
Abstract
Perfluorooctanesulfonic acid (PFOS) is a persistent organic pollutant with worldwide bioaccumulation due to a very long half-life. Perfluorooctanesulfonic acid exposure results in significant hepatic effects including steatosis, proliferation, hepatomegaly, and in rodents, carcinogenesis. The objective of this study was to determine whether PFOS exposure exacerbates nonalcoholic fatty liver disease and nonalcoholic steatohepatitis pathogenesis. Eight-week-old male C57BL/6 J mice (n = 5 per group) were fed ad libitum normal chow diet (ND) alone, 60% high-fat diet (HFD) alone, ND + PFOS, and HFD + PFOS (0.0001% w/w (1 mg/kg) of PFOS) for 6 weeks. Both HFD alone and the ND + PFOS treatment induced significant adiposity and hepatomegaly, but the HFD + PFOS treatment showed a marked protection. Oil Red O staining and quantitative analysis of hepatic lipid content revealed increased hepatic steatosis in ND + PFOS and in HFD alone fed mice, which was prevented in HFD + PFOS treatment. Further studies revealed that ND + PFOS treatment significantly affected expression of lipid trafficking genes to favor steatosis, but these changes were absent in HFD + PFOS group. Specifically, expression of CD36, the major lipid importer in the cells, and peroxisome proliferator-activated receptor gamma (PPARγ), its major regulator, were induced in HFD + no treatment (NT) and ND + PFOS-fed mice but remained unchanged in HFD + PFOS mice. In conclusion, these data indicate that coadministration of PFOS with HFD mitigates steatosis and hepatomegaly induced by HFD and that by PFOS fed in ND diet via regulation of cellular lipid import machinery. These findings suggest dietary lipid content be considered when performing risk management of PFOS in humans and the elucidation of PFOS-induced hepatotoxicity.
Introduction
Perfluorooctanesulfonic acid (PFOS) belongs to a class of chemicals known as perfluoroalkyl acids. These hydrophobic and lipophobic compounds are used in a variety of household and industrial applications due to their thermally and chemically stable properties. 1 Perfluorooctanesulfonic acid is present in 99.9% of human serum samples in the United States with a geometric mean serum concentration of 20.7 µg/L. 2 Perfluorooctanesulfonic acid can be found in humans and wildlife worldwide. 3 Perfluorooctanesulfonic acid undergoes bioaccumulation in humans due to the long serum half-life of 4.7 years. 4 Perfluorooctanesulfonic acid distributes primarily to the liver of rodent adults and fetuses exposed in utero, 5 where it induces hepatic steatosis, hepatomegaly, and hepatic carcinogenesis. 6 -8
Nonalcoholic fatty liver disease (NAFLD) is currently the major hepatic disorder with over 25% prevalence in the general population worldwide, including in the United States. 9 Patients with NAFLD are at higher risk of progressing to more severe forms of liver disease such as nonalcoholic steatohepatitis, fibrosis, and cirrhosis, ultimately resulting in hepatocellular carcinoma (HCC), which is the second most common cause of cancer-related deaths. 10,11 Nonalcoholic fatty liver disease is the primary hepatic manifestation of metabolic syndrome, which is constellation of symptoms including obesity, diabetes mellitus, and dyslipidemia. 12 Recently, toxicant-associated fatty liver disease (TAFLD) has been defined as the fatty liver disease associated with chemical exposure. 13 It is plausible that PFOS is a TAFLD-causing agent, given its ability to induce hepatic steatosis and ubiquitous presence in human serum samples with the potential for bioaccumulation.
In this study, we used an experimental model of NAFLD induced by high-fat diet (HFD) feeding to test the ability of human-relevant PFOS exposures to exacerbate the formation of fatty liver disease. Contrary to our hypothesis, we observed a protective effect against HFD-induced NAFLD when PFOS was coadministered with HFD. We examined expression of metabolic genes and regulators to investigate the potential mechanisms of protection. Our findings reveal diet-dependent toxicity of PFOS and provide evidence for its hormetic effects on the development of hepatic steatosis.
Materials and Methods
Animal Care and Tissue Preparation
All animal studies were approved by and performed in accordance with the Institutional Animal Care and Use Committee at the University of Kansas Medical Center. Eight-week-old male C57BL/6 J mice, purchased from Jackson Laboratories (Bar Harbor, Maine), received ad libitum access to normal chow diet (ND; 73% kcal carbohydrates, 11% kcal fat, 16% kcal protein; Custom Animal Diets, AD2001) or high-fat diet (HFD; 24.8% kcal carbohydrates, 59.2% kcal fat, 15% kcal protein; Custom Animal Diets, AD2002) with and without 0.0001% (1 mg/kg) perfluorooctanesulfonic acid (PFOS; Sigma Aldrich, St Louis, Missouri) for 6 weeks. Mice from each treatment cohort (n = 5) were group-housed in temperature-controlled conditions (23°C) with a 10-hour dark/14-hour light cycle. The dosing strategy was selected by extrapolating the dose used in a previous report when mice fed a diet containing 0.001% PFOS resulted in PFOS serum concentrations of 102 µmol/L. 14 Furthermore, a serum PFOS concentration of 11 µg/mL (∼23 µmol/L) was achieved by feeding 0.0001% PFOS-containing diet for 28 days. 15 Thus, the dose used in this study was expected to achieve serum concentrations nearing 10 µmol/L, which previously published literature review suggests is comparable to occupational levels of exposure. 16 To prepare the diet used in this study, PFOS was dissolved in water and thoroughly mixed into preweighed powdered ND or HFD to a final concentration of 1 mg/kg (0.0001%). Diets were left to dry overnight, and pellets were formed the next day. Untreated powdered diets were also mixed with water and formed into pellets to ensure feeding consistency. Liver and adipose tissues were harvested, weighed, and flash frozen in liquid nitrogen at the time of euthanasia. Blood glucose was measured with a glucometer using blood collected via retroorbital sinus at time of euthanasia.
