
Abstract
History has established that many drugs, such as the antibiotics, chemotherapies, and loop diuretics, are capable of inducing both nephrotoxicity and ototoxicity. The exact mechanisms by which cellular damage occurs remain to be fully elucidated. Monitoring the indices of renal function conducted in the Food and Drug Administration’s prescribed set of early investigational new drug (IND)–enabling studies may be the first signs of ototoxicity properties of the new drug candidate. In developing improved and efficacious new molecular entities, it is critically necessary to understand the cellular and molecular mechanisms underlying the potential ototoxic effects as early in the drug development program as possible. Elucidation of these mechanisms will facilitate the development of safe and effective clinical approaches for the prevention and amelioration of drug-induced ototoxicity prior to the first dose in man. Biomarkers for nephrotoxicity in early tier I or tier II nonclinical IND-enabling studies should raise an inquiry as to the need to conduct a full auditory function assay early in the game to clear the pipeline with a safer candidate that has a higher probability of continued therapeutic compliance once approved for distribution.
Introduction
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) has brought together drug regulatory authorities and the pharmaceutical industry to develop a strategy that delineates the scientific and technical aspects required for international registration of all new molecular entities (NMEs). Since its inception in 1990, the ICH has evolved to respond to the increasingly global face of drug development for both small-molecule and biological therapeutics. A series of industry and regulatory guidance documents have been crafted and adopted internationally that define a tiered testing approach to evaluate the relative safety of NMEs in animal subjects prior to testing them in healthy humans. 1 –3
The guidelines have established an initial set of targeted pharmacological and toxicological study designs (tier I) that generate sufficient data to define and build the next tier or stage of testing (tier II). The ICH has helped to define several factors that may identify the need for more information beyond a minimal data set established in the first phase of product development. Tiered approaches establish a regulatory scaffold upon which data are collected and analyzed and that are used to build a foundation for future staging of more complex, labor-intensive, and time-consuming study designs. Tiered testing helps to reduce the number of animals and nonclinical studies required for regulatory review prior to the first dose administration in man. Overall, the tiered system reduces the product development cycle time and overall drug development costs while maintaining a high standard of overall risk assessment in the process.
The ICH guidelines regulating safety pharmacology studies for human pharmaceuticals 2 describe a rationally based scientific approach to the design and conduct of a set of specific studies intend to establish the relative safety of a single dose administration in animals prior to dosing the new drug to humans. Under the ICH, a hierarchy of organ systems have been developed according to their importance with respect to life-supporting functions. Vital organs or systems, the functions of which are acutely critical for life (eg, cardiovascular system, respiratory system, and central nervous system [CNS]), are considered to be the most important (tier I) targets to assess in the early animal safety pharmacology studies. These tripartite critical organ safety protocols (cardiovascular system, respiratory system, and CNS) are required prior to the delivery of the first dose in man. Under the current strategy, it is presumed that other organ systems (eg, renal, sensory, hepatic, and gastrointestinal systems) can be transiently disrupted by adverse effects of a single dose without causing irreversible harm. These subordinated systems are targeted for a subsequent series of tier II testing if there are definitive adverse signs documented in the initial tripartite tier I or ancillary tier II study plans. Tiered testing approaches require the expertise of data integration, evaluation, and decision-making along each tier of the drug development program. As the primary data interpreter and single point of control of the ICH tier I, S7A, 2 S4, 1 and M3(R2) 3 study protocols, it is the responsibility of the study director (SD) to understand, predict, and suggest supportive tier II studies based on adverse findings in these initial nonclinical safety studies. At a minimum, auditory function testing is a tier II-level assay that is not commonly included in the nonclinical IND package, but it may be scientifically prudent to include the study in the next-in-line safety assessment plans.
A number of drugs that alter key parameters in standard renal function tests that help define a potential for an NME to induce renal kidney disease or acute renal toxicity are also known to selectively target auditory transduction mechanisms and sensory neuropathways. Humes 4 called out a striking association between the hair cells of the inner ear and cells of the proximal tubules of the kidney with regard to a common cellular susceptibility to iron-induced free radical formation following therapeutic use of aminoglycoside antibiotics and cellular programmed death pathways initiated by direct inhibition of ionic (sodium, potassium, and chloride) cotransport systems in the endolymphatic systems of both organ systems by therapeutic use of loop diuretics. The best known association between chronic kidney disease (CKD) and auditory threshold shifts is Alport syndrome that has genetic etiology. However, most hearing losses that occur in CKD are not genetic and have been linked to the anatomical, physiological, pathological, and pharmacological similarities between the nephron of the kidney and the microvascular system of the cochlea (stria vascularis). 5 The hearing impairments in patients with CKD are greater than in the general population, even in children, and is most severe in the high-frequency range. 6,7
Torban and Goodyer 8 reported that following a therapeutic dose of the chemotherapeutic, cisplatin, 50% of the drug is excreted in urine within 24 hours, but cells of the S3 segment of the renal proximal tubules 9 and marginal cells of the auditory stria vascularis 10 appear to accumulate the drug at concentrations greater than plasma and initiate cyclin-dependent kinase (CDK2) pathways to programmed cellular death. 11 –14 Within 48 hours of the first cisplatin dose administration, polyuria can be documented followed by a drop in glomerular filtration rates (GFRs) suggesting proximal tubule damage, but there is also distal renal tubular acidosis. The stria vascularis of the inner ear secretes the K+ into endolymph that bathes the apical poles of the hair cells with their mechanoreceptors. Selective damage to the stria vascularis of the inner ear is coincident with a reduction in endocochlear potentials and a loss of hearing within 24 hours of a single 16 mg/kg dose of cisplatin in animals.
A biomarker is a biologic specimen that may be a marker of exposure to a substance, its metabolism, or an integration of exposure and metabolism. Biomarkers may also reflect host characteristics related to toxicity and/or cell death. Because biomarkers are sometimes related to risk assessment, they are critically important in nonclinical toxicology screening. Pepe et al 15 presented a guideline for the development of biomarkers that was introduced by the National Cancer Institute in 2001 (https://epi.grants.cancer.gov/biomarkers/). While these guidelines were developed for cancer, they have been generalized by other federal regulating agencies to other risk assessment scenarios. Phase I, the preclinical exploratory stage, leads to identification of potentially useful biomarkers, and phase II, the validation stage, involves studies to determine the capacity of the biomarkers to distinguish between drug-induced and disease-induced states. Later stages of development of the biomarker focuses efforts to identify the potential predictive value of the marker and determine the overall impact (ie, risks and benefits) of screening. Predictive biomarkers represent sensitive assays that can measure subtle changes in inflammation, oxidative damage, and other pathways that suggest a subliminal process of more severe toxicity may be present, but just not yet identified.
