
Abstract
This research provides a cautionary example when evaluating changes in behavioral end points with respect to postulated pharmacologic activity. Various small molecule substrate mimetic protein tyrosine phosphatase 1B (PTP1B) inhibitors were investigated as pharmacologic agents for decreasing food consumption using intranasal (IN) dosing as a means for direct nose-to-brain delivery along the olfactory/trigeminal nerve pathways. Although food consumption was decreased in diet-induced obese (DIO) mice, nasal discharge was observed. Studies were conducted to investigate local effects on the nasal airway and to develop structure–activity relationships. Intranasal administration of PTP1B inhibitors at ≥0.03 mg/d to DIO mice produced dose-dependent injury to various cell types of the nasal epithelia. Protein tyrosine phosphatase 1B inhibitors with calculated log octanol >3.0 were the most toxic. Whereas a pharmacologically inactive analog of a PTP1B inhibitor produced nasal injury, along with decreased food consumption, the marketed IN drug ketorolac produced no lesions at the same dose of 0.3 mg/d and only minor changes at 3 mg/d. Rat skin fibroblast cells were exposed in vitro to PTP1B inhibitors, ketorolac, paraquat, and the detergent sodium dodecylbenzene sulfonate (NDS) followed by measures of cytotoxicity. The most potent PTP1B inhibitors were similar to NDS, whereas ketorolac was the least toxic compound. Cytotoxic potency in vitro was similar to in vivo. In conclusion, PTP1B inhibitors injured nasal epithelium through a mechanism independent of PTP1B inhibition and likely due to nonspecific cytotoxicity such as disruption of the cell membrane. Decreased food consumption in DIO mice was due to toxicity rather than a pharmacologic mode of action.
Introduction
Obesity is a disease affecting more than one third of the adult population in the United States and constitutes a major risk factor for morbidity and mortality due to cardiovascular disease, diabetes, and cancer. Although several US Food and Drug Administration (FDA)–approved pharmacologic therapies are available for treating obesity, they have limited efficacy and significant liabilities associated with their use. 1,2 Significant weight loss can be achieved with gastric surgery; however, this is not an option available to large numbers of patients. Thus, there is a significant need for safe and more effective drugs for obese patients.
Leptin is a hormone produced by adipose tissue that is present in serum with circulating levels that correlate with total body fat content. Leptin acts on the leptin receptor in the arcuate nucleus of the hypothalamus to decrease feeding behavior by stimulating anorexigenic signals mediated by proopiomelanocortin neurons and inhibiting orexigenic signals by neuropeptide Y and agouti-related protein neurons. Insulin acts in a similar fashion as leptin to elicit anorectic signals in the hypothalamus during positive nutrient conditions. Although serum levels of leptin are elevated in obese states, anorectic responses to leptin signaling are diminished, resulting in prolonged orexigenic/feeding behavior. 3,4
Tyrosine phosphorylation and dephosphorylation of proteins is a major component in signal transduction pathways that regulate various cellular processes in growth, differentiation, and metabolism. Protein tyrosine kinases and phosphatases control the level of tyrosine phosphorylation and dephosphorylation, respectively. Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of leptin signaling in the hypothalamus by dephosphorylating proteins downstream from the leptin receptor. 5,6 Protein tyrosine phosphatase 1B–deficient mice show enhanced insulin sensitivity and increased phosphorylation of the insulin receptor in liver and muscle tissue after insulin injection. On a high-fat diet, both homozygous and heterozygous knockout mice were resistant to weight gain and remained insulin sensitive, whereas wild-type mice rapidly gained weight and became insulin resistant. 7 Homozygous PTP1B null mice are protected against diet-induced obesity due to decreased adipose tissue, increased basal metabolic rate, and total energy expenditure. 8 Mice lacking neuronal PTP1B are resistant to high-fat diet-induced obesity and are protected from leptin resistance. 9 Protein tyrosine phosphatase 1B is expressed in hypothalamic regions containing leptin-responsive neurons and regulates leptin signaling by dephosphorylating the leptin receptor–associated kinase, Jak2 (Janus kinase 2). 10,11 These studies suggest that a major physiological effect of PTP1B is to act as a negative regulator for both the insulin and leptin signaling pathways; therefore, PTP1B could be a target for treating diabetes and/or obesity. 12 Additional research suggests that pharmacologic inhibition of PTP1B in the brain could be a target for the treatment of obesity and/or diabetes. 9
A research program was initiated at Novartis, Cambridge, MA to evaluate whether small molecule inhibitors of PTP1B could be identified which reverse leptin resistance as a pharmacologic strategy for treating obesity. The intranasal (IN) route of administration was investigated as a means for drug delivery to the hypothalamus, whereby systemic exposure may be minimized while still achieving sufficient central nervous system (CNS) exposure. 13 The IN administration may provide a means for delivering therapeutic compounds directly to the CNS, bypassing the blood–brain barrier, by transiting along olfactory- and trigeminal-associated extracellular neuronal pathways. 13 Olfactory neurons are unique in that they interface directly with the nasal airway surface in the olfactory epithelium. These neurons then travel through the cribriform plate into the brain where they terminate on dendrites of olfactory bulb neurons, which project to deeper structures. The nasal respiratory epithelium is innervated by branches of the trigeminal nerve, or fifth cranial nerve, which may provide another conduit for molecules to reach the CNS. Compounds with poor oral bioavailability and/or low blood to brain distribution may be bioavailable to the CNS via IN delivery, whereby direct absorption into the nasal vasculature and/or distribution along extraneuronal pathways may lead to sufficient CNS exposure. 13
Initial studies with specific PTP1B inhibitors in leptin-resistant diet-induced obese (DIO) C57BL/6 mice demonstrated decreased food consumption and body weight loss following IN delivery, but not following subcutaneous dosing despite higher systemic drug exposures. Increased levels of phosphorylated STAT3 (signal transducer and activator of transcription 3), a target for PTP1B dephosphorylation, were observed in the hypothalamus following IN delivery, suggesting pharmacologic activity. However, despite these initial promising results, nasal discharge was observed shortly after IN dosing of a PTP1B inhibitor. This observation prompted the design of combination pharmacology–toxicology studies to conduct anatomic evaluation of the nasal airway. A series of studies were conducted in DIO mice, along with in vitro cell culture assays, to examine structure–activity relationships (SARs) for the PTP1B inhibitors, evaluate in vivo to in vitro correlates, and identify a means for screening compounds for nasal toxicity.
