
Abstract
There is overwhelming evidence that the microbiome must be considered when evaluating the toxicity of chemicals. Disruption of the normal microbial flora is a known effect of toxic exposure, and these disruptions may lead to human health effects. In addition, the biotransformation of numerous compounds has been shown to be dependent on microbial enzymes, with the potential for different host health outcomes resulting from variations in the microbiome. Evidence suggests that such metabolism of environmental chemicals by enzymes from the host's microbiota can affect the toxicity of that chemical to the host. Chemical-microbial interactions can be categorized into two classes: Microbiome Modulation of Toxicity (MMT) and Toxicant Modulation of the Microbiome (TMM). MMT refers to transformation of a chemical by microbial enzymes or metabolites to modify the chemical in a way that makes it more or less toxic. TMM is a change in the microbiota that results from a chemical exposure. These changes span a large magnitude of effects and may vary from microbial gene regulation, to inhibition of a specific enzyme, to the death of the microbes. Certain microbiomes or microbiota may become associated with different health outcomes, such as resistance or susceptibility to exposure to certain toxic chemicals, the ability to recover following a chemical-induced injury, the presence of disease-associated phenotypes, and the effectiveness of immune responses. Future work in toxicology will require an understanding of how the microbiome interacts with toxicants to fully elucidate how a compound will affect a diverse, real-world population.
Keywords
Chapter I: The Microbiome, an Introduction
The Microbiome
The term “microbiome” generally refers to the collective genomes of all of the microorganisms that inhabit the human body, while “microbiota” and “microflora” typically refer to the actual community of microorganisms in a specific niche on or in the human body. The concept of the microbiota and microflora having an influence on human health dates at least to the early 1900s where it was studied in regard to its effect on nutrition and periodontal health. However, the term microbiome only came into common usage during the “omics” era with the advent of improved DNA sequencing technologies that allowed more thorough evaluation of the composition of the microbiota. Within the human gut microbiota, there are estimated to be between 10 and 100 trillion microbial cells, potentially exceeding the estimated 10 trillion cells that comprise the human body. 1 -4 In the human gut, there are estimated to be around 1,000 to 1,150 unique bacterial species, which, in totality, include 3.3 million nonredundant microbial genes, nearly 150 times greater than the total number of genes encoded in the human genome. 2 These microbial genes directly influence the levels of many metabolites in the human body, including amino acids, vitamins, antioxidants, isoflavanoids, and short-chain fatty acids. 5 They also metabolize xenobiotic compounds, which can affect the bioavailability or toxicity of these materials. Furthermore, humans are dependent on bacterial enzymatic processes to digest components of our diet, such as plant polysaccharides, highlighting the important and ancient symbiotic relationship. 6
The microbiome dynamically reacts to environmental cues and stressors. For example, recent investigations into the role of the host microbiome in health and disease have revealed insights into the significance of microbiome homeostasis. For example, a 2015 study of microbiome fluctuations associated with pigs raised on either sow milk or feed-based diet showed distinct differences in the fecal gut microbial compositions that were specific to the food being consumed. 7 In the absence of dietary changes, these microbial populations were relatively stable, and the microbial species associated with the different diets reflected an increase in specific metagenomes (a collection of genomes from a population or community) that encode enzymes necessary for the metabolic processing of milk-derived nutrients or those specific to the feed-based diet. 7 It has also been shown that sustained alteration to one’s diet can cause significant, and sometimes rapid, changes in the gut microbiota composition. Varying the levels of fruits and vegetables, carbohydrates, fats, proteins, and total calories consumed have all been shown to influence the gut microbiota composition in consistent, reproducible, ways. 8 A significant portion of interindividual gut microbiota variability is associated with diet and drugs. 8 It has also been shown that the gut microbiota composition correlates more strongly among genetically unrelated individuals who shared a household than between genetic relatives who did not cohabitate. 8
Studies also indicate that microbiota can act as a first line of defense against opportunistic pathogens. Commensal bacteria on the skin and in the gastrointestinal (GI) tract prevent colonization from invasive bacteria by competing for resources 9 and by releasing antimicrobial compounds, such as bacteriocins. 10,11 Additionally, microbiota can directly modulate the host’s immune response. For example, peptidoglycans from intestinal microbiota can enter circulation and “prime” neutrophils in the bone marrow that then exhibit an increased ability to defend against invading pathogens. 12 In addition, the interaction between Toll-like receptors of the host immune system and gut microbiota is essential to the maintenance of intestinal epithelial homeostasis and protection against gut injury. 13
The Human Microbiome
In the wake of the Human Genome Project, it was noted that the endeavor would be incomplete until our microbial coinhabitants were also characterized. 14 -16 The momentum of scientific interest in the microbiome is highlighted in Figure 1, showing more than a 400-fold increase in the total yearly publication of microbiome-related publications between 2007 and 2018.