Histology and Immunohistochemistry
Paraffin sections from mouse livers were stained for hematoxylin and eosin (H&E) and observed under light microscope. Paraffin sections from mouse livers were stained for proliferating cell nuclear antigen (PCNA) as described previously. 17 Fresh frozen liver sections were used to determine hepatic lipid content by staining with Oil Red O as previously described. 18
Protein Isolation and Western Blot Analysis
Protein isolation and Western blot analysis was performed as previously described. 16 Primary antibody used to detect PCNA was purchased from Cell Signaling Technology (Danvers, Massachusetts; Cat. #2586).
Hepatic Triglyceride Isolation and Quantification
Total hepatic triglycerides (TGs) were isolated from frozen liver using potassium hydroxide (KOH) digestion protocol as previously described. 19 Quantification of isolated TGs was performed using the Triglyceride Glyceride Phosphate Oxidase (GPO) Reagent Set (Pointe Scientific, Catalog #T7532-500) and calculated by comparison to a standard curve of known TG concentrations using the Triglyceride Standard Reagent (Pointe Scientific, Catalog #T7531-STD) according to manufacturer’s protocol. Triglyceride content measured in each sample was normalized by the weight of liver used for TG isolation.
Serum Lipid Quantification
Serum was prepared from centrifugation of fresh blood at 5,000 rpm for 10 minutes. Serum TG was measured using the Triglyceride (GPO) Reagent Set (Pointe Scientific, Catalog #T7532-500) and calculated by comparison to a standard curve of known TG concentrations using the Triglcyeride Standard Reagent (Pointe Scientific, Catalog #T7531-STD) according to manufacturer’s protocol. Serum Cholesterol concentration was measured using the Cholesterol Reagent Set (Pointe Scientific, Catalog #C7510) and calculated by comparison to a standard curve of known cholesterol concentration using the Cholesterol Standard Reagenet (Pointe Scientific, Catalog #C7509-STD) according to manufacturer’s protocol.
Real-Time Polymerase Chain Reaction
RNA isolation, conversion to cDNA, and real-time (RT) polymerase chain reaction (PCR) analysis was performed as described previously without modification. 20 Fold change in expression compared to control diet fed, untreated group was determined using the ddCt method 21 with 18 s used as a housekeeping control gene. Primer sequences for genes of interest are provided in Table 1.
Primer Sequences Used in This Study.

Statistical Analysis
All bar graphs depict the mean ± standard error of mean. A Shapiro–Wilk test was performed to check for normality of distribution, and Levene test was used to check for homogeneity of variance. All data met these requirements. Multiple comparisons were performed using a 1-way analysis of variance followed by a Duncan post hoc analysis with P < .05 considered significant. Statistical analysis was performed using IBM SPSS Statistics Version 25.
Results
Perfluorooctanesulfonic Acid Exacerbates Adiposity and Body Weight Gain in Normal Diet But Not in HFD Conditions
Mean food consumption per mouse did not differ between groups throughout the study (Figure 1A). Based on the body weight and food consumption data, we calculated that ND + PFOS-fed mice received a dose of 0.089 mg/kg/d and HFD + PFOS-fed mice received a dose of 0.087 mg/kg/d. There was no statistical difference in the dose received between these groups (Table 2). Figure 1B details the differences in body mass that developed between groups during the study. No significant differences in body mass existed between groups at day 0. At day 4, HFD-fed groups were significantly heavier than ND + NT mice but not heavier than ND + PFOS mice. At days 18 and 35, HFD-fed groups were significantly heavier than ND-fed groups. By day 42, while HFD + NT mice were significantly heavier than ND-fed groups, PFOS treatment increased weight in ND-fed mice and decreased weight in HFD-fed mice, resulting in no significant difference between ND + PFOS and HFD + PFOS treated mice. To determine the cause of differences in body weight, we determined adiposity by weighing epididymal fat pads (Figure 1C). The HFD-fed mice showed significant increase in epididymal fat mass as expected. However, while PFOS treatment increased epididymal fat mass in ND-fed mice, PFOS treatment did not exacerbate accumulation of epididymal fat mass in HFD-fed mice.