Materials and Methods
Experimental Compounds
Substrate mimetic PTP1B inhibitors (compounds A-G) and a pharmacologically inactive control (compound H) were prepared at Novartis. Ketorolac tromethamine and paraquat were obtained from the Novartis compound archive. Sodium dodecylbenzene sulfonate (NDS) was obtained from Aldrich, St. Louis, MO. The structures of compound G (which is representative of various PTP1B inhibitors), ketorolac tromethamine, paraquat, and NDS are presented in Figure 1. Calculated log octanol (cLogP) was obtained using the Moka program in FOCUS software (Molecular Discovery, Pinner, Middlesex, United Kingdom).
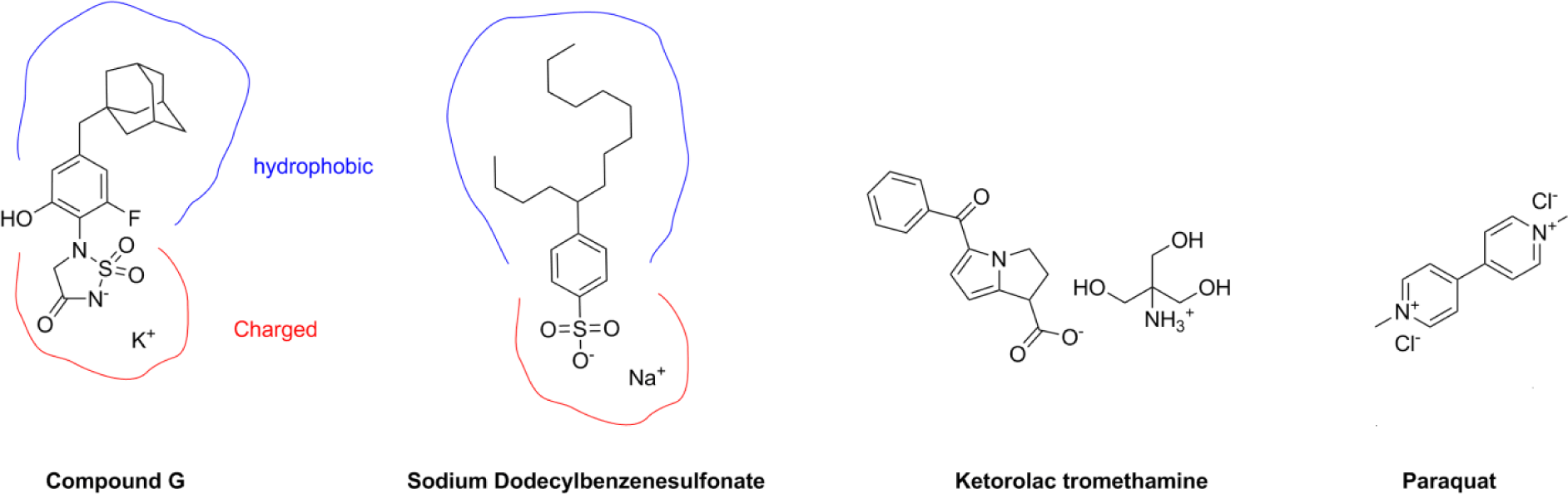
Chemical structures of compound G, ketorolac tromethamine, paraquat, and sodium dodecylbenzene sulfonate (NDS).
Animals and Dosing Procedures
Male C57BL/6 mice were obtained from Taconic Biosciences, Germantown, NY and fed ad libitum a high-fat diet (diet D12492i from Research Diets Inc, New Brunswick, New Jersey) from 6 weeks of age to become obese (DIO mice). Animals used in the studies were approximately 15 weeks old (20 weeks in the final study conducted). Animals were housed 1 per cage in a reverse light cycle room (dark cycle from 11:00 to 23:00) in an AAALAC accredited facility. All animals were anesthetized with isoflurane, placed on their backs, and dosed IN with a pipette at a total dose volume of 25 µL distributed between both naris (approximately 12.5 µL/nare). Animals remained anesthetized in a dorsal position for 3 minutes prior to being returned to their home cages. All procedures utilized were in compliance with the Novartis Institutional Animal Care and Use Committee.
Animal Studies
Following initial reports of nasal discharge occurring immediately postnasal dosing of a PTP1B inhibitor (compound A), an initial acute study was conducted in which compound A (both sodium and potassium salts) was administered as a single dose of 1.5 mg (60 mg/mL) or 2 doses of 0.3 mg/dose (12 mg/mL) 24 hours apart. Vehicle control (water alone) and untreated naive control groups were included, and all animals (n = 4/group) were necropsied immediately following the last dose for that group, with microscopic evaluation of nasal airways conducted on all animals.
A repeat dose study was next conducted in which compounds A and B were administered twice per week for 2 weeks at doses of 0.03 and 0.1 mg/dose (drug concentrations of 1.2 and 4 mg/mL, respectively) with a vehicle (water) control included (n = 8/group). Food consumption was measured 24 hours after each dose, and body weight was measured on dosing days. Animals were euthanized and necropsied 24 hours following the last dose and microscopic evaluation of nasal airways conducted on n = 4/group.
A study was conducted to examine whether a SAR exists across the PTP1B inhibitors evaluated for injury to the nasal epithelium. Animals (n = 4/group) received 2 doses of compounds B, C, D, E, F, or G at 0.3 mg/dose (drug concentrations of 12 mg/mL) approximately 24 hours apart. A vehicle control (phosphate-buffered saline [PBS]) was included. All animals were necropsied and had nasal airways collected for microscopic evaluation at 4 hours after the second dose. Phosphate-buffered saline was used as the vehicle to minimize any effects of the vehicle alone on nasal epithelium. The pH and osmolality of the dosing solutions were measured and are presented with PTP1B 50% inhibitory concentration (IC50) and compound cLogP in Table 1. Individual animal food consumption was measured 24 hours following the first dose.