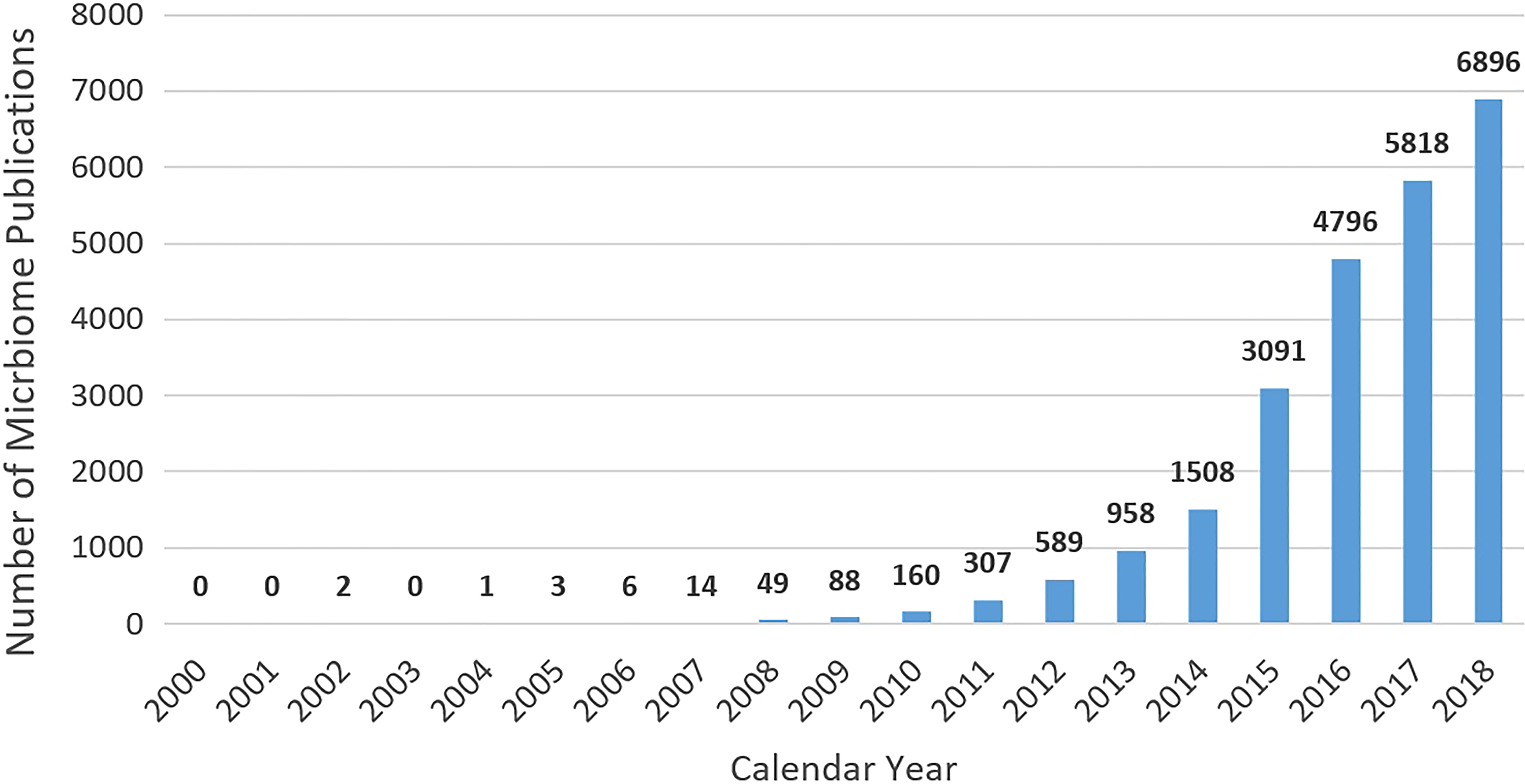
Quantification of publications containing the term microbiome (2000-2018). PubMed was utilized to search for the “Text Word” microbiome during the publication dates presented. Data are current as of December 12, 2018.
Through the efforts of the Human Microbiome Project, 4,788 total specimens from 242 patients were used to compare the structure, function, and diversity of the healthy human microbiome across 5 areas of the body (ie, oral cavity, nasal cavity, skin, GI tract, and urogenital tract) with a total of 18 sampling sublocations. 17 The major body sites defined primary patterns of variability. In general, oral and stool flora were quite diverse, whereas the bacterial community in the vagina was less diverse. Despite such diversity, the presence of core metabolic modules was consistent across many of the subsites, indicating conserved community functions, such as ribosome and adenosine triphosphate synthesis (Figure 2). These core metabolic modules are conserved across many bacterial species and confer similar functionality between the groups. These core modules can be carried out by different enzyme families at different body sites, but the core functionality is conserved. 18
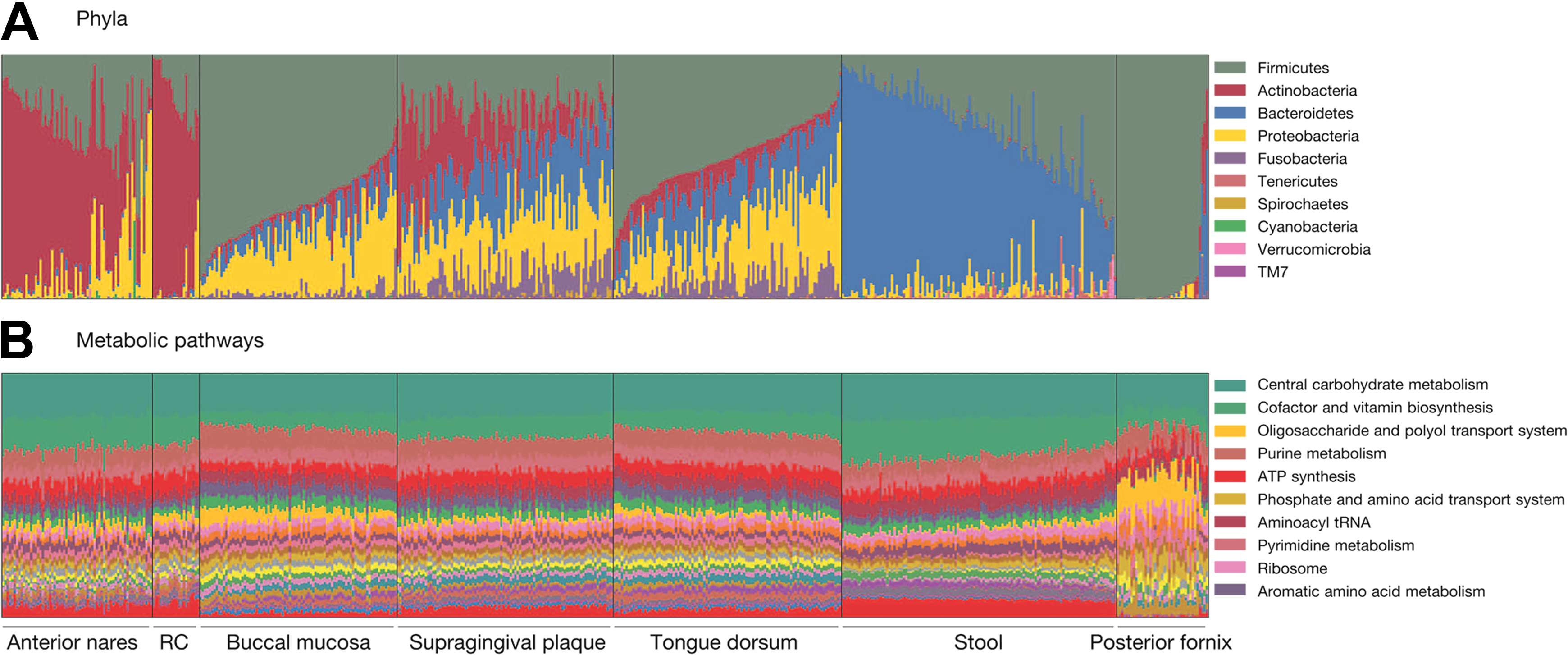
Abundance of bacteria (A) and genetic pathways (B) within human microbiotas. RC indicates retroauricular crease. Reprinted by permission from Macmillan Publishers Ltd: Nature. 2012;486(7402):207-214.
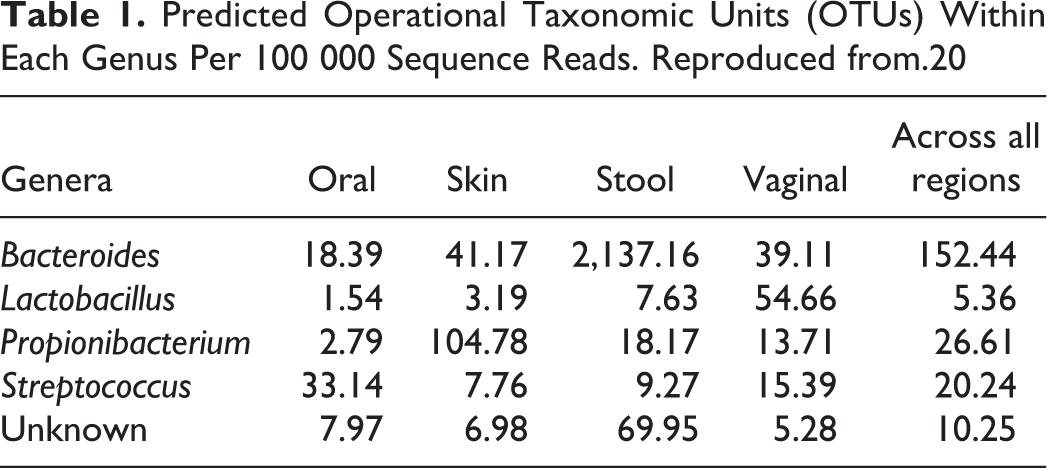
Although the particular genes that implement the core metabolic pathways vary, these functions are likely necessary for human-associated microbial life. 19 The variability is driven by genetics, events, and environmental factors, such as diet, exposures, and pharmaceuticals. 19 It is important to note, however, that when considering all identifiable taxonomic groups (ie, operational taxonomic units [OTUs], which are clusters of similar sequence variants), stool demonstrates the greatest α diversity (species diversity at one sampling site). 20 However, when considering only genera, the left and right antecubital fossae demonstrate the greatest mutually exclusive diversity between individuals. 20 Li et al identified systematic differences between microbial communities through the use of statistics (Tail statistic, “τ”) that takes into consideration the weighting of the OTUs by subsite (Table 1). 20
Predicted Operational Taxonomic Units (OTUs) Within Each Genus Per 100 000 Sequence Reads. Reproduced from.20