This laboratory recently published a “method paper” focused on the integration, evaluation, and decision-making processes in determining the necessity of including a tier II electroencephalography CNS study based on the data generated during the standard tripartite, tier I, safety assessments conducted under the ICH S7A guidelines. Giving the SD a set of predictive markers that suggest the new drug has a marked propensity to induce CNS neurotoxicity early in the drug development time line is intended to save time, energy, and money for the pharmaceutical developer to “shelve” the new drug in the pipeline. We now turn our attention to the critical factors SDs should be cognizant of in making recommendations for a significant financial and temporal investment in conducting a tier II ototoxicity study based on tier I or early tier II renal function study results.
Kidney and Ear: A Toxic “Folie à Deux”
Medically, a folie à deux is characterized by a complex psychodynamic-dependent relationship between 2 individuals. We use the term here to refer to a complex relationship of both developmental and physiological pathways that play a significant role in exacerbating irreversible end-organ toxicity in both animals and man. A balance must be struck between therapeutic efficacy and targeted toxicity; not all drugs that induce toxicity will necessarily “kill a compound.” The overall value of an efficacious NME may outweigh the negative aspects of any known end-organ toxicity that will result from dose administrations. Hearing loss in a diabetic patient treated with sildenafil for erectile dysfunction, or the patient with congestive heart failure who is administered a loop diuretic, or the child treated with an aminoglycoside for an antibiotic resistant infection may seem warranted. The overall good outweighs the overall potential for toxicity. But each drug treatment is based on a doctor–patient trust founded on the available evidence to allow for informed consent. The identification of biomarkers for site-specific cellular damage discovered in the early nonclinical safety study designs during product development is beneficial to both pharmaceutical manufacturers and, eventually, the drug’s end user.
A comprehensive review by Izzedine et al 16 brought together a number of renal and auditory pathways linked to common developmental, molecular, and genetic factors. A number of the essential developmental control genes seem to be involved in the coordination of the assembly and functional control of both renal and auditory systems. Table 1 reproduces some of the common genetic cofactors and their related functions that were highlighted by Izzedine et al 16 in 2004.
Some Identified Genes and Their Functions in Ear and Kidneys.a
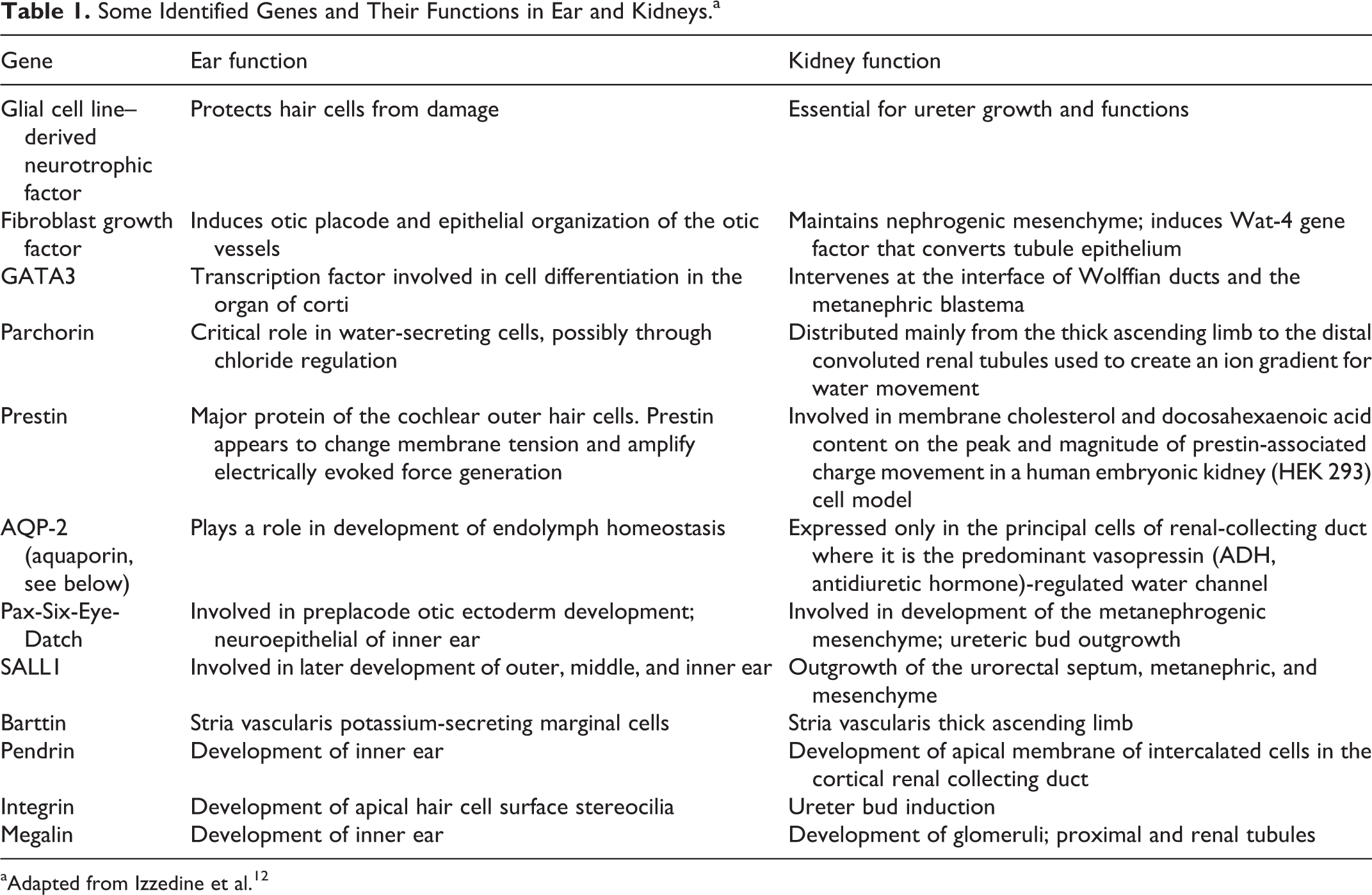
aAdapted from Izzedine et al. 12
The mammalian inner ear has an array of interconnected fluid compartments that are enclosed by highly specialized epithelial cells. The luminal fluid, endolymph, differs in composition between different parts of the inner ear. The epithelial cells enclosing the endolymph are diverse sensory inner and outer hair cells (OHCs) and stria vascularis. It is the sensory hair cells that convert (transduce) mechanical fluid wave movements into electrical signals and release neurotransmitters to activate other sensory neurons to the auditory nerve pathway. The stria vascularis secretes potassium ions into the endolymph and generates the endocochlear voltage potential, which drives the sensory signal to the cortex of the brain. Sensory transduction of the sound pressure waves in fluid within the cochlea provides the basis for hearing and, in the vestibular labyrinth, transduces the mechanical stimuli associated with head position and head movement. The vestibular neural output provides input to the cerebellar system that controls balance, posture, and eye movements. Both auditory and vestibular transduction critically depend on cycling of potassium between endolymph and perilymph of cochlea and semicircular canals, respectively.
The similarity between epithelial transport of potassium in the inner ear and the cellular transport of metabolic ions in the kidney was suggested when the loop diuretic, furosemide (Lasix), was linked to hearing loss.