In Vivo Structure–Activity Relationship Study.a
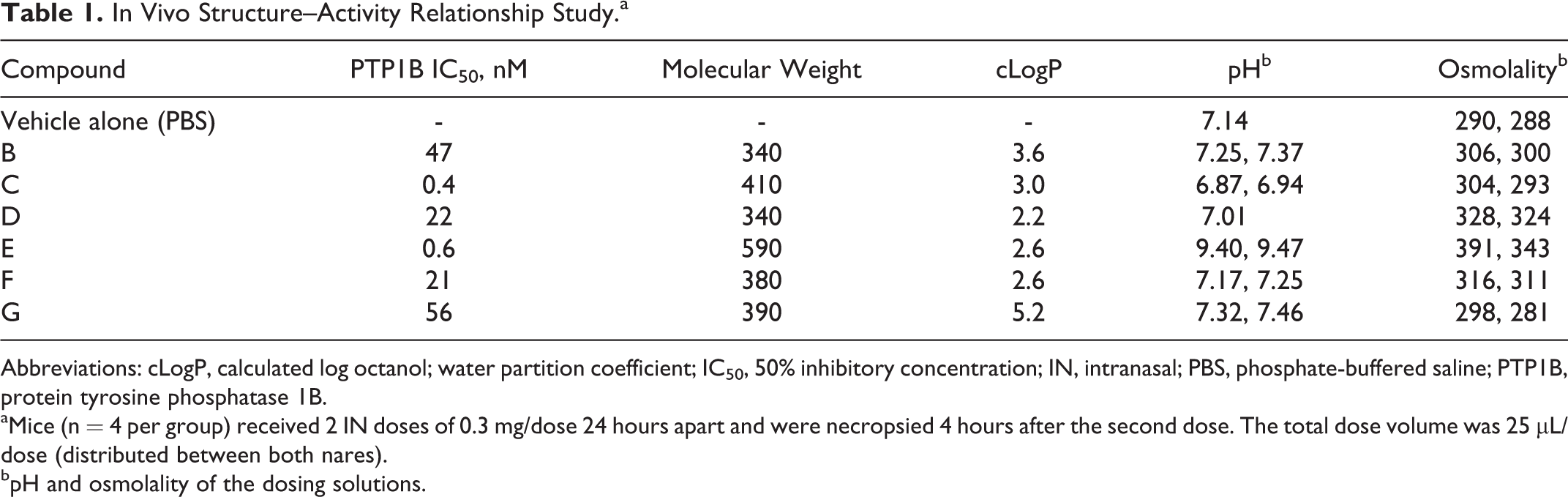
Abbreviations: cLogP, calculated log octanol; water partition coefficient; IC50, 50% inhibitory concentration; IN, intranasal; PBS, phosphate-buffered saline; PTP1B, protein tyrosine phosphatase 1B.
aMice (n = 4 per group) received 2 IN doses of 0.3 mg/dose 24 hours apart and were necropsied 4 hours after the second dose. The total dose volume was 25 µL/dose (distributed between both nares).
bpH and osmolality of the dosing solutions.
The final animal study compared the responses in the nasal airway following administration of compound A (PTP1B IC50 of 0.11 µM; cLogP = 2.6) versus a pharmacologically inactive analog, compound H (PTP1B IC50 of 213 µM; cLogP = 3.6), and to a FDA-approved and marketed IN drug, ketorolac tromethamine. Animals (n = 4/group) received 2 doses (approximately 24 hours apart) of compounds A and H at 0.3 mg/dose (drug concentrations of 12 mg/mL) and 2 doses of ketorolac tromethamine at 0.3 and 3 mg/dose (drug concentrations of 12 and 120 mg/mL, respectively). A vehicle control (PBS) was included. All animals were necropsied 4 hours after the second dose and had tissues collected for microscopic evaluation of the nasal airways. Individual animal food consumption was measured at 24 hours following the first dose.
Microscopic Evaluations
All animals were euthanized with CO2 and necropsied. Skulls were collected and fixed in 10% neutral-buffered formalin and decalcified, and 3 standard sections 14 were made for microscopic evaluation of the nasal airways that included sections from (1) immediately posterior to the incisors, (2) at the level of the incisive papilla, and (3) through the middle of the second molar. Tissue sections were stained with hematoxylin and eosin and evaluated by a veterinary pathologist (C.S.) using a 5-point scoring system, which included (1) minimal = 1 or few small foci of histopathologic change, (2) mild = small- to medium-sized foci, (3) moderate = frequent and moderate-sized foci, (4) marked = extensive confluent foci, and (5) severe = histopathologic changes in the majority of the tissue section.
In Vitro Cytotoxicity Assay
Rat skin fibroblast cells (ATCC, Manassas, Virginia; catalog # CRL1213) were plated at 15,000 cells per well in 200 µL culture medium in 96-well clear-bottom black-walled collagen I coated plates (BD Bioscience, San Jose, CA). This cell line was chosen based on prior laboratory experience, desire to avoid primary cells, and hypothesis that the compounds produce nonspecific cytotoxicity. Cells were cultured at 37°C/5% CO2 in Dulbecco's Modified Eagle's Medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Invitrogen) and 1% penicillin/streptomycin (Invitrogen) and used at passages 10 or 11. Two independent experiments were conducted to confirm reproducibility. All experiments consisted of 9- or 10-point dose–response curves using a 2-fold dilution series, with test compound concentrations ranging from 0.039 to 20 mM (or 25 mM for paraquat). Test compounds were prepared on the day of use and delivered to the cells in culture medium, followed by incubation for 24 hours. Triplicate wells were prepared for each concentration of a compound. Because compounds A and B produced nasal epithelial injury at dosage concentrations of 4 mM, the concentrations selected for the in vitro assay bracketed this concentration. After incubation, medium was collected and analyzed for lactate dehydrogenase (LDH) content. Cell viability was measured after an 1-hour incubation with CellTiter-Blue reagent (Promega, Madison, WI). For the second experiment, microscopic images were taken of representative wells for each dilution of all compounds prior to performing cytotoxicity assessments.