No taxa were universally present across subsites and individuals; however, a few signature taxa typically defined the diversity of the community at subsites (Figure 3). 17 The metabolic modules and clade abundances (frequency of the presence of bacteria from a specific monophyletic group), however, varied significantly with race or ethnicity, age, body mass index (BMI), and other factors such as pH in the vagina. Although these studies represent the most extensive sampling of the healthy human microbiome, only 800 reference strains from the human body had been sequenced in 2012. 17 Indeed, the vast majority of microbial species have yet to be isolated, cultured, or sequenced, primarily due to technical challenges. 21 Although not part of the Human Microbiome Project, the company, uBiome has collected over 250 000 microbiota samples and is on track to have over 1 million microbiota samples and 200 billion sequenced microbes by the end of 2019. 22
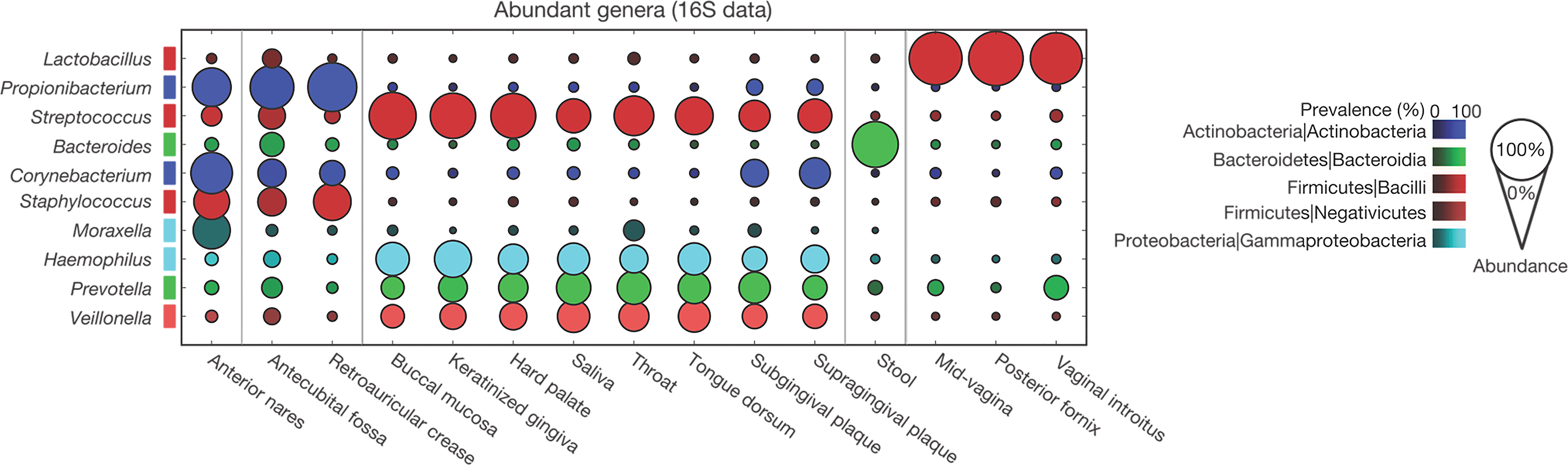
Distribution of bacterial genera within human body sites. Reprinted by permission from Macmillan Publishers Ltd: Nature. 2012;486(7402):207-214.
Significant variability has been observed between different individuals’ microbiomes. 23 -25 Many factors, including diet, 26 geographical location, 27 and host genetic constitution, 28 -31 contribute to this variability. Furthermore, temporal variability within the microbiome is strongly associated with the diversity of one’s microbiota composition. 24 Interpersonal variability has been shown to be greater than intrapersonal variability over time. 23 A positive correlation has been observed between diversity and stability of the microbiome, with increased stability with greater diversity and increased instability with lower diversity. 23,32 Specific microbiota habitats exhibit unique patterns of diversity, with the skin displaying the largest fluctuations. 24 In the case of the GI microbiota, temporal variability is associated with changes in the abundance of persistent species in the gut. 24 Overall, the concept of a “personalized” microbiome is supported by the large interindividual variability, as well as the variable rates of change within one’s microbial populations. 23,24
The Microbiome in Health and Disease
Dysbiosis of the microbiome can significantly impact human health and disease. As mentioned earlier, changes in the microbiome result from a variety of factors, including diet, 26,33 antibiotic use, 10,33,34 and other environmental exposures, including stress. 35 Dysregulation of one’s microbiome is associated with numerous human disorders, including obesity, 36 diabetes, 37 nonalcoholic steatohepatitis, 38 and coronary heart disease. 39 The gut microbiome can play a significant role in immunological and neurological diseases, such as allergic responses, 34 asthma, 40 atopic dermatitis, 41 and inflammatory bowel disease. 42,43