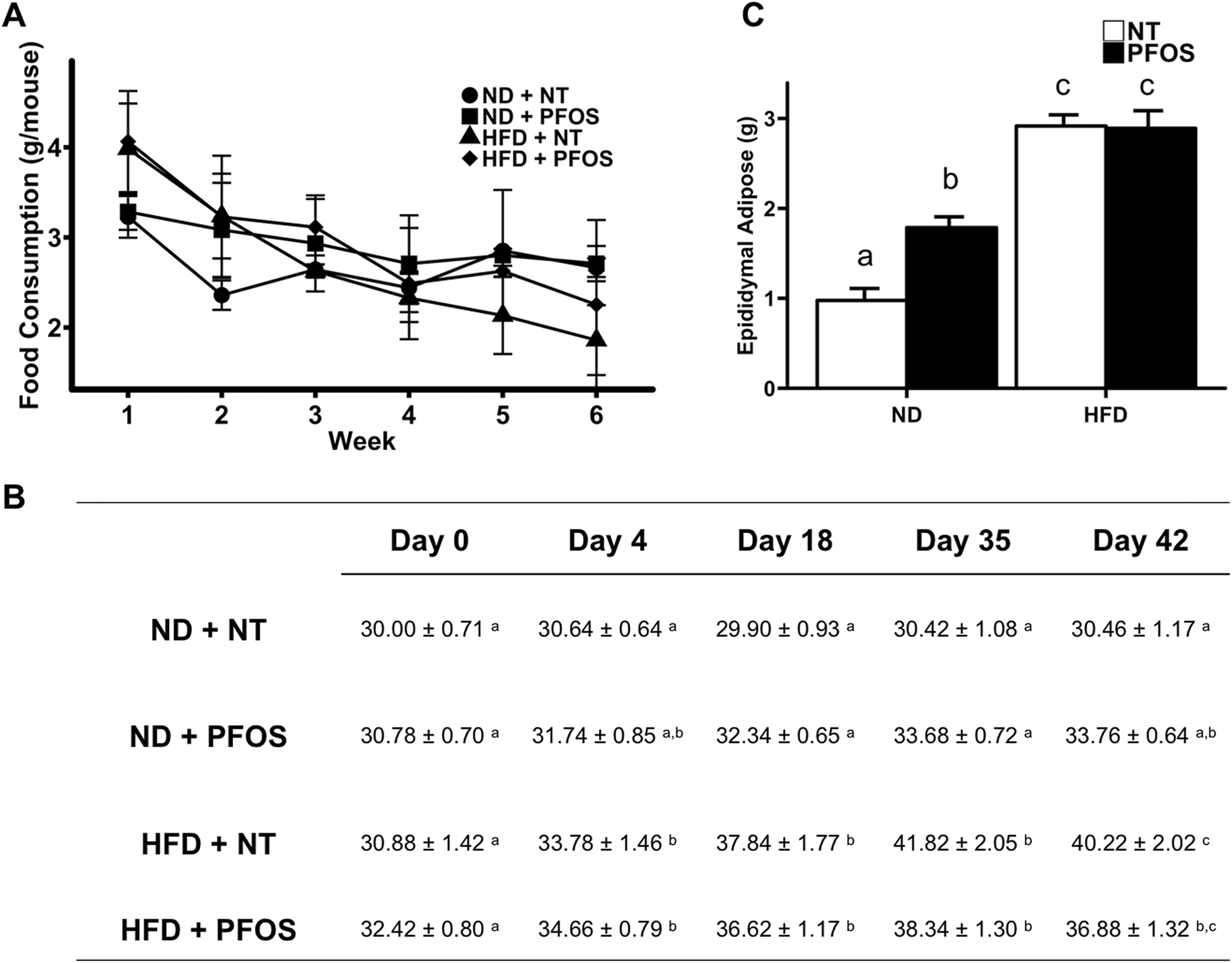
Perfluorooctanesulfonic acid (PFOS) exacerbates adiposity and body weight gain in normal diet but not in high-fat diet conditions. A, Average food consumption per mouse for each group throughout the study. B, Body mass for each group throughout the study. C, Average weight of epididymal adipose tissue collected per mouse. All values are expressed as mean ± standard error of mean. Letters indicate homogenous subsets with P < 0.05.
Dose of PFOS (mg/kg/d) Received by Each Treatment Group Calculated by Average Food Consumption Throughout the Feeding Period.
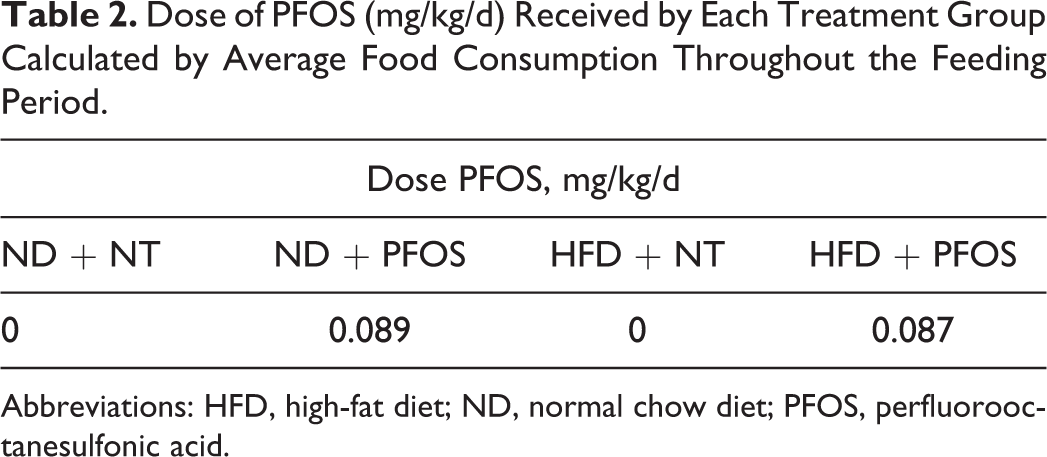
Abbreviations: HFD, high-fat diet; ND, normal chow diet; PFOS, perfluorooctanesulfonic acid.
Perfluorooctanesulfonic Acid Increases Hepatomegaly in Normal Diet But Not in HFD Conditions
Liver to body weight ratios were used to identify hepatomegaly. Normal chow diet + PFOS-fed mice showed significant increase in liver to body weight ratio when compared to ND-fed mice. Interestingly, a significant decline in liver to body weight ratio was observed in HFD-alone treatment group, which further decreased and was lowest in HFD + PFOS mice (Figure 2A).