The CellTiter-Blue assay was performed as described in the manufacturer’s instructions. All reagents were equilibrated to room temperature (RT) before use. After removal of the supernatant for the LDH assay, 100 µL of culture medium and 20 µL of CellTiter-Blue reagent were added to all sample wells, and plates were shaken for 10 seconds at RT. The plates were then incubated at 37°C/5% CO2 for 1 hour. Supernatants (100 µL per well) were collected and transferred to a 96-well black plate (Costar, No. 3915, Corning, NY), and the plate was shaken for 10 seconds. Fluorescence was measured using a SpectraMax M5 plate reader at 560/590 nm, top read.
Lactate dehydrogenase activity was measured as described in the manufacturer’s instructions (Roche, Indianapolis, IN). All reagents and samples were equilibrated to RT prior to performing the assay. Sample and LDH reaction mix (50 µL of each) were added to 96-well clear plates (Costar, No. 9017, Corning, NY) and shaken for 15 minutes at RT and protected from light. Absorbance was measured using a SpectraMax M5 plate reader at 490 nm. For total LDH content, 2% Triton X-100 was prepared in medium and added directly to the cells to induce lysis.
Data Analysis
Cytotoxicity data were analyzed using an Excel template for each end point. Average values of empty background wells were subtracted from values of all sample and control wells. For CellTiter-Blue, data were normalized to the average of control (untreated) values and multiplied by 100%. For LDH, the average released LDH from untreated cells was subtracted from the net data. A final normalization to the average LDH released from lysed cells was performed with this value being multiplied by 100%. The individual values of percentage of control from both experiments were graphed using GraphPad Prism 6 (version 6.02) software, and best fit curves and 50% toxic concentration (TC50) values determined using a 4-parameter nonlinear curve fit for both end points. Food consumption was determined by subtracting food weight 24 hours postdose from the weight prior to the dose. One-way analysis of variance with Dunnett test was performed on the food consumption data with statistical significance set at P < 0.05.
Results
Animal Studies
Following reports of nasal discharge observed in mice immediately after IN dosing of compound A, an initial investigative study was conducted in DIO mice in which compound A (in water vehicle) was administered either as a single 1.5 mg dose or 2 daily doses at 0.3 mg/dose. Compound A produced a dose-dependent (≥0.3 mg/dose) increase in the incidence and severity of necrosis affecting all types of nasal epithelia (olfactory, respiratory, transitional, and squamous epithelium). The changes observed were independent of the salt form (potassium or sodium) administered and occurred immediately following a single dose at 1.5 mg. Although water alone produced injury to the nasal epithelium (primarily olfactory), likely due to hypotonic stress, compound A produced epithelial injury to a much greater extent.
A second study was conducted to investigate whether 2 different PTP1B inhibitors, compounds A and B, when administered at lower doses, would injure the nasal epithelia. Compounds A and B (both potassium salts in water vehicle) were dosed twice a week for 2 weeks at 0.03 or 0.1 mg/dose. Microscopic changes were not observed in the mice dosed with water alone. Both compounds injured the nasal epithelium, with incidence and severity of the lesions increased in a dose-dependent fashion. In the olfactory epithelium, atrophy, single cell necrosis, eosinophilic globules, and respiratory metaplasia were seen. Degeneration/regeneration and eosinophilic globules of respiratory epithelium, squamous metaplasia in respiratory and transitional epithelium, and IN exudates were also observed. The incidence and severity of the lesions were greater for compound B (cLogP = 3.6) than for compound A (cLogP = 2.5). Food consumption was significantly reduced 24 hours after the first dose of either compound A or B at 0.1 mg (decreased 36% and 53%, respectively), but concurrent injury to the nasal epithelia introduced a confounding factor for this pharmacodynamic measure.
Based upon the results of the first 2 studies in DIO mice, it became apparent that IN administration of compounds A or B results in injury to the 4 major cell types of the nasal epithelium in a dose-dependent fashion. Injury occurred very rapidly, which suggested that disruption of cell membranes leading to rapid cell lysis was a potential mechanism. Compound B is more hydrophobic than compound A, suggesting that physical–chemical properties such as hydrophobicity may be a factor in this toxicity. A study was conducted in which 6 different PTP1B inhibitors of varying hydrophobicity (based on cLogP) and chemical structure on a common scaffold were administered to DIO mice. Animals received 2 doses of 0.3 mg/dose at approximately 24 hours apart and were necropsied 4 hours following the second dose (at which time acute injury was expected to be present). The compounds (Table 1) were dissolved in PBS vehicle at a pH and osmolality intended to minimize the potential for vehicle-induced changes to the nasal epithelium (ie, pH 3-10 and 200-400 mOsm/kg). 15,16 Microscopic lesions were not observed with PBS vehicle alone. Single-cell necrosis of the olfactory epithelium was produced by all compounds, with the greatest severity produced by compound B (Figure 2). One or more of the following changes were produced in the respiratory (Figure 3) and transitional (Figure 4) epithelium by all of the compounds: necrosis, degeneration and regeneration, or subepithelial neutrophilic infiltration. Segmental full-thickness necrosis of the respiratory and transitional epithelium was only observed with compounds B and G. Squamous epithelial necrosis was produced by compound B. Compounds C, D, E, and F produced injury to the nasal epithelium to a similar degree, with incidence and severity less than compounds B and G. Based on the microscopic changes in the nasal epithelium, the PTP1B inhibitors were rank-ordered based on toxicity with B > G > C, D, E, F. In general, compounds with cLogP > 3.0 were the most toxic. Twenty-four-hour food consumption was significantly decreased 50% to 85% by the compounds in comparison with PBS control; however, this effect was likely due to injury to the nasal airway, including effects on olfaction, rather than pharmacological activity such as PTP1B inhibition in the hypothalamus.