17,18
While the function and purpose of the inner ear and kidney are different, most of the genes encoding the epithelial transporters or channels in the inner ear are similarly expressed and sensitive to pharmacological interventions in renal tubular transport. Water transport is driven by osmotic gradients that are established by the transport, metabolism, or catabolism of ions in solution. Water moves through membranes freely with the exception of thick apical membrane in the ascending limb and cortical collecting ducts of the kidney in the absence of the antidiuretic hormone, vasopressin. The permeability of water through cell membranes is critically dependent on 13 known aquaporin channels. Many of the channels, carriers, and active pumps that transport fluid in the inner ear are expressed in the kidneys. For example: parchorin, a chloride intracellular channel family member, is distributed in the thick ascending limb of the kidney to the distal convoluted tubules and is found in the cochlea and semicircular canals of the ear; and prestin is a recently cloned gene protein that fits the pattern of OHC development in the inner ear and is expressed in normally nonmotile kidney cells.
16
Prestin is involved in cellular amplification in the cochlea
19
and has been proposed to be a biochemical predictive marker for early detection of acquired sensorineuronal hearing loss.
20,21
Nishizawa et al 22 cloned a new protein, parchorin, that demonstrated homology to the chloride intracellular channel family. Most of the chloride channels were considered to be intracellular vesicles. Parchorin is a soluble cytosolic phosphoprotein that can translocate to the plasma membrane under stimulation. When acid-secreting parietal cells are stimulated, tubule vesicles containing H+-K+-ATPase fuse with the apical membrane, and both K+ and Cl− permeability in the apical membrane are increased. As described by Mizukawa et al, 23 parchorin moves from the cytosol to the apical membrane and accelerates the rate of chloride efflux. Parchorin is preferentially expressed in tissues related to water movement, that is, choroid plexus, kidney, and cochlea. Parchorin plays an important role in the regulated movement of body fluid through the active chloride transport. 24
Parchorin has been found mainly in the thick ascending limb of the loop of the Henle and the distal convoluted tubules of the kidney. Various ion transporters and channels working in the kidney comprise the complex system for homeostatic control of urine concentration. Nonclinical studies have shown that chloride channel (CLC)-K2 is expressed more broadly in the distal nephron; expression levels are highest along the thick ascending limb of the loop of Henle and distal convoluted tubule. Expression of CLC-K1 (CLC-KA in humans) is upregulated by dehydration and downregulated by the diuretic furosemide (Lasix), whereas expression of CLC-K2 (CLC-KB in humans) is upregulated by furosemide and downregulated by high salt levels. CLC-KA is important for maintenance of the corticomedullary osmotic gradient and the kidney’s capacity to concentrate urine. Furosemide’s action on cochlear hair cells has also been linked to sensorineuronal loss in the cochlea through similar actions on the chloride flux.
Mizukawa et al 23 strongly suggest that parchorin involves physiological chloride absorption, since the corresponding regions of the kidney in which it has been found have a very high level of chloride absorption in the kidney. The inner ear has a highly controlled water movement system to ensure hearing and body balance. As described earlier, chloride transport channels are present in OHCs. Similar to the kidneys, parchorin is highly expressed in epithelial cells near the organ of Corti in the cochlea and plays a critical role in chloride ion transport in the cells that create an ion gradient for water movement in the endolymph.
Can Scheduled Early IND-Enabling Clinical Pathology Parameters Predict the Need for Ototoxicity Screening?
Before proposing any predictive index for tier II ototoxicity studies from early IND-enabling clinical pathology parameters (urine and hematology), we would like to set the stage with all apparent “bells and whistles” admonishing and acknowledging the pitfalls that may need to be addressed between the SD and the pharmaceutical sponsor in the decision to move forward with expensive ancillary tier II ototoxicity studies. Here, we propose that the relationship between renal and auditory functions provides a sufficient and valid foundation to base predictions for early identification of cytotoxicity in the organ system that is rarely analyzed in standard pathology tissue collection schemes in the pre-IND study protocols—the cochlea. With these facts in mind, we propose the following.
Clinical pathology data from standard protocol that required collections of urine and blood samples generated in early IND-enabling toxicology studies are voluminous, especially in rodent studies where more than 5,000 clinical chemistry values may be generated. 25 The majority of these data are obtained using automated biochemical analyzers with Good Laboratory Practice Guidelines (GLPs)-compliant validated hardware and software systems. According to Evans, 25 generating analytical results from early toxicology studies is defined by the protocol or institutional standard operating procedures (SOPs), and using standardized laboratory equipment and data storage software, it is possible to select “reflex” testing in which certain identified or “tagged” parameters can automatically initiate follow-up testing when certain threshold limits are obtained on the first series of analysis requests. Once a sample is re-assayed, the SD must decide whether the first or second result is correct. Biological variations occur within and between animals on the same study. The animals in the standard IND-enabling toxicology study are purposely bred with known genetic backgrounds and matured in carefully controlled environments with special references to pathogens and usually with free access to a well-controlled and certified diet.
In studies conducted under the GLPs of the US Food and Drug Administration (FDA), it is the responsibility of the SD to identify aberrant sample values that may have been affected by poor sample aspiration or collection techniques leading to a “short” sample (insufficient sample volume required for the test, small blood clots, or analyzer flags for insufficient samples). Some data may look suspect, for example, very high potassium concentration with low calcium concentrations that may suggest the incorrect use of anticoagulants. The SD must decide whether to repeat the analysis for confirmation; however, in many small animal toxicology studies, the allowable blood sampling volumes are restricted and there may not be enough of the original aliquot left to re-assay. If a parameter value is outside the flagged reference ranges preselected on the analyzer, or by SOP, or historical control values, the SD must decide the prudence of repeating just one sample, especially when all other surrounding sample values appear to be within their ranges. Despite the management of many current Contract Research Organization’s attempt to direct SDs away from using the terminology, the term “normal range” is often used instead of “reference range.” According to the International Federation of Clinical Chemistry, 26 “reference ranges” are determined from samples collected from healthy experimentally and drug-naive animals. 27 Reference ranges are generally based on data published in peer-reviewed scientific journals or determined within the institution and qualified by: (1) strain, age, and gender of animal; (2) route of administration; (3) source of the animal (breeder supply); (4) sample collection method; (5) analytical equipment used and methodologies of sample processing; (5) exclusion criteria of the equipment sensitivity; and (6) the number of animals used for comparative range and the specific calendar periods of data collections.
Using a discretionary “bootstrap” set point (group mean ± 3 standard deviation values) as the threshold for conducting a time-consuming ototoxicity study (cytohistopathology, cytocochleograms, and some measure of shifts in auditory sensitivity (Auditory Brainstem Response [ABR] or Distortion Product Otoacoustic Emissions [DPOE])) should be tempered with the knowledge that many clinical pathology parameters do not follow a normal Gaussian distribution. Parametric testing that is generally used for predictive discriminations generally assumes the parameters are “normally distributed,” the distributions are similar in all groups being compared (ie, generational, breeder, age differences), and there are no extreme values. The SD that relies heavily on (renal function) reference ranges to identify or predict toxicology in subordinate or ancillary (auditory) organ systems and is without cognizance of the influence of all these confounding factors is not acting in the best interest of the sponsor, the public health, or the FDA. Many colleagues have drawn attention to the variations observed in distributions for the same analyte in control groups between differing studies of rats in the same laboratory. 28 –33 With all these caveats in mind, we propose the following decision tree or logic for early identification and prediction of ototoxicity liability of a new drug candidate in the standard ICH nonclinical safety assessment pathway.