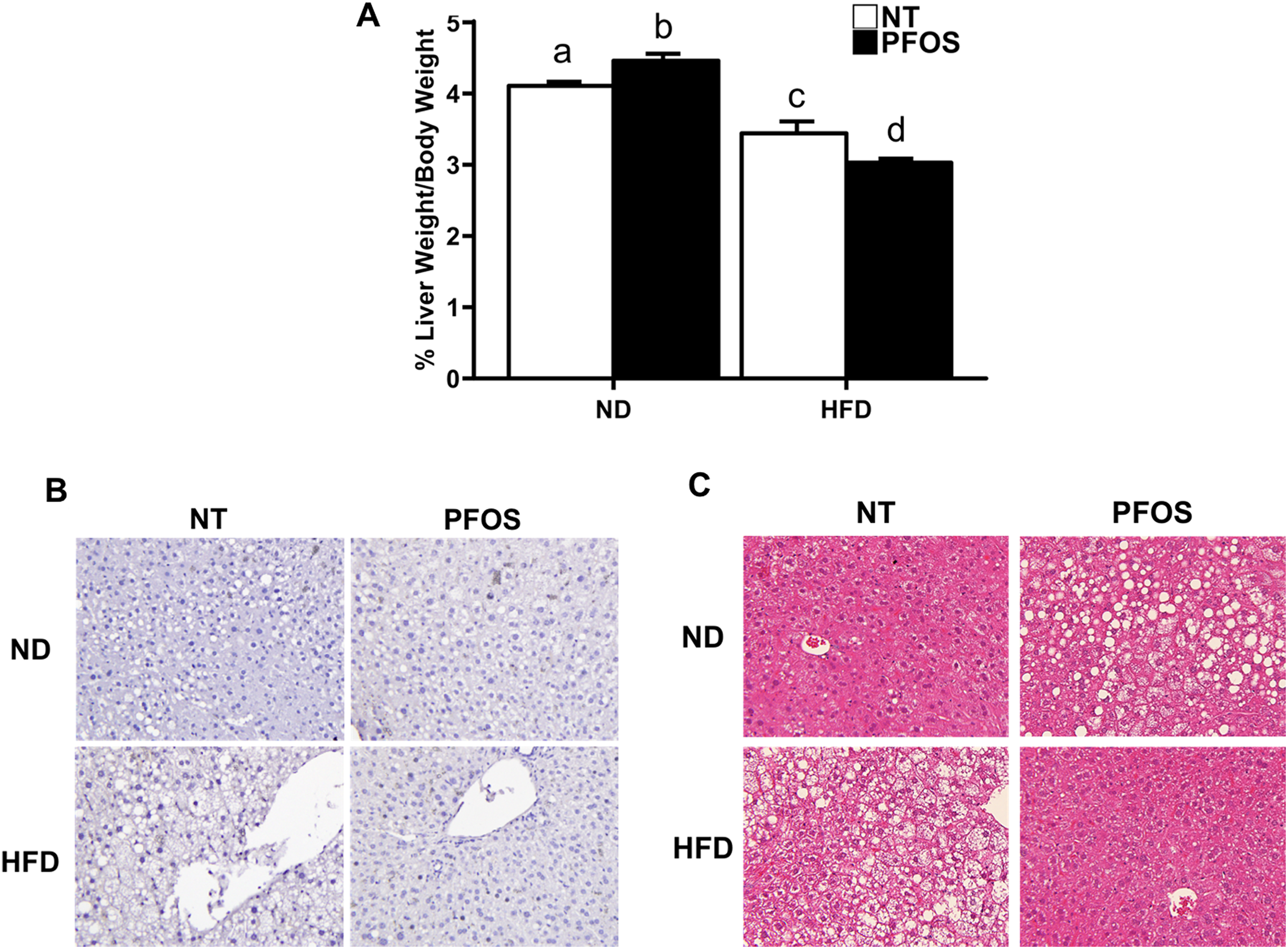
Perfluorooctanesulfonic acid (PFOS) increases hepatomegaly in normal diet but not in high-fat diet conditions. A, Liver mass to body mass ratios calculated at the time of euthanasia. Representative photomicrographs (×40) of liver sections stained for (B) proliferating cell nuclear antigen (PCNA) and (C) Hematoxylin and eosin (H7E) from NT or PFOS-treated mice fed normal or high-fat diet. All values are expressed as mean ± standard error of mean. Letters indicate homogenous subsets with P < 0.05.
Absence of Hepatocyte Proliferation After PFOS Treatment
Immunohistochemical analysis of PCNA was performed to determine whether the differences in liver size were due to changes in hepatocyte proliferation. However, none of the treatment groups exhibited any PCNA-positive nuclei (Figure 2B). Western Blot analysis further confirmed no differences in PCNA protein expression between all groups (data not shown).
Perfluorooctanesulfonic Acid Protects Against HFD-Induced Hepatic Steatosis
Hematoxylin and eosin stained liver sections were observed to identify differences in gross liver histology (Figure 2C). Significant micro and macrovesicular steatosis was evident in ND-fed mice following PFOS treatment. Similarly, a significant increase in hepatic steatosis was observed in HFD + NT mice. However, the HFD + PFOS mice showed normal liver histology without any steatosis resembling that of ND alone fed mice.
Next, we stained frozen liver sections for Oil Red O to determine hepatic lipid accumulation (Figure 3A). Extensive hepatic steatosis was observed in ND + PFOS mice. Hepatic steatosis was also observed in HFD + NT mice. However, HFD + PFOS mice were protected from hepatic steatosis. The degree of hepatic steatosis was determined by quantification of TGs isolated from frozen liver (Figure 3B). This analysis confirmed the results of Oil Red O staining. Hepatic TG were significantly induced in ND + PFOS and HFD + NT mice, but livers from HFD + PFOS mice contained the same amount of TG as ND + NT mice. Serum glucose was significantly elevated by PFOS in ND-fed mice but was not affected by PFOS in HFD-fed mice (Table 3). No significant changes in serum TG or serum cholesterol were observed in any groups (Table 3).