Multiple studies have shown the microbiome’s utility as a predictor of health and disease. The gut microbiota composition is a better indicator of BMI, blood glucose levels, cholesterol levels, and cardiac health than genetic factors. 44 The composition of the microbiota of amniotic fluid is a predictor of preterm birth and chorioamnionitis. 45 The gut microbiota composition is predictive of hospitalization risk and disease severity in patients with cirrhosis. 46 Taken together, these studies suggest that our microbiota is not only our first line of defense against disease but can also be used as an indicator of health status and disease susceptibility.
The deliberate modulation of one’s microbiome is also an important concept. In the clinical procedure of fecal microbiota transplantation (FMT), 47 the supplementation and restoral of one’s microbiota with a “healthy” fecal specimen from a donor can be an effective means of treating diseases like recurrent Clostridium difficile infections, 48 inflammatory bowel disease, 49,50 and diabetes. 51,52 In the case of C difficile infections, FMT is effective in >90% of cases. 47 Further illustrating the link between health status and the microbiome is a case where the microbiota from an obese but otherwise healthy donor was used in an FMT procedure for a lean individual with a C difficile infection. 53 In this particular circumstance, the patient was ultimately cured of the infection but developed a post-FMT onset of obesity, providing anecdotal evidence of the important, yet poorly understood, roles that our microbiomes may play in our lives. 53 Despite the side effects observed in the aforementioned study, FMT is now an established therapy to cure C difficile infections and is currently being studied as a potential treatment for metabolic syndrome and some psychological disorders. 54
Chapter II: Interactions Between Chemicals and the Microbiome
Chemical–microbial interactions can be categorized into 2 classes: microbiome modulation of toxicity (MMT) and toxicant modulation of the microbiome (TMM; Figure 4). Microbiome modulation of toxicity refers to transformation of a chemical by microbial enzymes or metabolites to modify the chemical in a way to make it more or less toxic. Toxicant modulation of the microbiome is a change in the microbiota that results from a chemical exposure. These changes span a large magnitude of effects and may vary from microbial gene regulation or inhibition of a specific enzyme to the death of microbes. For the purpose of this review, we have focused on direct effectors, but indirect effects have been included when relevant or when the order of effect is unknown. An example of a second-order, indirect-effect TMM would be modulation of the host immune system by a chemical that could lead to dysbiosis (eg, see ethanol below), while a second-order, indirect-effect MMT could occur by induction of host-detoxifying enzymes by microbial metabolites that results in a shift in the metabolic pathway for a chemical and result in differential levels of toxicity.