Predicting Ototoxicity Liability Based on Clinical Renal Pathology Parameters
Early IND-enabling drug studies harmonized under the International Council’s Harmonized Guidelines 3 established a process of development of a new pharmaceutical product using a stepwise process involving an evaluation of both animal and human efficacy and safety information. The goals of the nonclinical safety evaluation generally include characterization of toxic effects with respect to target organs, such as the kidney, liver, heart, and so on. The focus of early IND-enabling animal studies is to determine a dose-dependent relationship with drug exposures, and, when appropriate, potential reversibility of those effects during postdose washout periods. This information is used to estimate an initial safe starting dose and dose range for the human trials and to identify parameters for clinical monitoring for potential adverse effects. The nonclinical safety studies are designed to characterize potential adverse effects that might occur under the conditions of the clinical trial to be supported. Historically, acute toxicity information has been obtained from single-dose toxicity studies in 2 mammalian species using both the clinical and a parenteral route of administration. Under the ICH guidelines, lethality should not be an intended end point in studies assessing acute toxicity, but acute toxicity induction is the purpose of these initial study protocols.
The tiered system of toxicity screening adopted by drug approval agencies has adopted a hierarchy of organ systems according to their importance with respect to life-supporting functions. Vital organs or systems, the functions of which are acutely critical for life, such as the cardiovascular system, respiratory system, and CNS, are considered to be the most important ones to assess safety in pharmacology studies. Other organ systems, such as the renal or gastrointestinal system, can be transiently disrupted by adverse pharmacodynamic effects without causing irreversible harm. These biological systems are of less immediate investigative concerns for the IND-enabling study plan for the new molecule. However, effects of the test article on renal function parameters are still required to be assessed.
The kidney is one of the main toxic targets for screening of all new drug candidates. Urinary and blood sampling are included in the early toxicology study protocols, but accurate detection of nephrotoxicity remains difficult in these early stages of drug development. 34 Urinalysis and urine chemistry testing of urine volume, specific gravity, protein, glucose, the presence of ketones, urobilinogen, bilirubin, occult blood, and sediment and quantitative electrolyte concentrations of sodium, potassium, and chloride are included in the standard renal function profile of routine toxicology study protocols, but these are rarely definitively meaningful or useful information for a diagnosis of renal impairment. 35 Serum creatine and blood urea nitrogen have been used as biomarkers for the detection of drug-induced acute kidney injury (AKI); however, these are insensitive or nonspecific to diagnose AKI because both parameters are easily influenced by many other physiological functions, for example, gastrointestinal bleeding, without negative impact on the kidneys. 36 –39 Traditionally, AKI has been indicated by a diminished urine output coincident with an elevated serum creatinine concentration. For example, Waring and Moonie 40 proposed a diagnostic threshold of an elevated serum creatinine concentration of ≥150 µmol/L (1.69 mg/dL) or ≥50% increase from baseline for a diagnosis of AKI, but the rate of increase in serum concentrations after AKI is quite variable, and the thresholds may not be detected until 2 or more days from chemical insult.
There are several urinary biomarkers that appear in peer-reviewed scientific journals that are recommended to be used as noninvasive indicators of AKI, 24,41 including N-acetyl-β-D-glucosaminidase (NAG 42 ), kidney injury molecule-1 (KIM-1 43 –46 ), neutrophil gelatinase-associated lipocalin (NGAL 47 –49 ), and vanin-1. 40,50 Kidney injury molecule-1 is a transmembrane protein that is not normally expressed in urine but is highly expressed in proximal tubule epithelial cells following toxic injury. 43,45,46 Human urinary KIM-1 has been reported to increase following peaks of urinary NAG and NGAL following AKI and cardiac surgery. 51 Identification of urinary NGAL precedes that of other biomarkers such as NAG and β2-microglobulin (B2M) in human kidney injury, but it is also elevated under other pathological conditions such as pneumonia 52 and inflammatory bowel disease. 53 Therefore, at the early stages of the rodent nonclinical toxicology studies, these biomarkers may not provide a robust predictive index for a definitive case of AKI and, in turn, not be predictive of ototoxicity, as well. Urinary concentrations of the epithelial glycosylphosphatidylinositol-anchored pantetheinase, vanin-1, 54,55 are expressed in the metabolic process associated with oxidative stress. 56 Vanin-1 has been reported to increase prior to detectable increases in serum creatinine, NAG, NGAL, B2M, and KIM-1 in both rats 50 and nonhuman primates 41 following chemically induced renal tubular injury (ie, cisplatin and gentamicin).
The European Medicine Agency (EMA) and the US FDA have “qualified” 7 urinary/kidney/renal function biomarkers: (1) total protein (TP), (2) albumin (ALB), (3) B2M, (4) clusterin (CLU), (5) cystatin C (Cys C), (6) KIM-1, and (7) trefoil factor 3 (TFF3).
The preferred species for early toxicology studies remains the standard purpose-bred laboratory rat. Expected urine output from rats over a standard collection period of 16 to 24 hours lends suspect to the accuracy and predictability of any set of parameters predictive of drug-induced kidney injury. However, according to Uchino et al, 36 these 7 biomarkers are considered “acceptable” in the context of nonclinical drug development programs for the detection of drug-induced acute toxicity in renal tubule and glomeruli in rats. The EMA and FDA have accepted KIM-1, ALB, CLU, and TFF3 changes as potential markers for drug-induced acute renal tubular alterations, while TP, B2M, and Cys C changes may suggest acute glomerular alterations/damage and/or impairment of kidney tubular or impairment of renal tubular absorption in rats (EMA 57 –60 ; FDA 61,62 ). More recently, the EMA and FDA have also “qualified” renal papillary antigen 1 for detecting renal tubular alterations, particularly in the collecting ducts in rats (EMA 58 ; FDA 62 ). Additionally, increases in NGAL (lipcalin-2) and osteopontin have been linked to proximal tubular degeneration/necrosis in rats. 59,60,63,64 Since there are common threads in development, genetic cofactors, and anatomy between renal tubule damage and cochlear damage, the most likely set of biomarkers to predict ototoxicity in early IND-enabling studies may be the “qualified” biomarkers that covary with proximal and distal kidney tubule damage already “qualified” by drug approval agencies. Table 2 shows the 7 biomarkers qualified by both EMA and FDA which suggest AKI through proximal or distal renal tubule damage that produce both nephrotoxicity and ototoxicity confirmed by postmortem histopathology (cisplatin and aminoglycosides).
EMA- and FDA-Qualified Rodent Renal Function Biomarkers Induced by Drug Treatments That Induce Both Nephrotoxicity and Ototoxicity.