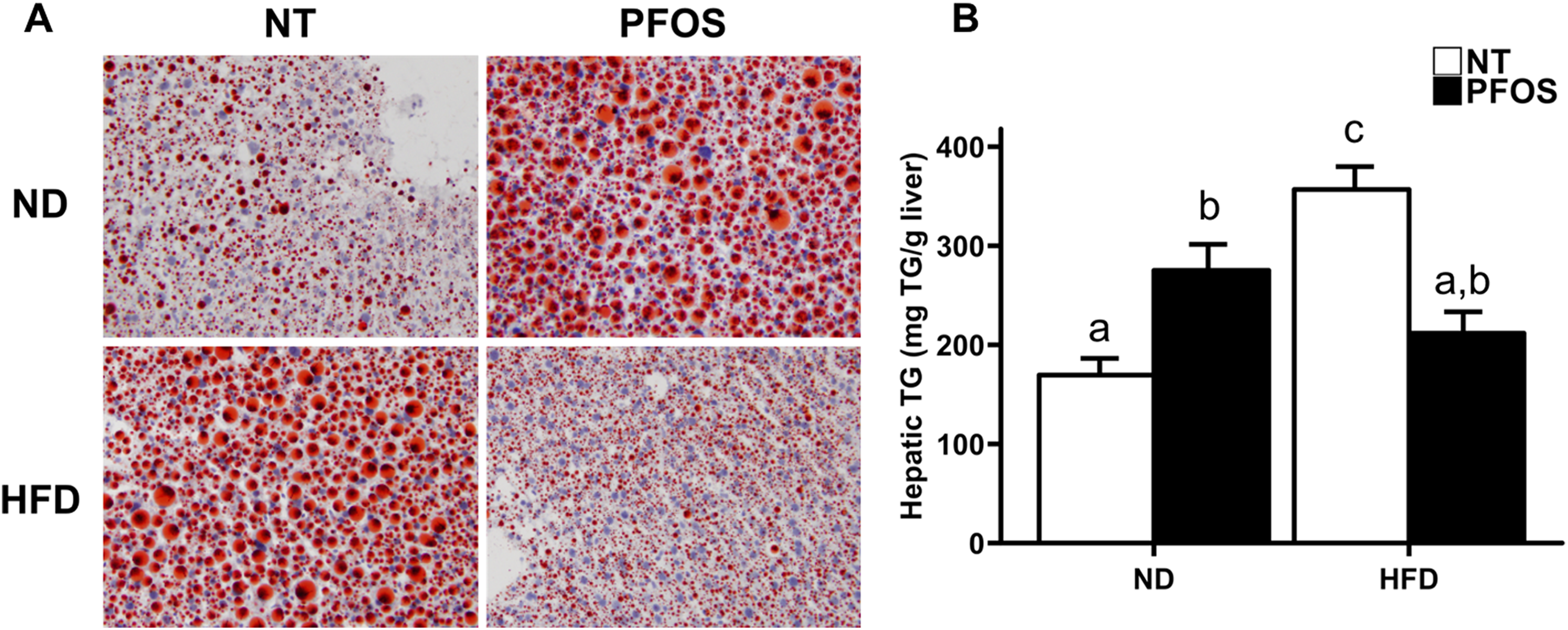
Perfluorooctanesulfonic acid (PFOS) protects against high-fat diet–induced hepatic steatosis. A, Oil Red O stained liver sections from NT- or PFOS-treated mice fed normal or high-fat diet. B, Quantification of hepatic triglycerides isolated from NT- or PFOS-treated mice fed normal or high-fat diet. All values are expressed as mean ± standard error of mean. Letters indicate homogenous subsets with P < 0.05.
Average Concentration of Serum Triglyceride (TG), Serum Cholesterol and Blood Glucose for Each Treatment Group Measured at the Time of Euthanasia.a
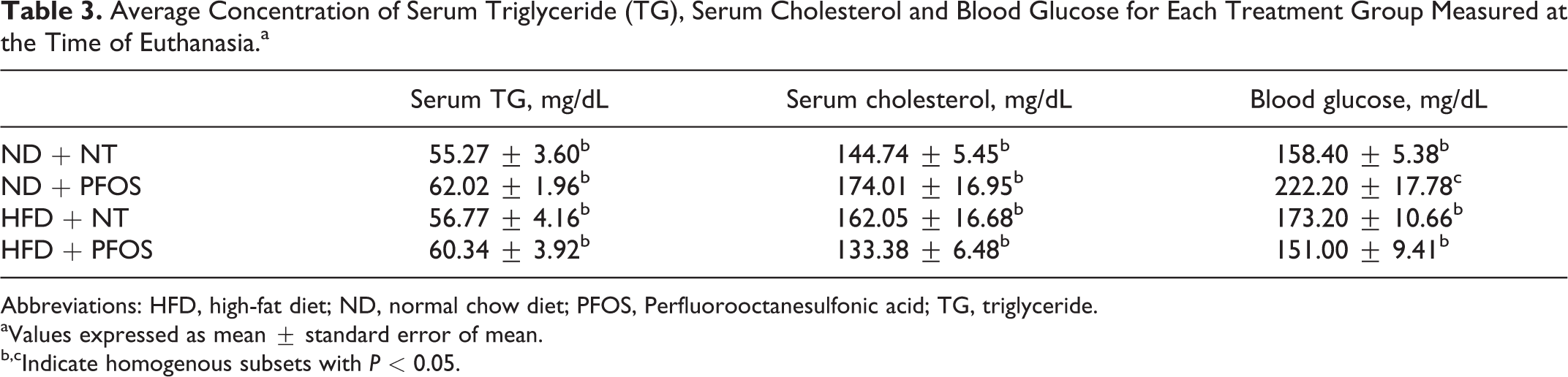
Abbreviations: HFD, high-fat diet; ND, normal chow diet; PFOS, Perfluorooctanesulfonic acid; TG, triglyceride.
aValues expressed as mean ± standard error of mean.
b,cIndicate homogenous subsets with P < 0.05.
Perfluorooctanesulfonic Acid Changes Expression of Metabolism Genes in Diet-Dependent Manner
To explore potential mechanisms for the diet-dependent effects of PFOS, we quantified mRNA of several genes associated with lipid and carbohydrate metabolism (Figure 4). In ND + PFOS-fed mice, expression of apolipoprotein genes ApoA1, ApoA2, ApoB, and the apolipoprotein packaging gene microsomal triglyceride transfer protein (MTTP) was reduced. In HFD + PFOS-fed mice, expression of these genes remained unaffected when compared to ND or HFD + NT. Expression of genes involved in gluconeogenesis, such ase phosphoenolpyruvate(PEPCK and glucose 6-phosphatase (G-6-Pase), were inhibited by PFOS in ND-fed mice and were inhibited in both HFD-fed groups compared to ND + NT. In HFD-fed mice, PFOS did not affect expression of these genes. Finally, we measured expression of 2 genes involved in lipid and sterol metabolism, such as carboxylesterase 3 (CES3sterol regulatory element binding transcription factor 1 (SREBF1). CES3 expression was reduced by PFOS in ND-fed mice. Perfluorooctanesulfonic Acid did not affect CES3 expression in HFD-fed mice. SREBF1 was nonsignificantly induced by PFOS in ND-fed mice and was significantly induced by PFOS in HFD-fed mice.