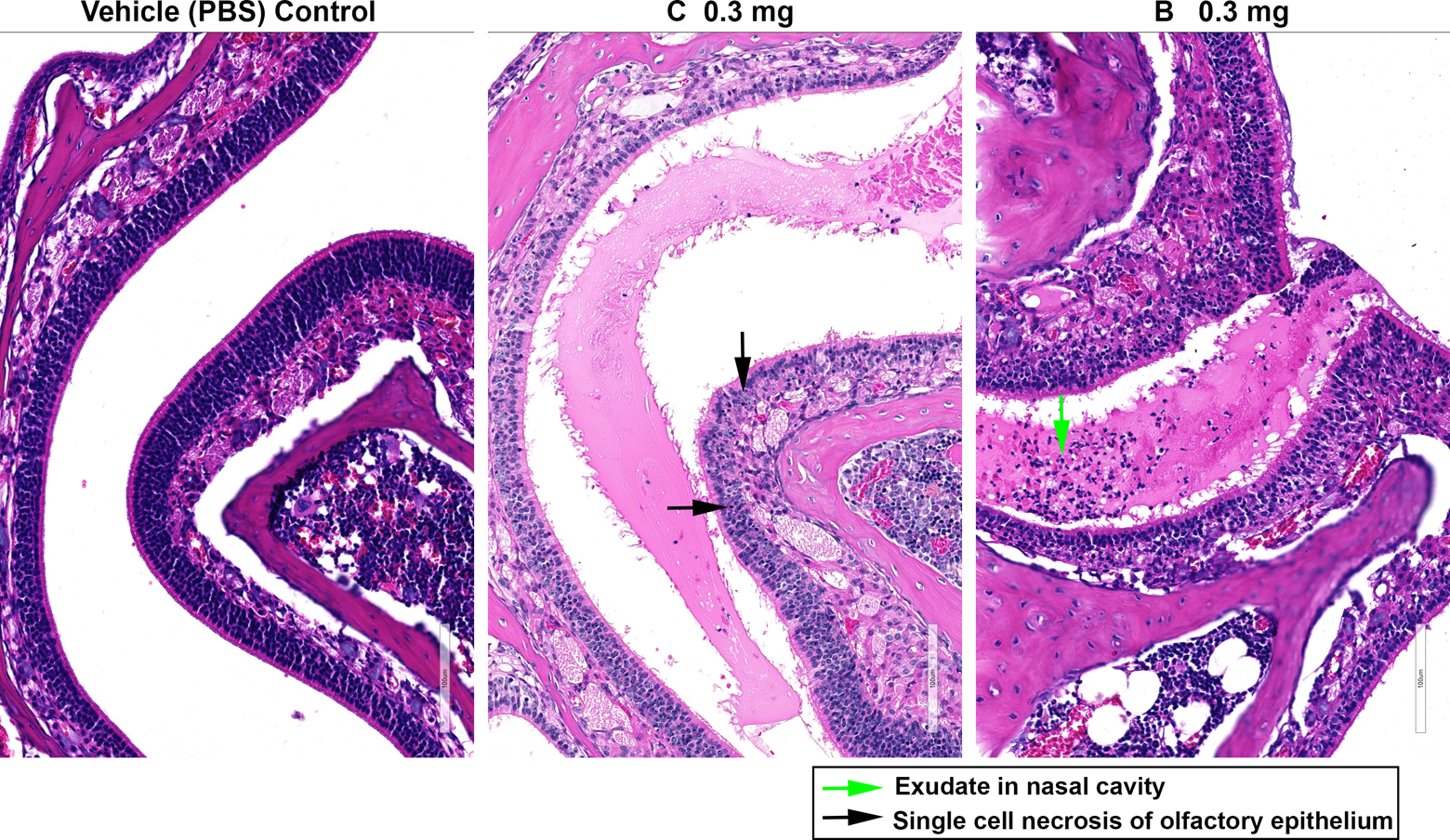
Changes to olfactory epithelium produced by protein tyrosine phosphatase 1B (PTP1B) inhibitors. Single-cell necrosis of the olfactory epithelium and exudate in nasal cavity were produced in mice by compounds C and B following intranasal (IN) administration (0.3 mg/dose). Compound B elicited a more severe response compared to compound C. No changes were evident following IN administration of phosphate-buffered saline (PBS) vehicle alone.