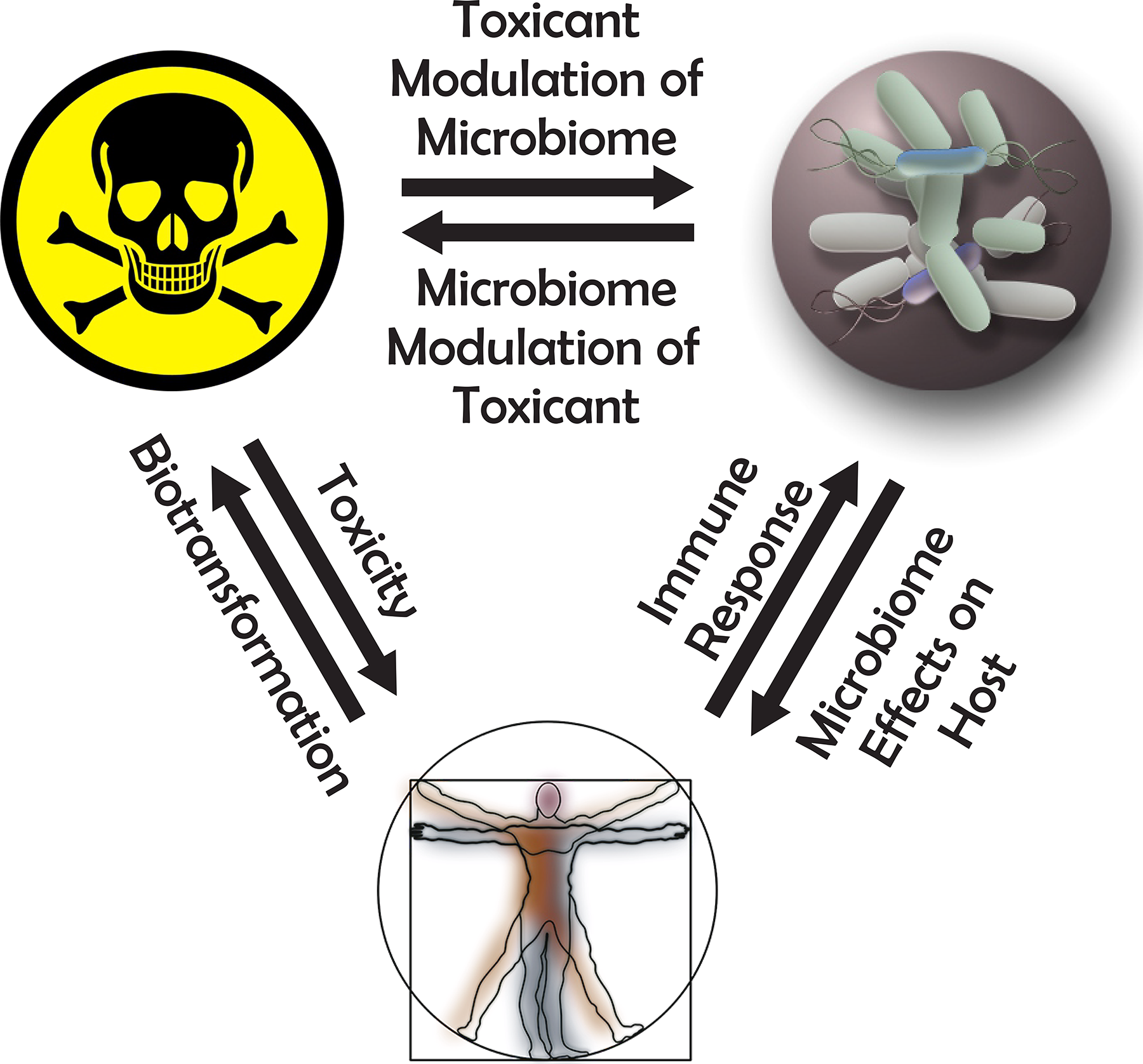
The dynamic relationship between the microbiome, toxicants, and human health and disease. Reproduced from Arcidiacono et al, 2018.
Microbiome Modulation of Toxicity
Microbes inherently exhibit the ability to interact with and metabolize chemicals as a characteristic of life, and thus it is not surprising that they are performing this function in humans. Five main categories of bacterial metabolizing compounds have been identified: azoreductases, nitroreductases, β-glucuronidases, sulfatases, and β-lyases, although other functionalities do exist. 55 Evidence suggests that the metabolism of environmental chemicals by enzymes from the host’s microbiota can affect the toxicity of that chemical to the host. 55 The first mechanism by which toxicity could be affected is by enzymatic modification of a compound to a form that is more or less toxic. Reports indicate that the intestinal microbiota metabolizes xenobiotics more extensively than any other organ. 56 -58 Changing the bioavailability of a compound is a second mechanism by which the microbiome may affect toxicity, making the toxin more or less accessible to the host’s body. A third mechanism by which the microbiome may affect toxicity is interference with the host’s detoxification mechanisms. Interactions between the gut microbiota and the host’s endogenous detoxification enzymes are still not well elucidated; therefore, further research will be important to determine the impact of the gut microbiota and endogenous xenobiotic metabolism. 55 While considering the potential for the microbiome to affect toxicity, one should remember that interindividual variability results in the presence of different species of bacteria. This interindividual variability may result in differences in how the toxicant is modified by the microbiota and thus may result in significant differences in toxicity between individuals for a single toxicant.