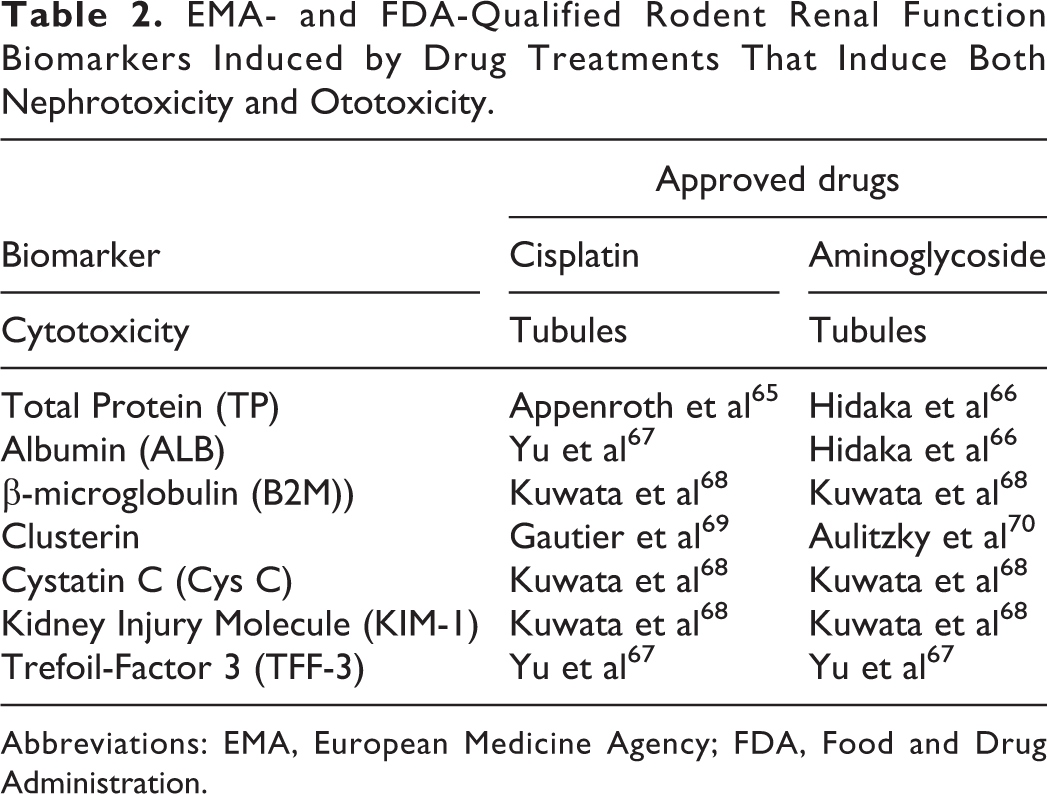
Abbreviations: EMA, European Medicine Agency; FDA, Food and Drug Administration.
The correlation between nephrotoxicity and ototoxicity is not perfect (<1.0). The cytotoxicity pathways are not identical, and systemic exposure of a proposed toxic agent would be predicted to be both dose- and time dependent. The first principle of pharmacology and toxicology is “the dose makes the poison” (https://chemm.nlm.nih.gov/toxprinciples.htm#principle3). The clinical effects of any drug on either kidney or ear are based on the amount of exposure. The dose is the total amount of chemical absorbed during an exposure. Dose depends on the concentration of the chemical and duration (contact time) of the exposure. The sheer volume, concentration, and exposure duration should predict that renal function would show greater susceptibility compared to inner ear cochlear hair cells. But this is not always the case. The initial use of cisplatin in patients with cancer was associated with 70% of the patients experiencing renal failure and 66% of the patients having hearing loss. 9 According to Torban and Goodyer, 8 following a therapeutic dose of cisplatin, approximately 50% of the drug is excreted through the kidneys within 24 hours. However, cisplatin is specifically taken up by cells of the S1 segment of the renal proximal tubules and marginal cells of the stria vascularis, and it accumulates in these cells to levels many times greater than plasma concentrations. 10 Within 48 hours of dose administration, cisplatin patients experience polyuria and a decrease in GFR. In the ear, animal studies show selective cell damage and hearing loss within 24 hours of a high dose.
For comparisons, 2 other major ototoxic agents, furosemide (Lasix) and sildenafil (Viagra), do not induce AKI. Neither drug has yet been reported to induce any of the 7 biomarkers during dose administration in nonclinical IND-enabling study designs. Drug-induced ototoxicity is dependent on the dose and duration of exposure. Hearing loss can worsen if treatment is continued and sometimes even after the drug is withdrawn. 71 The cumulative dose and duration of aminoglycoside and cisplatin therapies are more important than peak serum concentrations. Auditory toxicity has been reported to occur after only 5 days of a relative therapeutic dose of aminoglycoside (gentamicin), but significant auditory threshold shifts have been reported after a single dose of furosemide. 72
Furosemide (ie, Lasix) can induce auditory threshold shifts following a single dose administration in the rat with no ancillary damage to the kidneys. Furosemide has been reported to modify GFRs, 73 but no deleterious effects on proximal or distal tubule functions. 74,75 The majority of administered furosemide (>97%) is bound to plasma or ALB in humans and other laboratory animals (ie, rabbit), but it is highly bound to rat tissues. 76 Furosemide can increase urine output to >100 mL/h, an indicator of a GFR of >20 mL/min. This increased urinary output almost certainly excludes progression to AKI. 77 These findings suggest differential mechanisms of toxicity following drugs that induced AKI (eg, cisplatin and aminoglycosides) and the other 2 ototoxic drugs, furosemide and sildenafil. 78
Animal experiments have shown that loop diuretics (furosemide) act directly on the stria vascularis, producing edema of these tissues and a temporary loss of function, resulting in a decrease of the endocochlear potential. The inner hair cells (IHCs) and OHCs require the presence of a normal endocochlear potential for transduction. Furosemide has been reported to induce changes in the oxygen tension in the perilymphatic fluid and directly on the stria vascularis to induce ischemia. 79 Consistent with this ischemia, there is a direct action on OHC motility as well (chinchilla 80 and guinea pigs 81 ). The implication of impaired IHC and OHC function suggests that furosemide cochlear damage may be mediated by different mechanisms than cisplatin and aminoglycosides. 82 Furosemide-induced ototoxicity has also been linked to oxidative stress pathways that induce apoptotic cascades and cell death. 83 Glutathione derivatives serve as a primary antioxidant and block these oxidative stress pathways. Furosemide is a potent inhibitor of glutathione S-aryltransferase, glutathione S-alkyltransferase, and glutathione S-epoxide transferase. 84 Other diuretics giving the greatest inhibition of these reactions are chemically related to ethacrynic acid. Salicylic acid (aspirin) reduces the impact of ethacrynic acid drugs on glutathione-related antioxidative mechanisms and serves as a hydroxyl radical scavenger. 85 Sensorineuronal hearing loss can be induced by injecting salicylic acid, which may suggest another neurotoxic pathway other than through direct reactive oxygen species (ROS) that involve glutathione protection in producing the hearing loss. 86 Salicylic acid binds to the motor protein, prestin, in the OHCs, 87 –90 which may suggest that furosemide-induced hearing loss is not mediated by ROS as implicated with renal damage induced by cisplatin and aminoglycosides. 90 While renal function biomarkers may not be affected by furosemide treatments that induce direct ototoxicity, one of the biomarkers, CLU, has been reported to be affected by both salicylic acid and furosemide that does induce ototoxicity.