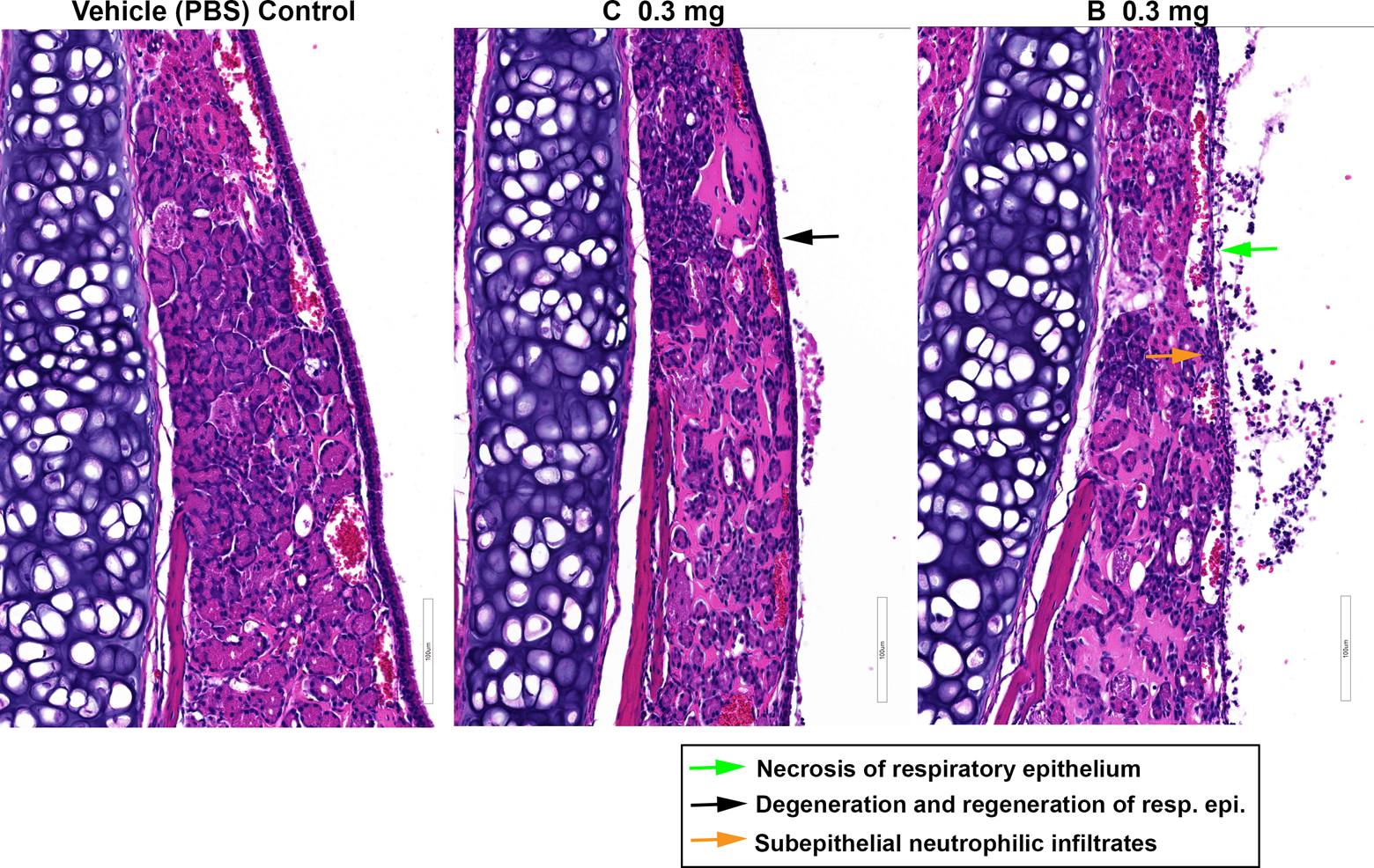
Changes to nasal respiratory epithelium produced by protein tyrosine phosphatase 1B (PTP1B) inhibitors. Necrosis, degeneration, and regeneration of the nasal respiratory epithelium were produced in mice by compounds C and B following intranasal (IN) administration (0.3 mg/dose). Compound B elicited a more severe response compared to compound C. No changes were evident following IN administration of phosphate-buffered saline (PBS) vehicle alone.
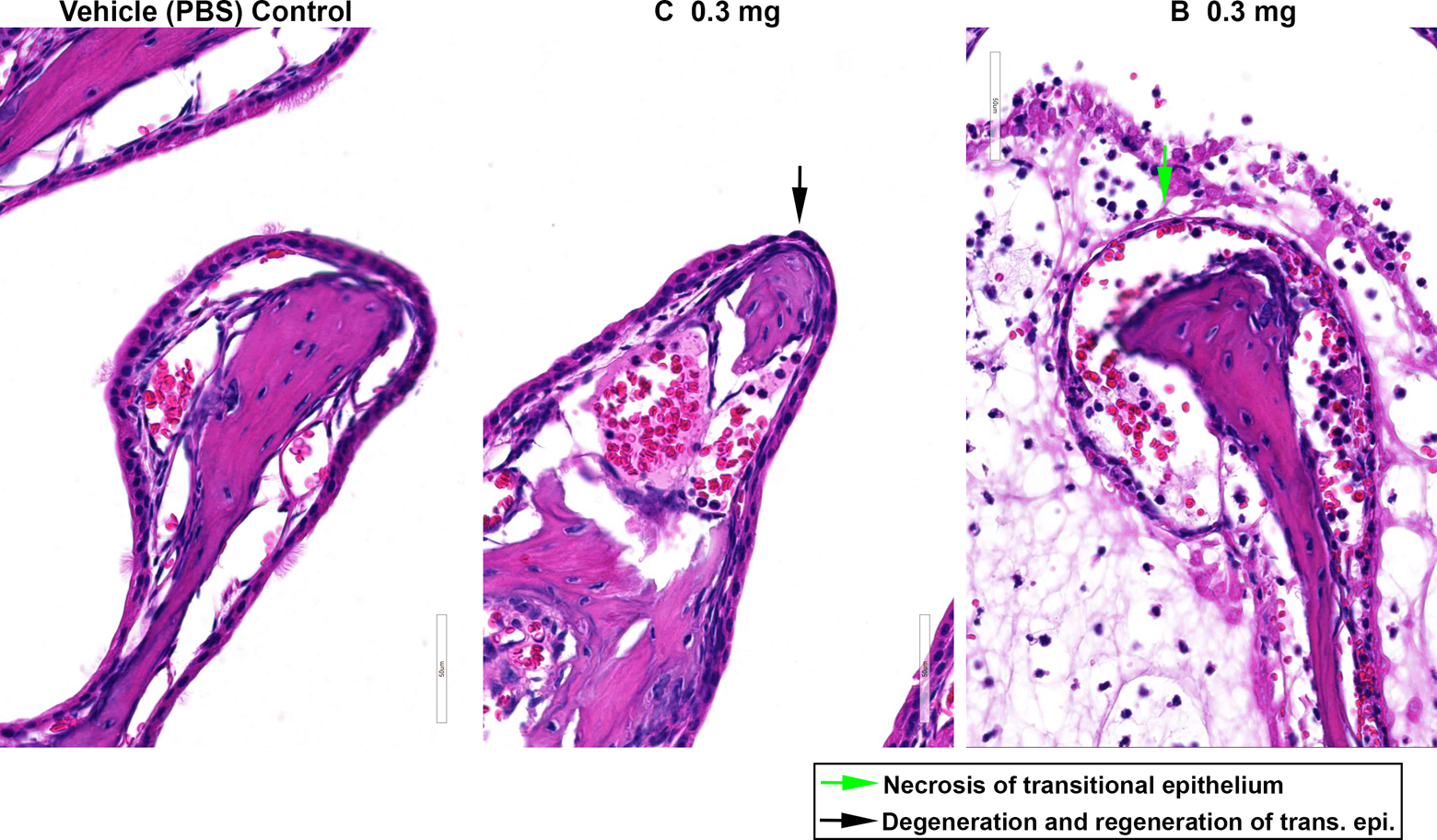
Changes to nasal transitional epithelium produced by protein tyrosine phosphatase 1B (PTP1B) inhibitors. Necrosis, degeneration, and regeneration of the nasal transitional epithelium were produced in mice by compounds C and B following intranasal (IN) administration (0.3 mg/dose). Compound B elicited a more severe response compared to compound C. No changes were evident following IN administration of phosphate-buffered saline (PBS) vehicle alone.
