. 105 Several HLA genotypes were associated with liver damage from amoxicillin-clavulanate. 106 These associations have been reviewed in Fan et al, 2017, 107 and support the hypothesis that an activated immune system underlies some IDILI reactions.
Drugs associated with IDILI in humans in general do not cause liver damage in experimental animals unless the animals are genetically modified or have been exposed to an inflammagen. The most commonly used inflammagen for these studies is LPS, although others have been used, including cytokines, products from gram-positive bacteria, and agents that activate toll-like receptor 3. 108 -113 Trovafloxacin is a drug associated with IDILI. Developed as a broad-spectrum antibiotic, it was released in 1998. By 1999, several cases of liver injury, including 14 cases of acute liver failure and 6 deaths, were linked to its use. Trovafloxacin did not produce liver damage in rats or mice; however, when coupled with exposure to a nontoxic dose of LPS, trovafloxacin caused liver injury in both species. 114,115 Interestingly, a drug in the same class as trovafloxacin but with little association with IDILI, levofloxacin, did not cause liver damage in LPS-treated animals. 116
Several elements of inflammation are critical to liver damage in these models. TNFα and IFNγ are cytokines integral to immune responses. During inflammation, they play critical roles in promoting leukocyte adhesion and altering cell proliferation. Both TNFα and IFNγ can also affect cell killing. Tumor necrosis factor α can signal through its receptors to initiate apoptotic cell death that involves diverse signaling pathways including MAPKs. 117,118 Important pathways in cell death from IFNγ include Fas/Fas ligand and STAT-1 (signal transducer and activator of transcription 1), which can signal through IFN regulatory factor 1. 119 Interaction between TNFα and IFNγ also occurs to enhance cell killing.
Both TNFα and IFNγ play an essential role in animal models of inflammation-mediated DILI. For example, exposure of mice to LPS caused an increase in the concentration of IFNγ in plasma. Although trovafloxacin given by itself did not lead to release of IFNγ into the plasma, the concentration of IFNγ was greater in LPS-treated mice given trovafloxacin than in mice treated only with LPS. 114 Likewise, the LPS-induced increase in TNFα in plasma persisted longer after treatment with trovafloxacin. Importantly, interference with TNFα or IFNγ through genetic or pharmacologic means ameliorated or abolished liver injury in mice treated with trovafloxacin and LPS. Similar results have been observed with a number of IDILI-associated drugs. 108 -113
Synergy between cytokines and drugs to cause cell death occurs in vitro as well. Many drugs associated with IDILI do not kill hepatocytes in vitro at clinically relevant concentrations; however, they become cytotoxic in the presence of nontoxic concentrations of TNFα and/or IFNγ. 120 -123 This synergy occurs in isolated murine and human hepatocytes as well as in transformed human hepatocytes (HepG2 cells). We hypothesized that these drugs induce cellular stress in hepatocytes, rendering the cells vulnerable to killing by cytokines.
Trovafloxacin was only cytotoxic to hepatocytes in vitro in the presence of TNF. 121,123 In HepG2 cells, treatment with trovafloxacin led to DNA strand breaks, inhibition of cell division, and replication stress, presumably due to inhibition of topoisomerase II. 124,125 The trovafloxacin-induced DNA damage was greater in magnitude and occurred earlier in the presence of TNFα, although TNFα alone was without effect. Trovafloxacin caused activation of ataxia telangiectasia and Rad3 related (ATR), a protein involved in cellular responses to DNA damage and replication stress. 125 Inhibition of ATR ameliorated the cytotoxicity caused by trovafloxacin/TNFα exposure. The MAPK c-Jun-N-terminal kinase (JNK) can be activated in response to replication stress. c-Jun-N-terminal kinase was activated in trovafloxacin-treated hepatocytes, and TNF coexposure led to persistent JNK activation. Inhibition of JNK reduced cytotoxicity induced by trovafloxacin in the presence of TNFα. 121 Collectively, these results suggest that trovafloxacin causes replication stress, and this cellular stress makes cells vulnerable to cell death in the presence of TNFα.
Diclofenac is a nonsteroidal, anti-inflammatory drug that has been associated with IDILI. Like trovafloxacin, diclofenac was not cytotoxic to HepG2 cells except in the presence of TNFα. 126,127 Coexposure to IFNγ further increased cytotoxicity in the presence of TNFα and diclofenac. 123 As was observed with trovafloxacin, diclofenac treatment led to activation of JNK that was greater in the presence of TNFα. Inhibition of JNK abolished cytotoxicity induced by diclofenac/TNFα or diclofenac/TNFα plus IFNγ. Extracellular-regulated kinase (ERK) was also activated by treatment with diclofenac, an effect that was mediated in part by JNK. The activation of ERK was essential for the augmentation of cytotoxicity by IFNγ, likely due to the participation of ERK in the downstream activation of STAT1 128 (Figure 2).