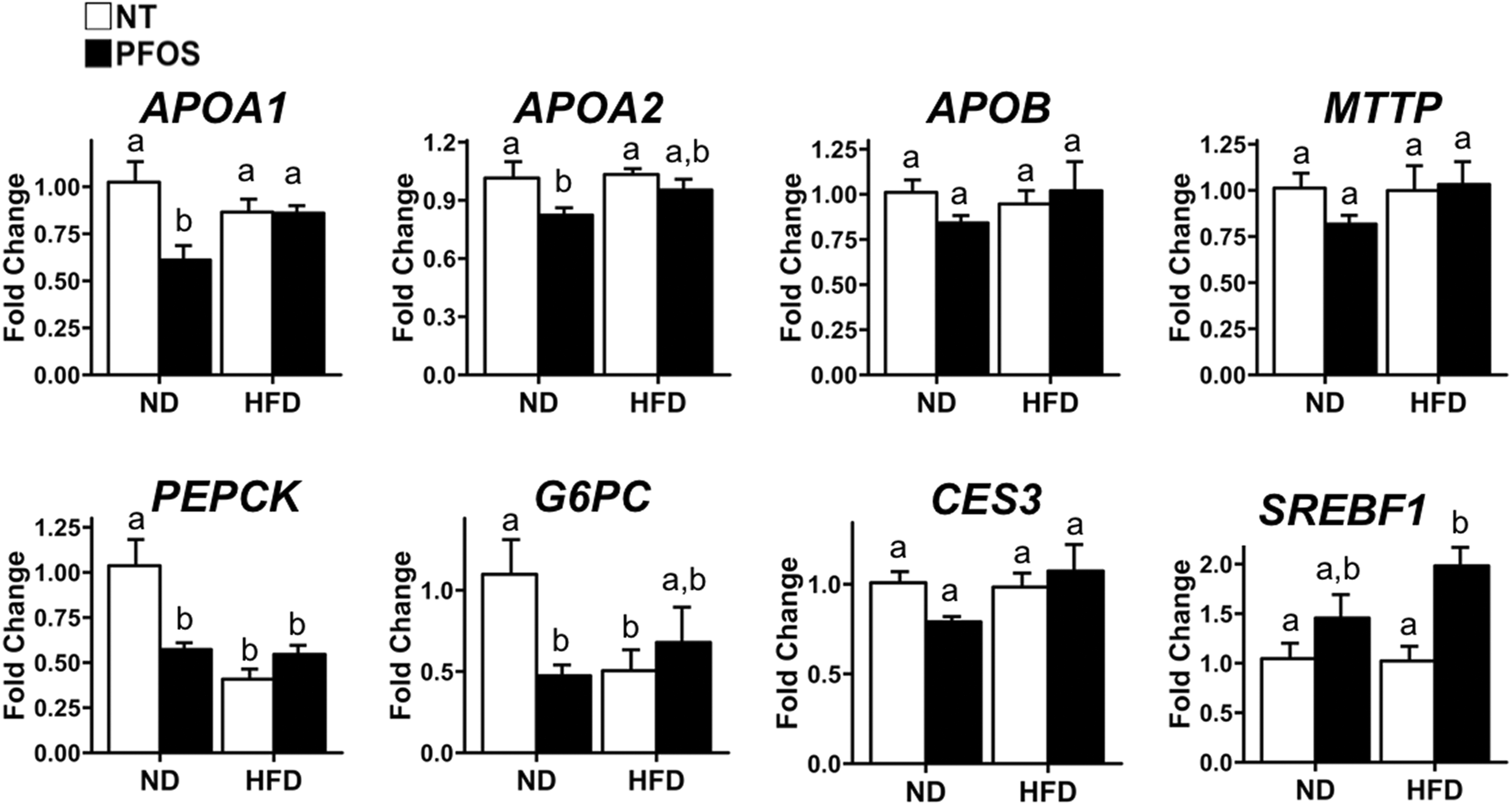
Perfluorooctanesulfonic acid (PFOS) changes expression of metabolism genes in diet-dependent manner. Real-time polymerase chain reaction (PCR) analysis of lipid and carbohydrate metabolism genes. All values are expressed as mean ± standard error of mean. Letters indicate homogenous subsets with P < 0.05.
Protection From PFOS-Induced Hepatic Steatosis Correlates With Changes in Hepatic Lipid Import
Finally, we measured mRNA expression for CD36, the major lipid importer in the cell (Figure 5A). Normal chow diet + PFOS-fed and HFD alone-fed mice had significantly elevated CD36 expression. However, CD36 expression did not change in HFD + PFOS-treated mouse livers. Next, we quantified mRNA of 2 nuclear receptors known to regulate CD36 expression including PPARα and PPARγ (Figure 5A). There was no change in PPARα expression in any groups. Expression of PPARγ exhibited similar pattern as that of CD36. There was a significant increase in PPARγ expression in HFD alone and ND + PFOS groups but no change in HFD + PFOS group.