The final animal study compared nasal epithelial changes produced by compound A (PTP1B IC50 = 0.11 µM) versus a pharmacologically inactive structural analog (compound H; PTP1B IC50 = 213 µM) to investigate whether toxicity was due to the chemical scaffold or due to PTP1B inhibition (ie, on-target effects). In addition, the FDA-approved IN drug ketorolac tromethamine (a nonsteroidal anti-inflammatory drug) was included in the study for comparison (Table 2, Figure 5). One or more of the following changes were present in the transitional and respiratory epithelium in all mice dosed with compound A: acute necrosis, erosion/ulceration, mucosal and submucosal neutrophilic infiltration, and degeneration/regeneration accompanied by IN neutrophilic exudates, consistent with the prior studies. In addition, compound A produced mild, acute, olfactory epithelial necrosis. The severity and incidence of these changes were comparable between compounds A and H (PTP1B active and inactive analogs, respectively), as indicated in Table 2. Ketorolac tromethamine did not produce any nasal epithelial changes at 0.3 mg/dose, but transitional and respiratory epithelial changes were present at 3 mg/dose. The incidence and severity of nasal epithelial changes with ketorolac tromethamine at 3 mg/dose were low compared to compounds A or H at 0.3 mg/dose (Table 2). Following the first dose, 24-hour food consumption was significantly reduced 70% and 54% by compounds A and H at 0.3 mg/dose, respectively, and 79% by ketorolac tromethamine at 3 mg/dose (but not at the lower dose). These data provide further evidence that the food consumption changes initially observed with the PTP1B inhibitors were due to nasal toxicity rather than pharmacological activity.
Microscopic Changes in Nasal Epithelium: Incidence and Severity Scores.a
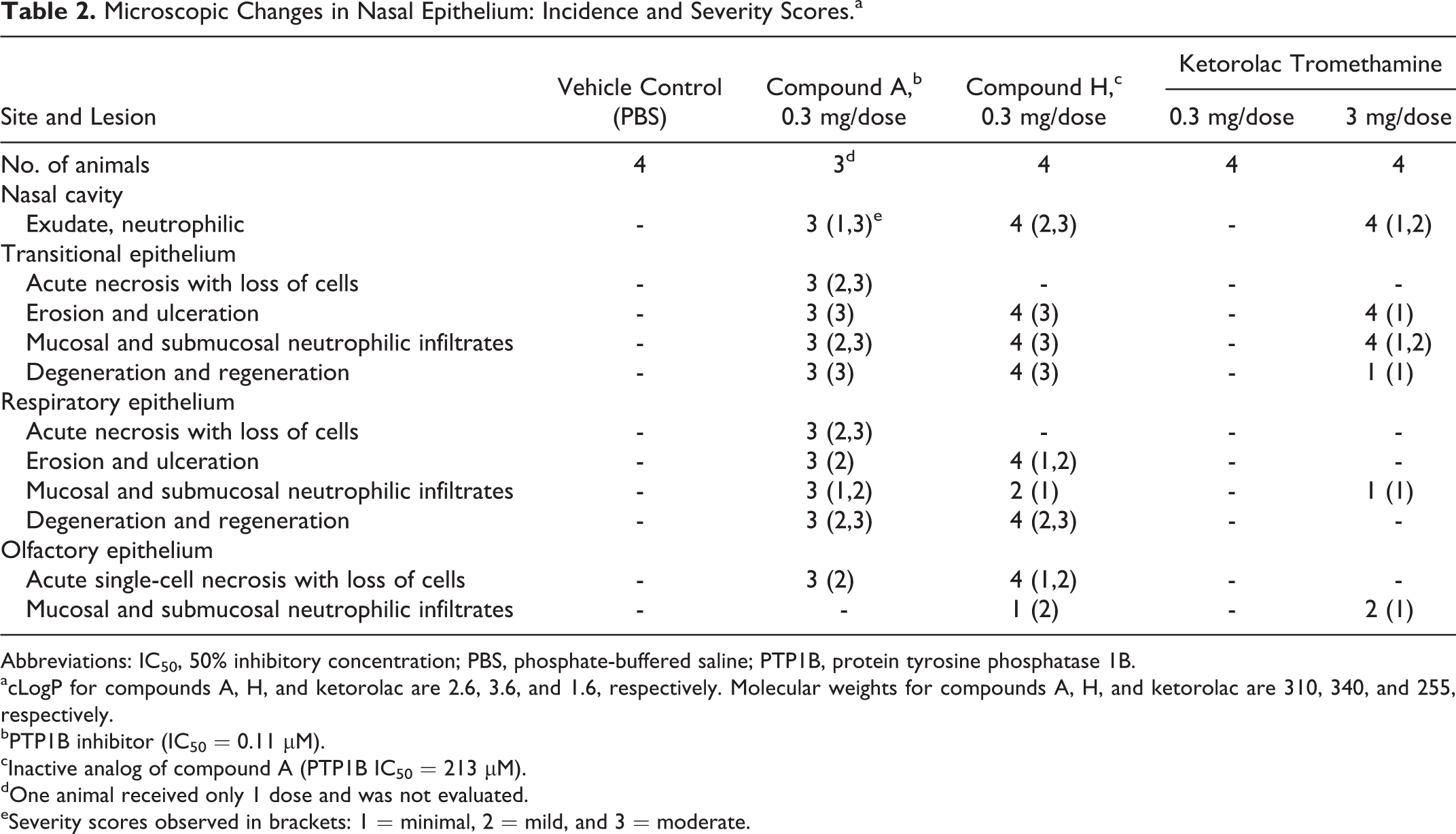
Abbreviations: IC50, 50% inhibitory concentration; PBS, phosphate-buffered saline; PTP1B, protein tyrosine phosphatase 1B.