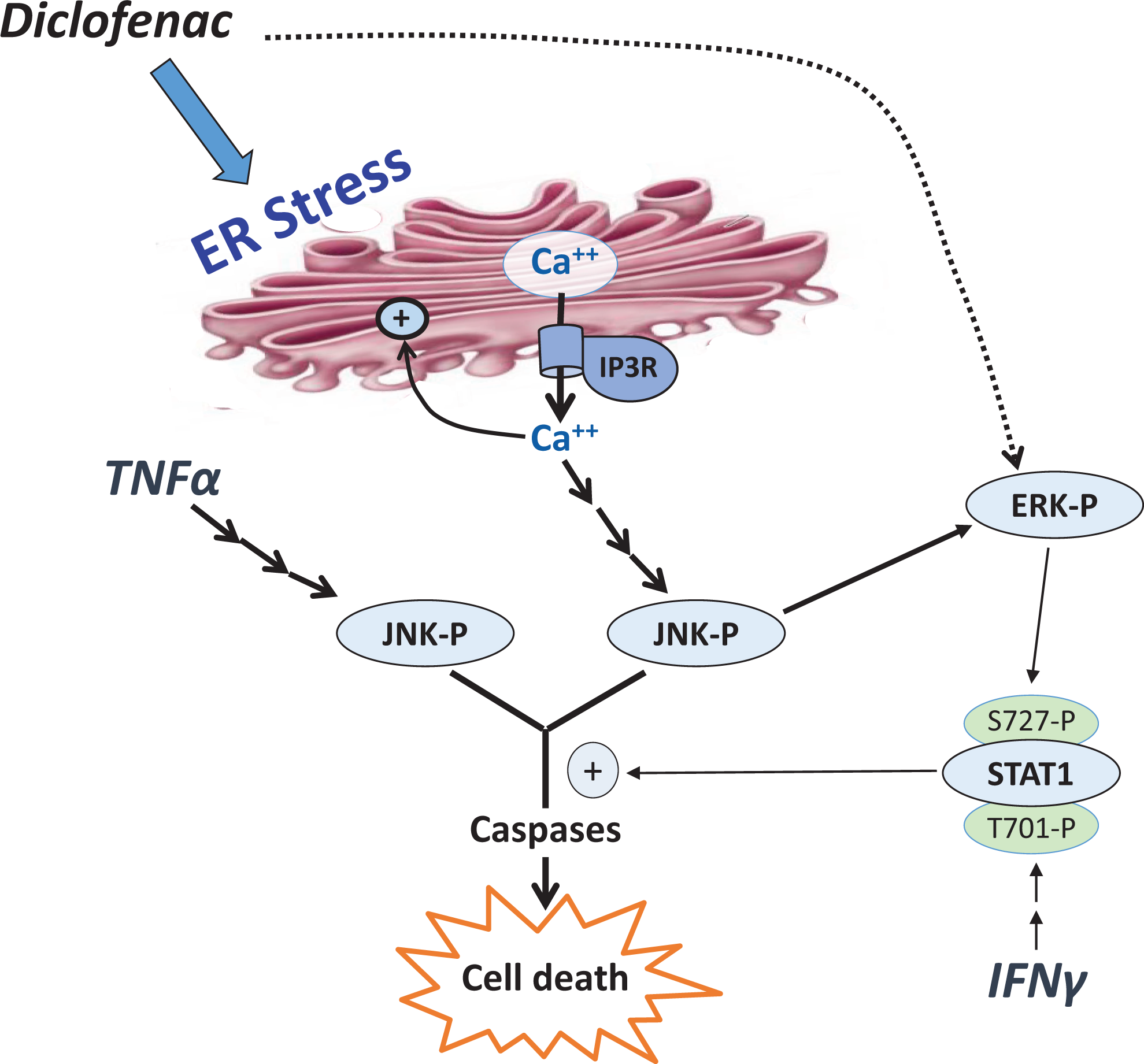
Proposed mechanism of diclofenac hepatotoxicity, with induction of endoplasmic reticulum (ER) stress in HepG2 cells, causing an increase in intracellular free calcium and rendering the cells susceptible to the cytotoxic effects of tumor necrosis factor α (TNFα) and interferon γ (IFNγ).
The activation of ERK and JNK in the presence of diclofenac was mediated by drug-induced endoplasmic reticulum (ER) stress. Endoplasmic reticulum stress is associated with activation of protein kinase R-like ER kinase and upregulation of CCAAT/-C/EBP homologous protein. These events were induced by diclofenac in HepG2 cells and were unaffected by addition of cytokines. 126,127 Endoplasmic reticulum stress is also characterized by an increase in intracellular free calcium. Calcium was increased in diclofenac-treated cells, and this effect was augmented by addition of TNFα plus IFNγ. 128 Addition of an intracellular calcium chelator or an antagonist of the IP3 receptor responsible for calcium efflux from the ER diminished cytotoxicity, ER stress, and activation of ERK and JNK in cells exposed to diclofenac and cytokines. These data suggest that diclofenac induces ER stress in HepG2 cells, causing an increase in intracellular free calcium and rendering the cells susceptible to the cytotoxic effects of TNFα and IFNγ (Figure 2).