The ability of microbial life to reduce the toxicity of compounds has been repeatedly shown through numerous bioremediation strategies. There are over 1,200 compounds, over 800 enzymes, almost 1,300 reactions, and almost 500 microorganism entries listed in the University of Minnesota Biocatalysis/Biodegradation Database that demonstrate this principle. 59 However, these examples do not provide direct evidence that these reactions are occurring within humans. Human gut microbiota interacts with over 40 different drug substrates, by metabolizing, sequestering, or altering the substrate, demonstrating that they can have an impact on the metabolism of the host. 60
The majority of published work looking at microbiome modulation of xenobiotics within humans is in the context of the metabolism of pharmaceuticals in the gut (Table 2). 58,61,62 Although many of these studies were focused on the efficacy of the drug, several demonstrated microbe-induced changes that increase the toxicity of the compound. Acetaminophen, 94 balsalazide, 65 chloramphenicol, 64 indomethacin, 73 irinotecan, 61 nitrazepam, 112 olsalazine, 69 phenacetin, 71,72 risperidone, 98 sorivudine, 87 and sulfasalazin 70 are all converted by microbes into metabolites with elevated toxicity. Although some of these altered metabolites have been shown to be more toxic than their parent compounds, further work is needed to determine whether these microbiome modulated metabolites have a differential effect on the host during an actual exposure of the parent compound.
Microbiome-Mediated Changes on Substances That may Affect Toxicity.a

aWhile the microbiome mediated modification to the toxic substance is known, further work is needed to better characterize the extent of the impact of those changes to the substance’s toxicity.
The data on MMT outside of pharmaceutical agents are sparse, but they indicate that human microbes are modulating the toxicity of substances by altering the parent compound. A study of the polyaromatic hydrocarbons, including naphthalene, phenanthrene, pyrene, and benzo(a)pyrene, showed that colon bacteria formed estrogenic metabolites from these compounds. 111 Another group identified bacteria from skin samples that fully degraded benzo(a)pyrene, 113 though some of the intermediate metabolites may be more toxic. 62
Another mechanism by which the microbiome modulates toxicity is by changing the bioavailability of toxicants. This is particularly relevant for metals, which can be sulfonated, methylated, oxidized, or reduced to change uptake from the gut. The reduction of Hg(II) to Hg(0) 102 and Cr (VI) to Cr (III) 99,114 by gut bacteria reduces the absorption of these metals. 100 However, the issue is more complex, as some bacteria (eg, Desulfovibrio desulfuricans ND132 and Geobacter sulfurreducens PCA) are also able to methylate mercury, 93 creating a more toxic form that is more readily absorbed from the gut. 115 The gut microbiota is also capable of reducing arsenate to arsenite and methylating arsenic. 92,116,117 However, the effects for arsenic are converse to those of mercury, as the reduction increases uptake and the methylation reduces it. 118 In a real-world example, pregnant women in Tanzania who were given a probiotic containing Lactobacillus rhamnosus had lower blood levels of arsenic and mercury. 119 The effects on bioavailability can extend beyond metals. A study by Jin et al showed a decrease in plasma concentrations of geniposide, a compound found in traditional remedies, in antibiotic-treated rats, suggesting that the gut microbiota is impacting the bioavailability of geniposide. 120
Effects of the microbiome on the host detoxification system have also been reported, which may result in MMT. Two direct mechanisms that have been observed are: (1) microbial metabolites causing regulation of phase I and phase II enzymes 121 and (2) disruption of host excretion pathways by removal of phase II conjugates in the intestines. 122 An example of microbe-derived metabolites affecting host-based detoxification pathways was shown by Lhoste et al, where dietary polyphenols differentially influenced expression of several phase I and phase II enzymes in germ-free rats when compared to rats with flora from a human donor. 123 Compounds that are conjugated in the liver by phase II enzymes are excreted in the bile to the intestines. It is now well known that bacterial enzymes are responsible for deconjugating these compounds, allowing them to be reabsorbed. This process is called enterohepatic circulation and is considered a normal process. 124,125 Changes in the rates of this deconjugation, due to variation in the microbiome, may greatly affect the biological half-life of a compound and, consequently, its toxicity.
The size of the microbiome’s gene pool provides for an abundance of enzymes that can mediate biotransformation of compounds and their toxicities. Although this concept has been established for decades, with our greater understanding of the complexity of the microbiome, it has become apparent that MMT must be considered in future toxicological studies. The challenges lie in the diversity of the microbiome and the differences between microbiomes of different subsites, individuals, and even species. Studies must be designed that take this diversity into account if we hope to fully understand the toxic potential of any compound.
Toxicant Modulation of the Microbiome
With the important role of the microbiome in human health (see Introduction), perturbation of the microbiome by some toxicant exposures may result in adverse health effects. Over the years, numerous human diseases have been attributed to low-dose chronic exposures to toxicants with little empirical evidence. Variations in the gut microbiome have been associated with a variety of disorders, including allergies, anorexia, autism spectrum disorder, Celiac disease, Crohn disease, obesity, and type 2 diabetes. 126 Evidence is emerging that shows some of these diseases may be mediated by low-dose chronic toxic effects on the microbiome, such as obesity from the use of antibiotics, 127 and obesity and diabetes from exposure to environmental toxicants. 128 Obesity resulting from antibiotic-derived perturbations in the microbiome was shown to be transferrable in a mouse model. 129 It should be noted that in the cases of human disease, these linkages are correlative and have not yet definitively been shown to be causative.