The CLU is an extracellular chaperone protein that is implicated in diverse physiological and pathophysiological effects in both kidney and auditory cellular processes. Expression of CLU is upregulated in response to oxidative cellular stress and under other conditions such as neurodegeneration and cancer. The CLU primarily functions as a chaperone that exerts cytoprotective effects by removing cellular debris and misfolded proteins and also acts as a signaling molecule that regulates prosurvival pathways (antiapoptosis: BAX, BCl2, and so on). Gregory et al 91 have suggested that elevation in chaperone levels can protect against neurotoxicity resulting from the effects of pathological protein misfolding in cell culture and in transgenic animal models. Most chaperones are localized in the intracellular compartment, although some are secreted into the extracellular environment. The CLU is present in both plasma and cerebrospinal fluid. It is a cytoprotective chaperone whose expression level is increased in response to a diverse range of stresses including heat, proapoptotic insults, oxidative stress, ionizing radiation, and proteotoxicity. Considering its cytoprotective effects, CLU has been suggested to play a role in mediating cellular defense mechanisms against hearing loss due to local cellular oxidative damage. Lee et al 73 using a quantitative real-time polymerase chain reaction analysis recently showed that CLU messenger RNA (mRNA) levels in the inner ear are increased during embryogenesis and remain constantly expressed in the adult. In the mature inner ear, CLU expression has been observed in Deiters’ cells, pillar cells of the organ of Corti, outer sulcus, and in basal cells of the stria vascularis in the cochlea. 73 These specific patterns of expression suggest a possible role for CLU within the inner ear in maintaining proper hearing functions. With this protective mechanism of ototoxicity, it may suggest that presence of CLU proteins in urine or plasma may predict the early processes of drug-induced ototoxicity.
Prestin is a motor protein of cochlear OHCs. It was identified 18 years ago. Prestin-based OHC motility is responsible for the high sensitivity and frequency selectivity seen in the mammalian cochlea. Prestin is the fifth member of an 11-member membrane transporter superfamily of SLC26A proteins. 92 Unlike its paralogs (ie, CLU), which are capable of transporting anions across the cell membrane, prestin primarily functions as an OHC (cilia) motor protein with unique capability of performing direct and reciprocal electromechanical conversion on a microsecond time scale. 93 Prestin is a voltage-sensitive transmembrane protein containing several negatively charged residues on both intra- and extracellular surfaces of the OHC. It is the OHCs that are most susceptible to damage and are affected gradually from the base of the cochlea to its apex, producing a pattern of hearing loss that begins in high frequencies and progresses to lower frequencies. The OHCs are “effector cells” that enhance tuning and sensitivity of the cochlea. In comparison, the IHCs are the “sensory mechanoreceptor” responsible for encoding the traveling fluid wave of the basilar membrane. These functions appear to be directly related to electromotile properties of the OHCs. The actual length of OHCs changes as a function of the membrane polarization. It is the membrane protein, prestin, that acts as the motor responsible for this endocochlear voltage changes in OHCs. Targeted deletion of prestin results in loss of OHC motility in vitro and a 40 to 60 dB loss of cochlear sensitivity in vivo, without disruption of mechanoelectrical transduction in the OHCs. 94 Increases in serum prestin concentrations following cisplatin and amikacin have been associated with the greatest increases in cochlear cell damage scores. 16,95 Naples et al 96 recently reported that changes in serum prestin levels were detectable prior to shifts in ABR thresholds in guinea pigs treated with cisplatin. Parham 21 and Parham and Dyhrfjeld-Johnsen 97 demonstrated that noise-induced cochlear injury leads to an initial increase in prestin serum concentrations with subsequent decreases in prestin concentrations as hearing impairment became permanent. Interestingly, Wang et al 98 reported that aminoglycosides (streptomycin and gentamicin) have no direct effect on OHC somatic motility. These results suggest that serum prestin concentration may be a promising early indicator of cochlear damage that is produced by mechanisms that are not present in aminoglycoside-induced ototoxicity.
Parham 21 has suggested that the inherent homeostatic cellular processes of the cochlear OHCs, baseline detection of prestin, is expected at low levels. Within hours of hair cell damage from exposure to noise or ototoxins, prestin levels are expected to rise above baseline. But approximately 3 to 5 days after exposure to the drug, insult prestin levels will peak and then decline. Because of increased expression in the surviving OHCs, Parham 21 predicts that prestin levels are expected to remain above baseline for at least 1 month after exposure.
The presence of CLU or prestin in test article–induced clinical pathology parameters found in early rodent IND-enabling toxicology studies may suggest a similar expression in the microcirculation of the inner ear. Urinary or blood prestin (serum) or CLU (plasma) changes may not be a necessary condition for ototoxicity, but its presence may be a condition sufficient for predicting cochlear damage to be forthcoming. We propose that any new drug that has a serum prestin or CLU-positive renal function test in early IND-enabling toxicology studies should at least initiate a conversation regarding further tier II testing of auditory function.
Sildenafil (Viagra), avanafil (Stendra), tadalafil (Cialis), and vardenafil (Levitra) are a family of phosphodiesterase (PDE) 5 inhibitors (PDEIs) used for the treatment of sexual dysfunction. 99 While it is nitric oxide (NO) that is involved in the increased blood flow associated with penile erection, it is the PDE that is involved in the outflow of blood. To retain an erection, NO-related mechanisms must outweigh the PDE mechanisms. Therefore, PDEIs are administered. While all 4 drugs are PDE-5 inhibitors, there are pharmacological differences between them. For example, vardenafil is more potent and more selective than sildenafil at inhibiting PDE-5, and vardenafil does not inhibit PDE-6 which is involved in visual color perception. Altered color vision is a rare side effect that sometimes occurs with sildenafil. A reported adverse effect of PDEI is sudden sensorineural hearing loss (SSHL; https://wayback.archive-it.org/7993/20170406044840/ https://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm106525.htm). According to the National Institute on Deafness and Other Communication Disorders 100 (https://www.nidcd.nih.gov/health/sudden-deafness), the drug-induced SSHL is characterized by a sudden or rapid loss of hearing and occurs in approximately 4,000 cases per year with an unknown etiology. 101 Liu et al 102 recently reported that the use of PDE-5 inhibitors is associated with a small but significantly increased risk of SSHL. Compared to nonuse, Liu et al 102 reported an adjusted hazard ratio of 1.25 (1.01-1.55) for current use with a risk difference of 1.97 (1.12-2.82) per 10,000 person-years. For recent use, the adjusted hazard ratio was reported to be 1.60 (1.33-1.94) and risk difference was 3.19 (2.24-4.14). In the most recent review of the literature, Steyger et al 103 concluded that while there are some small case series reporting SSHL by patients prescribed PDE-5 inhibitors for erectile dysfunction loss, the etiology and pathophysiology remain uncertain.