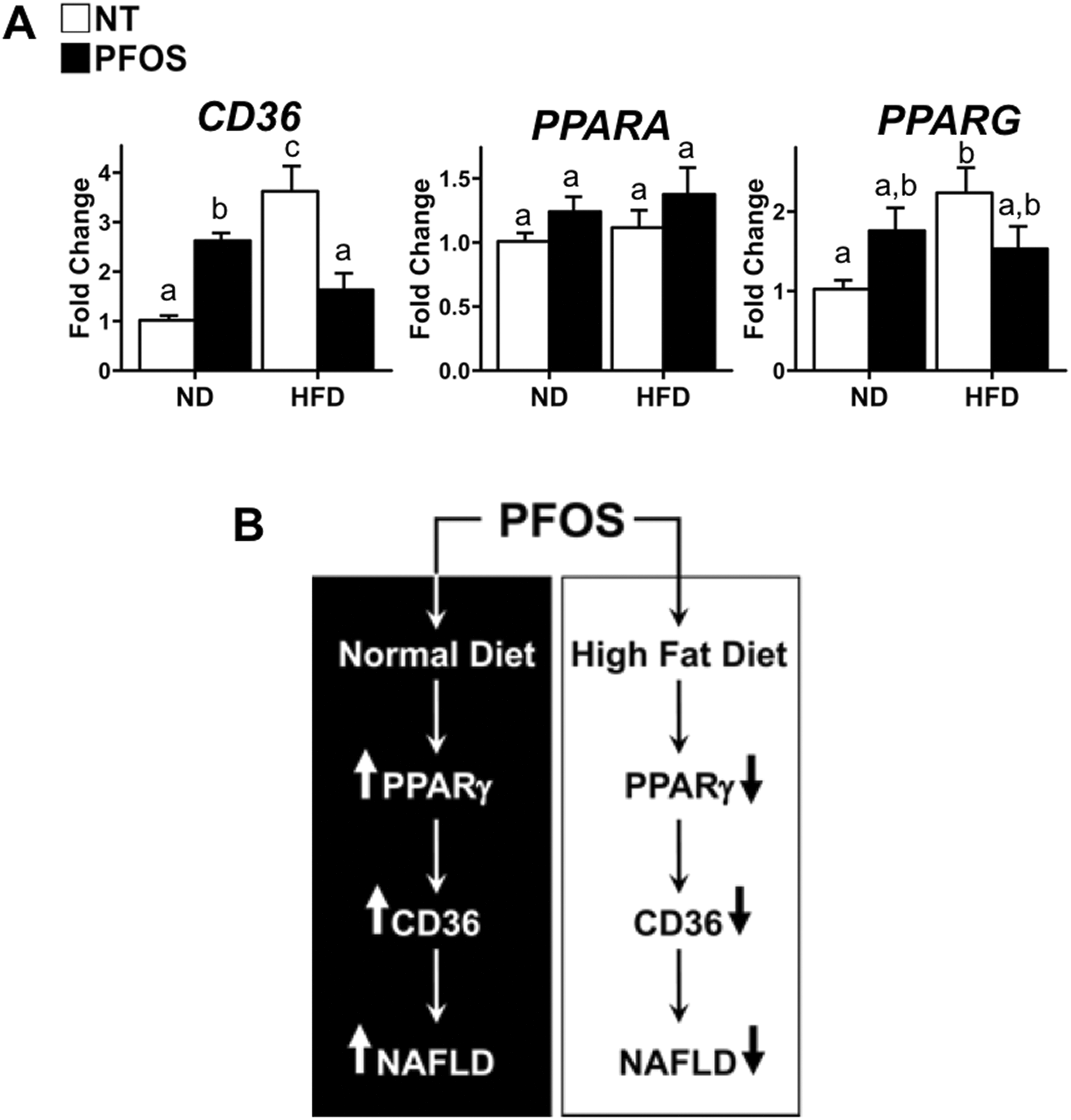
Protection from perfluorooctanesulfonic acid (PFOS)-induced hepatic steatosis correlates with changes in hepatic lipid import. A, Real-time polymerase chain reaction (PCR) analysis of hepatic lipid import transporter CD36 and known PFOS targets PPARα and PPARγ. B, Representative scheme of proposed pathways. In normal chow diet (ND)–fed mice, PFOS activates PPARγ leading to excess hepatic lipid import through CD36 resulting in nonalcoholic fatty liver disease (NAFLD). In high-fat diet (HFD)–fed mice, PFOS suppresses PPARγ leading to decreased hepatic lipid import by decreased expression of CD36 and protection from HFD-induced NAFLD. All values are expressed as mean ± standard error of mean. Letters indicate homogenous subsets with P < 0.05.
Discussion
Perfluorooctanesulfonic acid has been used extensively for industrial and consumer applications 1 and is found in human serum samples worldwide. 2,3 Perfluorooctanesulfonic acid toxicity manifests in the liver as hepatomegaly, hepatic steatosis, and hepatic carcinogenesis in rodents. 6 -8 We hypothesized that PFOS exposure would exacerbate NAFLD-associated HFD consumption. To test this, we used a well-established mouse model of 60% HFD. 22 Male mice were fed normal or HFD with and without PFOS for 6 weeks. The intent of the study was to test exposures of PFOS similar to those observed in humans. Literature survey revealed the highest levels of serum PFOS concentrations in humans with occupational exposures approaching 10 μmol/L PFOS in blood. 16 Previous studies have achieved 100 μmol/L serum PFOS concentrations by feeding mice 0.001% PFOS diet. We reduced this dose by 1 order of magnitude (0.0001% PFOS) to achieve an estimated serum concentration within range of occupational exposure levels. Previous studies have achieved serum PFOS concentrations of 11 µg/mL (∼23 µmol/L) after feeding 0.0001% PFOS for 28 day. 15 Our data indicate that dose of PFOS in ND + PFOS and HFD + PFOS is similar based on the bodyweight and food consumption calculation. Thus, the diet-dependent responses to PFOS in body size, adiposity, hepatic steatosis, and hepatic gene expression cannot be attributed to a difference in the dose of PFOS received between diets.