Direct experimental evidence for xenobiotic-induced perturbation of the gut microbiome has only been recently demonstrated. Two independent research groups have shown that gut microbial diversity was significantly altered in an inbred mouse model after exposures to arsenic, cadmium, or lead. 130,131 Our own study confirmed these effects, showing significant changes in the composition of the gut microbiota, particularly at the family level, in Sprague-Dawley rats exposed to arsenic, cadmium, cobalt, chromium, and nickel. 132 In the past several years, studies showing the perturbation of the microbiome by toxic exposures have been published for cadmium, 133 lead, 119 nickel, 134 arsenic, 131,135 antibacterial mouthwash, 136 valproic acid, 137 silver nanoparticles, 138 and antibiotics. 129
An important application of TMM is the utility of the microbiota as a tool for identifying exposure to certain toxicants. It is common for clinical symptoms to develop in a patient who may not have any knowledge of the causative exposure. Work by us and others demonstrated that exposure to different toxicants results in different changes to the microbiota. 132 Our studies found dose-dependent changes in rat gut microbiota in response to oral exposures that differed for arsenic, cadmium, and nickel. 132 Although further analysis is still needed to determine a microbiota profile indicative of each unique toxicant exposure, these data suggest that such profiling is possible. Another study exposed zebrafish to triclosan-laden food and found predictable changes in the gut microbiota taxa, based on the presence or absence of specific operational taxonomic units. 139 Not only can their data be used to predict which zebrafish will be susceptible to triclosan based on their gut microbiome, but it could also discriminate whether or not an individual had been exposed. Another group reported changes in the abundance of certain phyla of bacteria following epoxiconazole exposure 140 and suggest that these changes could be used as early indicators of toxic exposure and potentially for predicting health risk to the host. 140
The explosion of recent work looking for toxic exposure-induced changes in the microbiome is a reaction to concerns of the microbiome in human health, but more importantly, it indicates that TMM is likely to be a broader effect than has previously been observed. The effects of toxicants on bacteria and other microbes have been well studied outside an animal host. Studying these effects in vivo is expected to show similar effects and a resulting perturbation of the microbiome. An important aspect of future work will be to exhibit a causality linkage, rather than correlative, to health outcomes.
Case Studies
The toxicity of glyphosate is an interesting case to consider when evaluating human toxicity and interactions with the microbiome. Glyphosate is a broad-spectrum herbicide that targets the shikimate pathway for the biosynthesis of aromatic amino acids. 141 Because of the numerous toxicity studies and the knowledge that animals lack this pathway, glyphosate is considered to have an extremely low toxicity in animals, 142,143 though controversy still remains as to the potential carcinogenicity of the compound despite lack of clear evidence. It is widely used in agricultural settings due to the commercial availability of genetically modified crops that are resistant to this compound. However, the shikimate pathway is present in most bacteria and has the potential to be inhibited by glyphosate. 144,145 It was shown that glyphosate exposure caused sex-dependent changes in the gut microbiota composition in rats. 146 Additionally, exposure to glyphosate-based herbicides caused depression- and anxiety-like behavioral impairments in mice, which correlated with changes in the effected mice’s gut microbiota. 147 There have been several recent publications that suggest linkage between increased glyphosate usage and increased rates of human diseases, in part mediated through disruption of normal microbial function. 148 Although association of glyphosate with human diseases is tenuous, it provides an example of why toxicity testing needs to take into account both host and microbial metabolism.
Perhaps one of the best-studied examples of the interplay between toxic exposure and the microbiome is ethanol. The widespread consumption of alcohol, and the resultant health implications, has led to a wealth of research on this exposure. The primary paradigm for alcoholic injury is through injury resulting from oxidative metabolites that are generated during its degradation in the liver. The injury is known to be enhanced by gut leakage and translocation of bacteria and their metabolites (eg, lipopolysaccharide) from the gut, which heightens the inflammatory state in the liver. It has been known, at least since the mid-1980s, and repeated in many more recent studies, 149 -152 that chronic alcohol consumption leads to bacterial overgrowth and dysbiosis in the intestines, which contribute to the observed health effects. The direct cause of the dybiosis is complex, as there are some direct effects of the toxicant on certain microbial species. However, immune dysfunction also contributes to the bacterial overgrowth. Stimulation of the epithelial mucosa to overproduce mucin (Muc2) in response to the toxicant appears to hinder the immune system from modulating gut microbes. 153 Furthermore, the oxidative metabolism of ethanol by gut bacteria 95,154,155 generates acetaldehyde, which has been linked to the development of rectal cancer, another known risk of chronic alcohol consumption. This example demonstrates how both MMT and TMM affect a single toxic exposure, illustrating the complexity of the interactions between the host system, the microbiome, and the toxicant.
Arsenic exposure is another model of a well-studied toxicant that is worth further review. As stated above, there is experimental evidence for both MMT and TMM effects during arsenic exposure. The MMT includes reduction and methylation of arsenate to a more or less toxic form, respectively. 93 A thiolated version with unknown toxicity was also observed to be a direct metabolite of gut microbes. 117 Furthermore, a study using an interleukin 10 knockout strain of mice showed significant differences from wild type in both the composition of the gut microbiome and in the metabolic profile of arsenic. 28 As each of these species of arsenic has different absorption, distribution, metabolism, excretion, and toxicity profiles, these studies provide overwhelming evidence for the critical importance of the microbiome in the determination of arsenic toxicity. In addition, however, the microbiome is also a target of toxicity. Studies by our lab 132 and others 131,135 showed dysbiosis as a direct result of arsenic exposure. Taken together with recent human disease, it will be very important to consider the influence that the microbiome has on arsenic-induced toxicity and its role in the prevalence of type 2 diabetes mellitus and cardiovascular disease in arsenic-exposed populations. 156 -159
Chapter III: Future Directions of Research
When contemplating future directions of research, independent strategies will be required to delineate the MMT and TMM effects. The role of each is important in fully understanding the toxicity of a compound, so it is likely that studying toxicity from both perspectives will be required for a complete understanding of the toxicity of any given compound.