The PDE-5 inhibitors block the breakdown of 30,50-cyclic guanosine monophosphate, a signaling molecule generated by the NO-dependent soluble guanylate cyclase. In turn, cyclic guanosine monophosphate activates cyclic guanosine monophosphate–dependent protein kinase G, which leads to vascular tone modulation. 104 Several G protein–coupled receptors are present in the cochlea. Sheth et al 105 have described a distribution of adenosine receptors (A1 ARs) in the stria vascularis, spiral ganglion cells, and organ of Corti. 106 In the organ of Corti, the largest distribution of these ARs is in IHCs, Deiters’ cells, and lower levels of the OHCs. Localized application of adenosine has been reported to increase the activities of the major antioxidative enzymes glutathione and superoxide dismutase. 107 One of the active ROS in the cochlea is xanthine oxidase. This enzyme converts hypoxanthine, a metabolite derived from the breakdown of adenosine by adenosine deaminase, to uric acid. The cochlea contains a number of other endogenous antioxidant enzymes and antioxidants, such as heat shock proteins, KIM-1, antiapoptotic protein transcription factors, which may protect the cells from the influence of PDE-5 inhibitors.
The 4 approved PDE-5 inhibitors have different affinities for PDE-5 and different affinities for the 11 available PDE isoenzymes. Yafi et al 104 suggest that these differential affinities lead to divergent side effect profiles. For example, sildenafil and vardenafil cross-react with PDE-6, which is predominantly located in the retina, and tadalafil cross-reacts with PDE-11. As stated previously, PDEIs are administered for the treatment of erectile dysfunction, which covaries with other systemic disease processes such as diabetes, cardiac disease, and normal aging. Skeith et al 108 reported synergistic adverse relationships for ototoxicity between sildenafil and furosemide and CYP3A4 inhibitors such as diltiazem, especially in cases of renal failure and high peak drug levels. Diltiazem is a calcium channel blocker used to treat high blood pressure and to control angina (chest pain). This interaction may suggest a common cellular ROS pathway is involved in all drug-induced ototoxicity including cisplatin, aminoglycosides, furosemide, and sildenafil. Except the actual ROS pathway, the mechanisms involved in ototoxicity by PDEIs remain elusive and intimately tied to other disease states and normal aging processes that play a role in hearing loss.
Auditory testing may be required under a number of premarketing scenarios: (1) if there is clear evidence of auditory dysfunction in nonclinical toxicology studies (eg, functional observational battery, detailed clinical observations, or postmortem examinations), (2) if there are adverse events reported in the early-phase clinical trials, (3) if the pharmacological class has a known risk potential for ototoxicity (aminoglycosides, platinum-based chemotherapeutics), or (4) the intended route of administration of the drug is into the ear canal. There are 2 regulatory guidance documents that may help to characterize the pharmacological impact of the new drug on auditory functions. The Center for Drug Evaluation and Research has concluded that auditory function must be assessed for all drugs that are administered in or through the ear canal. 109 Based on this requirement, ototoxicity evaluation must be used to characterize all new drugs that are functionally or structurally similar to a known class of ototoxic-inducing agent or if there is demonstrative evidence in nonclinical or clinical trials that suggest or predict impairments in auditory function. Further discussions of second-tier safety assessments of the auditory system should be discussed in the follow-up studies delineated in the early IND-enabling animal studies that show renal toxicity or in the core battery section of the Safety Pharmacology Studies for Human Pharmaceuticals of the ICH S7A guidance document adopted by the FDA in 2001. 2
Recommended Clinical Pathology Parameters That May Be the Best Predictors of Ototoxicity in Early IND-Enabling Studies
The kidney is the second most common target in drug-induced organ injury in nonclinical drug development. 110 Many drug candidates are excreted through the urine, the kidney has an extensive blood supply, and the glomeruli have a relatively large surface area available for drug exposure. The kidney’s functional ability to concentrate solutes and drug entities also enhances the susceptibility to drug-induced injury. Drug-induced AKI in animal studies is of great importance, since, in humans, acute nephrotoxicity usually presents minimal early function changes and the progression to chronic kidney failure is silent. 110 One of the most common sites of drug-induced kidney injury is the proximal tubules, since most blood flow is delivered to this area of the kidney, which functions to concentrate and reabsorb solutes. The biomarkers for proximal and distal tubular damage may be an early biological indicator of a potential for ototoxicity based on factors described earlier. 111,112 The drug approval regulatory agencies seek for the pharmaceutical industry to police itself—they want mechanistic information on test compounds to aid in their own risk assessment reviews. This often encourages additional end points not specifically required under the ICH or FDA harmonized guidelines. In response, many SDs are adding end points to the standard multidose or single dose early IND-enabling study packages. Such end points are being added on a case-by-case basis and will continue to grow in necessity. The earliest detection of AKI that may be associated with or predictive of subsequent ototoxicity will provide an opportunity to minimize the risks of severe hearing loss during phase II or phase III clinical trials. However, since not all known ototoxic drugs produce changes in FDA’s “qualified” renal biomarkers, other nonrenal parameters may be required. Experiments in rats and other animals used in IND-enabling studies rely on histopathology as the “gold standard” for nephrotoxicity. Since tissue harvesting, processing, and histology of auditory sensory organ systems (middle and inner ear organs) are not part of standard tissue/organ lists in IND-enabling animal study protocols, it is imperative that the SD remain vigilant in identifying when more detailed tier II auditory toxicity screening studies should be scheduled.
Rybak et al 113 have highlighted that the inner ear itself expresses a unique isoform of Nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), NOX3. This enzyme may be the primary source of ROS generation in the cochlea. Rybak et al 113 reported that the knockdown of NOX3 by pretreatment with small-interfering RNA (siRNA) prevented ototoxicity, as demonstrated by preservation of hearing thresholds and inner ear sensory cells. 113 Trans-tympanic NOX3 siRNA reduced the expression of NOX3 and biomarkers of cochlear damage, including transient receptor vanilloid 1 channel 114 and KIM-1 in cochlear tissues themselves. 46,115 As a marker of oxidative stress in the cochlea, KIM-1 is upregulated in the rat cochlea following systemic dose administration (cisplatin). KIM-1 mRNA expression was shown to be upregulated in the organ of Corti, the lateral cochlear wall tissues, and the spiral ganglion cells. Furthermore, KIM-1 immunoreactivity was increased in OHCs themselves.