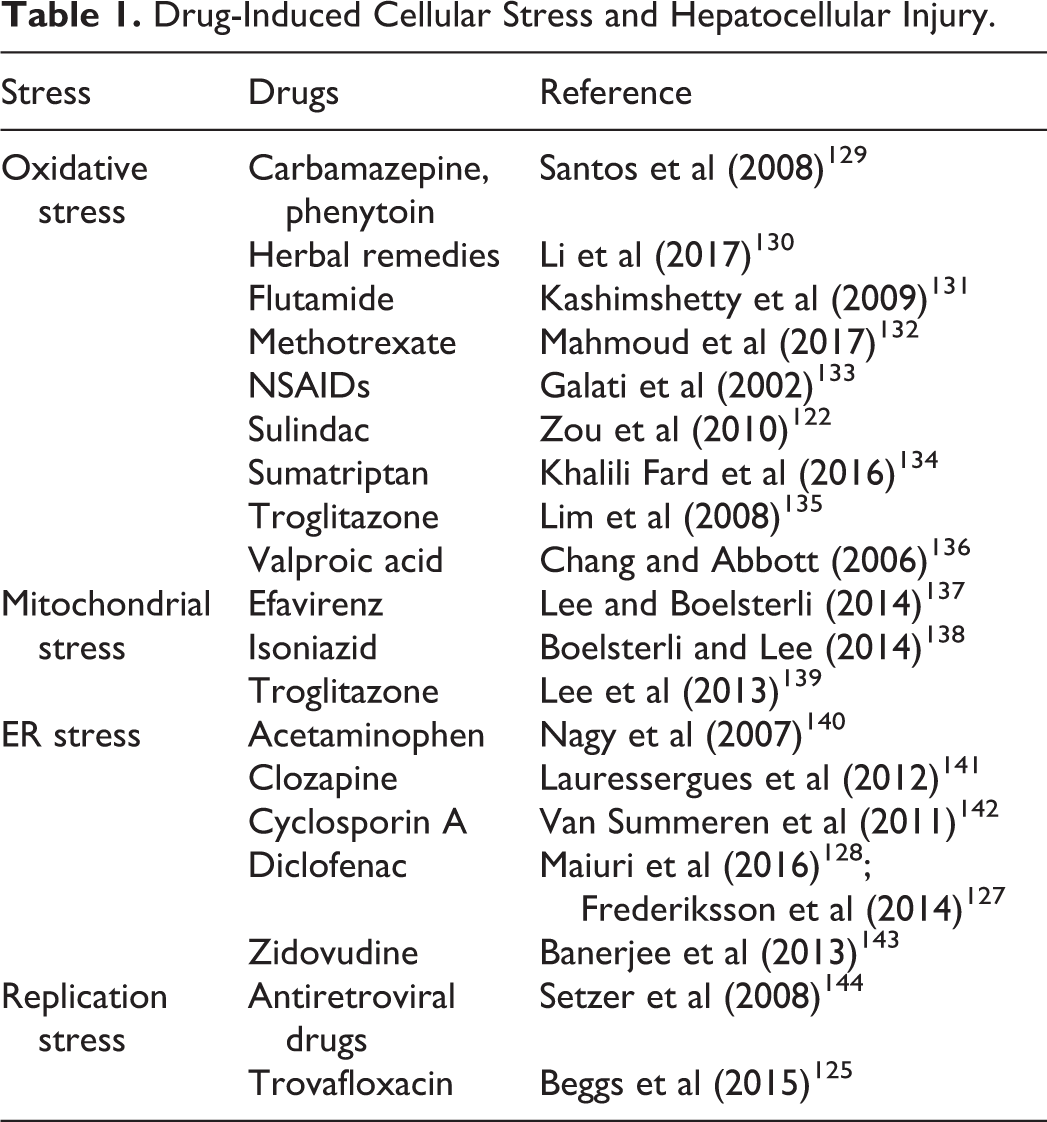
There are many examples for which hepatocellular injury has been ascribed to cellular stress induced by drugs or other agents. A partial listing is presented in Table 1. These findings provide insight into mechanisms by which drugs can interact with cytokines to lead to liver injury.
Drug-Induced Cellular Stress and Hepatocellular Injury.
Footnotes
Author Contribution
Hastings, K.L., Green, M.D., Gao, B., Ganey P.E., and Roth R.A. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and drafted the manuscript; Burleson, G.R. substantially contributed to conception or design, contributed to acquisition, analysis, or interpretation of data, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.