Tools for Studying the Microbiome
Tools for studying the microbiome range from computational models through human studies. In silico predictions: Numerous models have been developed to predict metabolism of a compound based on imputed enzymatic function from gene sequences. These tools utilize results from metagenomic analyses to create a pool of the genes, both host and microbiota, that encode enzymes that are likely to play a role in metabolism, and then predict potential metabolites of a given compound. These tools are relatively quick and inexpensive but have reduced accuracy compared to more advanced models. In vitro/cell cultures: Microbial cells isolated from various subsites can be cultured under a variety of conditions to mimic the microbiome of interest. Gut culture systems have become relatively advanced and often include multiple compartments in sequence to more realistically mimic the GI tract. These systems can be used for both MMT and TMM studies. The primary limitation of these systems is that not all microbes can be cultured, so these systems do not represent the entire microbiome of an organism. Additionally, these systems lack the full host–microbiome interaction, which may be important for the overall toxicity of a compound (eg, enterohepatic circulation). These systems are primarily used to determine potential metabolites of compounds. Traditional animal models: Animals have been the model of choice for toxicologist for decades. They allow both MMT and TMM studies. Unlike cell culture, they allow host–microbe interaction and include all the microbes in the host (ie, nonculturable microbes). As expected, the primary limitation of these systems is in determining how well the animal’s microbiome represents the microbiome of the species of interest (usually human). Traditional laboratory animal models are reared in uniquely clean environments with no exposure to pathogens. The immune systems of these animals have not been “tested” in a real-world environment, so they may not represent a realistic response to toxicant challenges. Furthermore, the analyzed microbiome systems in these animals are relatively invariant. To overcome this, some studies have been conducted in agricultural animals that have been reared outside, have likely been exposed to pathogens, and have a more diverse microbiome.
160
Gnotobiotic animal models: A variety of gnotobiotic animal models have been developed over the recent years. These animals are typically reared germ free and then associated with a specific microflora of interest. Multiple studies have used human microflora for these animals, which likely better represent human exposures than traditional animal models. However, there are 2 significant concerns with these models. The first is that animals raised in germ-free environments have some subtle differences in development, particularly in the intestines and the immune system
161
The developmental issue can be somewhat overcome by using F1 progeny for studies, but the stability of the introduced microflora must be monitored to ensure consistency.
162
The second area of concern is whether a microbiome from a different species can be adequately represented in an animal with a different immune system, considering the importance that the immune system plays in modulating the microbiome. Human studies: The use of human patients for studies of toxic effects is typically limited to observational studies. There are rare opportunities in certain occupational settings to perform longitudinal studies. For compounds with known toxicity profiles, human studies can be performed below toxic thresholds to help elucidate metabolism pathways. However, due to obvious ethical concerns, the use of human patients for toxicity studies is very limited.
Suggested Strategies for Future Studies
We propose that studies for determination of MMT should begin at the computational level, which will allow for a better understanding of the potential outcomes. The results can be used to tailor cell culture experiments to ensure that the most feasible culture systems are being used and that the proper analytical techniques are being employed to capture the predicted metabolites. Animal models, traditional or gnotobiotic, may be used as another tier of analysis, but care should be taken if the goal is MMT, as it may not be possible to distinguish microbial from host metabolism.
For determination of TMM, the use of animal models is the preferred approach because of the importance of host–microbiome interactions. The use of gnotobiotic models is significantly more costly, but it offers the unique advantage of studying the human microbiome (with aforementioned concerns of different immune systems and host interactions 163 ) and is preferential if budget allows. Furthermore, flora from multiple individuals can be utilized to begin to ascertain the differences in human toxicity for a given compound.
Depending on the toxicity and metabolic profile of the compound of interest, iterative studies may be warranted. For example, the secondary toxicities of compounds that show significant acute toxicity can be further studied by utilizing microbiome-derived metabolites in TMM studies. In addition, microbial flora from exposed animals can be introduced into germ-free animals. These studies can help to delineate the effects of the primary toxicant from the microbiome-mediated effects. 164
Additional studies are also needed to better associate certain microbiota with susceptibility or resistance to different exposures. Modulating the microbiome for therapeutic use has the potential for conferring resistance to or aiding recovery after toxic exposures. Some work has started to investigate the possibility of using engineered or custom microbiomes as therapeutics; however, more work is needed to better understand the therapeutic role of certain microbiota and to develop better techniques for the stable manipulation and transfer of such engineered microbiomes. Just as the microbiome is being used to predict certain health and disease states, it could also be used to predict the likelihood of injury following a toxic exposure or to predict whether a toxic exposure had occurred.
Conclusion
Future work in toxicology will require an understanding of how toxicants interact with the microbiome to fully elucidate how a compound will affect a diverse, real-world population. There is overwhelming evidence that the microbiome must be considered when evaluating the toxicity of chemicals. Disruption of the normal microbial flora is a known effect of toxic exposure, and these disruptions may lead to human health effects. In addition, the biotransformation of numerous compounds has been shown to be dependent on microbial enzymes, with the potential for different health outcomes resulting from variations in the microbiome. Certain microbiomes or microbiota will start to be associated with different health outcomes, such as resistance or susceptibility to exposure to certain toxic chemicals, ability to recover following a chemical-induced injury, the presence of disease-associated phenotypes, and the effectiveness of immune responses.
Footnotes
Authors’ Note
The views, opinions, and/or findings contained in this report are those of the author(s) and should not be construed as official Department of the Army position, policy, or decision, unless so designated by other official documentation.
Citations of commercial organizations or trade names in this report do not constitute an official Department of the Army endorsement or approval of the products or services of these organizations.
Author Contribution
Jason M. Koontz and Blair C. R. Dancy contributed to conception and design, analysis and interpretation, drafted manuscript, and critically revised manuscript; Cassandra L. Horton and Jonathan D. Stallings contributed to analysis and interpretation and critically revised manuscript; Valerie T. DiVito contributed to analysis and interpretation and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.