Perfluorooctanesulfonic acid has been associated with obesogenic effects in humans, 23 and exposure has been positively correlated with weight regain. 24 In our study, PFOS treatment significantly increased body mass and adiposity in ND-fed mice which was not observed when PFOS was treated along with HFD. Consistent with these data, we observed a significant increase in PPARγ mRNA, a known adipogenic nuclear receptor only in mice fed ND + PFOS group. Not only did PFOS fail to induce PPARγ expression when fed with HFD, it reduced expression in HFD group. The mechanism behind this observation is not completely known.
Consistent with previous studies, PFOS treatment in normal diet–fed mice resulted in hepatomegaly, but these effects were lost in HFD-fed conditions. Previous reports have shown that hepatomegaly in PFOS-treated rodents was caused by increased hepatocyte proliferation due to activation of PPARα and constitutive androstane receptor (CAR). 25 However, we observed no hepatocyte proliferation in any of the treatment groups. This suggests the differences in liver size were caused by changes in hepatic lipid content rather than hepatocyte proliferation. The discrepancies in hepatocyte proliferation could be attributed to the much higher PFOS doses used in previous studies, 25 since the lower dose used in this study did not lead to activation of the PPARα gene. Similarly, the perfluoroalkyl acid (PFAA) perfluorooctanoic acid induces PPARα-mediated hepatocyte proliferation at high but not at low doses. 26
The H&E and Oil Red O staining confirmed extensive micro- and macrovescicular steatosis in ND + PFOS and HFD alone groups. Strikingly, HFD + PFOS treatment showed a complete protection from steatosis. These changes seemed to be isolated to the liver, as the diet-dependent effects of PFOS did not directly translate to major changes to serum TG or cholesterol levels. However, we did observe hyperglycemia in ND-fed mice with PFOS treatment. This was not due to increased hepatic gluconeogenesis according to expression of PEPCK and G6PC. Previous studies have reported in utero exposure to PFOS resulting in steatosis, insulin resistance, increased blood glucose, and increased adiposity in the absence of major changes in serum TG or cholesterol. 27 We observed a similar phenotype in ND mice treated with PFOS. Why this does not occur in HFD conditions is of future interest.
We examined hepatic expression of genes involved in lipid metabolism which could be involved in the diet-dependent effects of PFOS. Previous reports have shown that PFOS can cause hepatic steatosis by inhibiting expression of apolipoproteins. 6 We observed decreased expression of ApoA1, ApoA2, and ApoB in ND + PFOS mice. However, there was no change in expression of these genes between treatments in HFD-fed mice. The same pattern was observed for MTTP, a gene important for packaging of lipids into apolipoproteins. Ces3, known to be involved in regulation of hepatic cholesterol metabolism 28 and of major importance in very low-density lipoprotein (VLDL) assembly, 29,30 was inhibited by PFOS in ND but not in HFD. Finally, SREBF1, a major regulator of hepatic lipogenesis, 31 was induced by ND + PFOS group, and HFD + PFOS treatment led to further increase in expression. Together, these results suggest protection from hepatic steatosis in HFD conditions is due to restored lipid flux rather than induction of lipid synthesis.
It has previously been reported that PFOS can activate PPARα. 25,32,33 In this study, we did not observe significant activation of PPARα at the mRNA or target gene level (ACOX1, PDK4, CPT1, and data not shown) in any of our treatment groups, which could be due to the single low-dose used. However, the hepatotoxic effects of PFOS can also occur in a PPARα-independent manner, 34,35 and there is evidence that PFOS activates other nuclear receptors. 36,37 Our previous work has suggested PFAAs can activate PPARγ 16 and has been further supported by transcriptional profiling studies. 38 We observed increased PPARγ mRNA expression following PFOS treatment in ND-fed mice but a decrease in expression following PFOS treatment in HFD-fed mice. Hepatocyte-specific deletion of PPARγ protects mice from HFD-induced steatosis and results in downregulation of CD36, MTTP, and PEPCK. 39 This is consistent with our observations and suggests activated PPARγ may be contributing to the steatosis observed in ND-fed mice treated with PFOS. Furthermore, CD36, the major hepatocyte lipid importer, was induced by PFOS in ND-fed mice but was suppressed to ND + NT levels by PFOS in HFD-fed mice suggesting hepatic lipid import is accelerated by PFOS in ND conditions and inhibited in HFD conditions. CD36 is a known target of PPARγ. 40 Previous reports have also found induction of CD36 following PFOS exposure. 41 CD36 is induced in livers with severe steatosis 42 and hepatocyte-specific deletion of CD36 can attenuate HFD-induced steatosis. 43
The schematic presented in Figure 4B represents our current understanding of the diet-dependent effects of PFOS on hepatic steatosis. It is not known at this time why PFOS could lead to activation of PPARγ and CD36 in ND conditions but suppress expression of these genes in HFD conditions. Potential mechanisms include a direct interaction between PPARγ or other transcription factors known to respond to PFOS exposure such as hepatocyte nuclear factor 4 alpha (HNF4α), pregnane X receptor (PXR), or CAR 16,36 and is further complicated when the activity of these transcription factors is dependent on metabolic conditions.
Together, these data report human-relevant exposures of PFOS offering protection against hepatic steatosis in a diet-dependent manner likely by affecting hepatic lipid import. These results impact our understanding of PFOS toxicity and other perfluorinated alkyls by recognizing the role of nutritional status when determining mechanism of toxicity. Finally, in combination with existing literature, these results suggest PFOS simultaneously activates multiple pathways of adverse outcome, and the interplay between these pathways and environmental conditions determines final outcome.
Footnotes
Authors’ Contributions
Ian Huck contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted manuscript, and critically revised manuscript; Kevin Beggs contributed to conception and design, contributed to acquisition and interpretation, and critically revised manuscript; Udayan Apte contributed to conception and design, contributed to interpretation, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: These studies were supported by NIH-COBRE (P20 RR021940-03, P30 GM118247), NIEHS Toxicology Training Grant (T32ES007079-34) and NIH R01DK 0198414.