In searching for novel predictive biomarkers, we begin with a broad-based screening because at the present time there is a paucity of confirmatory cytotoxicity pathways involved in ototoxicity. However, Table 3 lists the 7 FDA “qualified” biomarkers of renal injury that have also been reported in subjects with hearing loss or ototoxicity. Based on the totality of currently available data extracted from reports appearing in peer-reviewed scientific journals, we recommend that for all test articles that show (1) clear evidence of auditory dysfunction in nonclinical IND-enabling toxicology studies (eg, functional observational battery, detailed clinical observations, or postmortem examinations); (2) are from a pharmacological class that has a known risk potential for ototoxicity (aminoglycosides, platinum-based chemotherapeutics); or (3) show the intended route of administration of the test article is into the ear canal. The value of adding the following clinical pathology parameters should be thoughtfully considered by the sponsor and SD:
Method Details for EMA- and FDA-Qualified Rodent Renal Function Biomarkers.
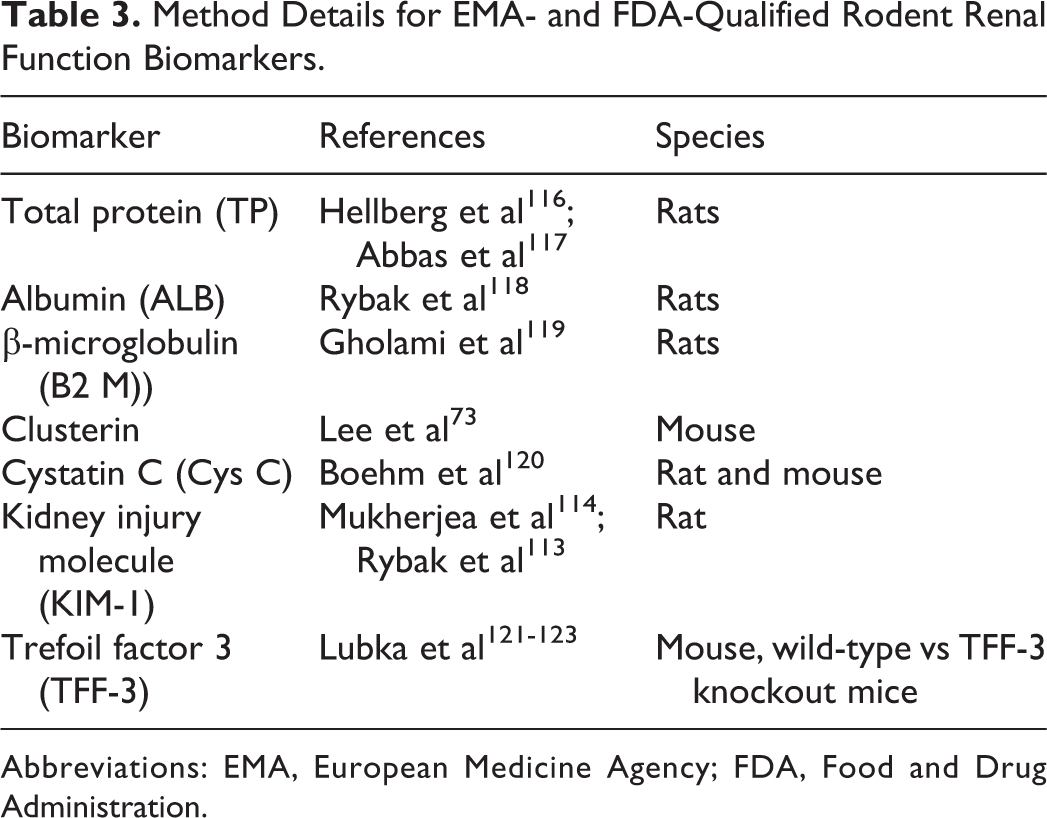
Abbreviations: EMA, European Medicine Agency; FDA, Food and Drug Administration.
The 7, or subset of the 7, “qualified” biomarkers of renal tubule damage should be incorporated in at least the terminal urinalysis to be scheduled in the protocol: TP, ALB, B2M, CLUS, Cys C, KIM-1, and TFF-3. If any of these biomarkers are altered by test article administration, a conversation between the sponsor and SD should be scheduled. Additionally, terminal blood sampling (prior to euthanasia) that will allow for a larger blood aliquot to be processed from even small animal subjects (rats) to serum or plasma should be considered. For drug candidates that do not pose a threat of nephrotoxicity from these 7 “qualified” biomarkers, we recommend that the serum or plasma aliquots be assayed for concentrations of CLUS, prestin, and KIM-1—3 significant candidates of local cellular ROS damage in the inner ear. These ototoxicity biomarkers may be present for as long as 1 month following drug-induced cell death and appear to be the best target for discussions in protocol development for all drug.
21
The additional costs to a study protocol to add these potentially valuable clinical pathology parameters would be minimal in comparison to the costs of public relations and legal ramifications of ototoxicity in phase III clinical trials or following New Drug Application (NDA) approval. The predictive accuracy of our recommendations will take time. “The proof will be in the pudding.” Whether these clinical pathology parameters will translate into nonclinical utility will depend on the ability to develop high-throughput assays for them in the standard small and large animal species of purpose-bred laboratory stocks used in current IND-enabling studies. The goal is to relate findings back to biology and to characterize the molecular mechanisms of apoptotic and necrotic pathways early in the drug development process, as possible. Proteins, their mRNA, their functionally important modifications, and binding characteristics must be placed within the context and current understanding of cellular “death pathways.”
As previously suggested by Yorgason et al, 124 drug-induced ototoxicity occurs with efficacious doses of FDA-approved drugs already on the market. These drugs are recommended to be used judiciously to treat patients who are not responsive to safer alternative medications. Care should be given when prescribing other known ototoxic medications. Blood levels of these drugs should be monitored to avoid toxic levels, and early detection of ototoxicity symptoms should be emphasized to minimize these toxic effects. Professional awareness and improved clinical decision-making are used to promote prevention of such drug-induced ototoxicity. However, in developing improved and efficacious NMEs, it is critically necessary to understand the cellular and molecular mechanisms underlying the potential ototoxic effects as early as possible. Elucidation of these mechanisms will facilitate the development of safe and effective clinical approaches for the prevention and amelioration of drug-induced ototoxicity prior to the first dose in man.
It is our hope that over time these clinical pathology parameters may prove to have high predictive validity for ototoxicity prevention, maybe not. In our opinion, the patient who is administered the drug remains the winner here in either case. Novel drugs that fail the test will be replaced by another candidate, or the federal agency’s risk-to-benefit analysis will subsequently determine and weigh efficacy over hazard. The future patient can feel confident that the regulatory approval process of new drug candidates remains vigilant as the pharmaceutical manufacturers “policing themselves” based on critically relevant data as it is generated in “real time.”
Footnotes
Acknowledgments
This review represents a “work-for-hire” product, conducted at no cost to the authors and no external funding sources.
Author Contribution
D. Gauvin substantially contributed to conception or design, acquisition, analysis, or interpretation of data and drafted the manuscript. Z. Zimmermann and R. Tapp substantially contributed to conception or design, acquisition, analysis, or interpretation of data and critically revised the manuscript. J. Yoder substantially contributed to conception or design and drafted the manuscript. T. J. Baird contributed to acquisition, analysis, or interpretation of data and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.