acLogP for compounds A, H, and ketorolac are 2.6, 3.6, and 1.6, respectively. Molecular weights for compounds A, H, and ketorolac are 310, 340, and 255, respectively.
bPTP1B inhibitor (IC50 = 0.11 µM).
cInactive analog of compound A (PTP1B IC50 = 213 µM).
dOne animal received only 1 dose and was not evaluated.
eSeverity scores observed in brackets: 1 = minimal, 2 = mild, and 3 = moderate.
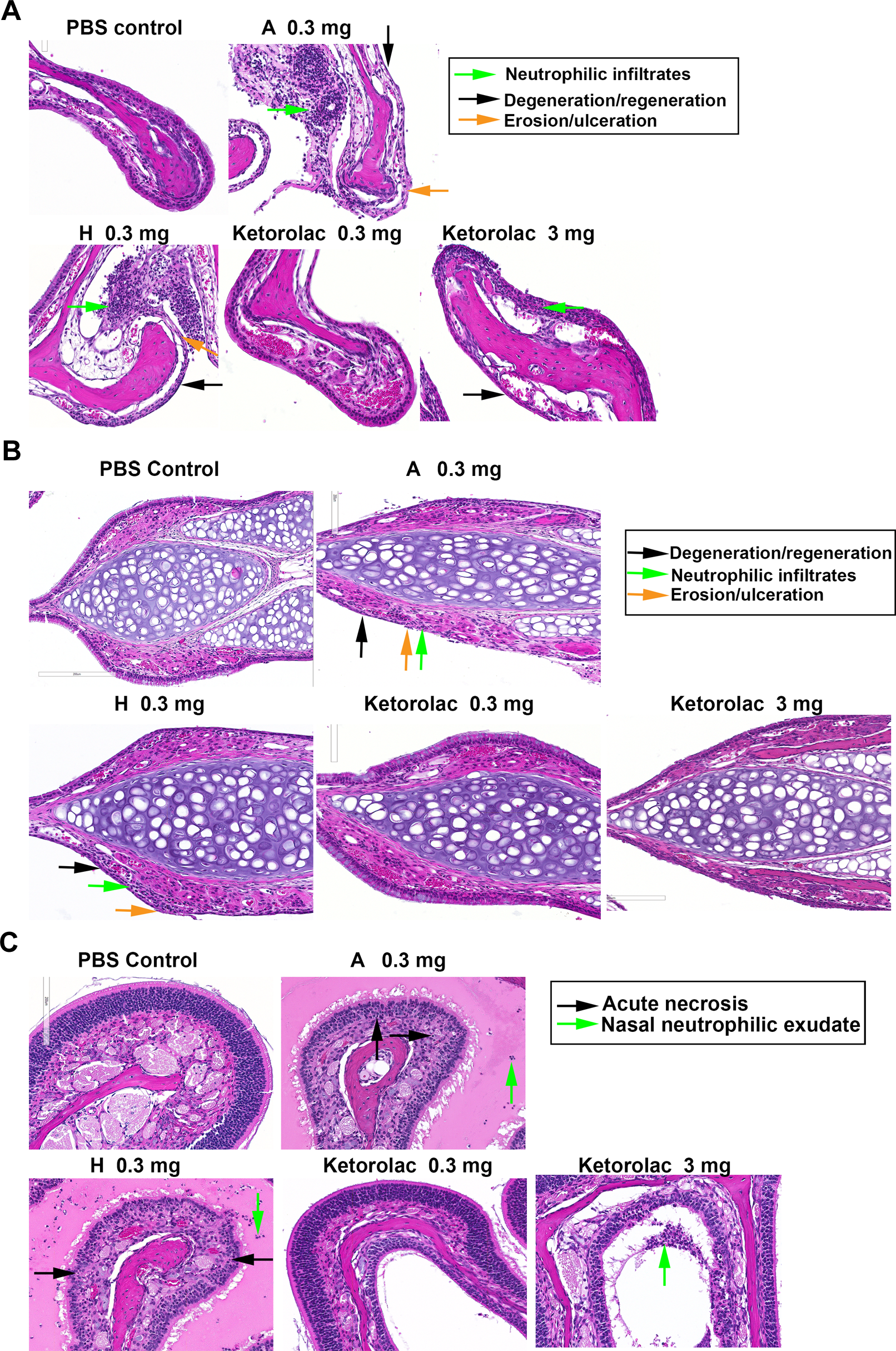
Comparison of microscopic changes produced by the protein tyrosine phosphatase 1B (PTP1B) inhibitor (compound A; 0.3 mg/dose), the pharmacologically inactive analog (compound H; 0.3 mg/dose), and ketorolac tromethamine (0.3 and 3 mg/dose). A, Erosion and ulceration, mucosal and submucosal neutrophilic infiltrates, and degeneration and regeneration of the nasal transitional epithelium were produced by the PTP1B inhibitor and pharmacologically inactive analog to a similar extent. No changes were produced by ketorolac tromethamine (0.3 mg/dose), with minimal changes produced at a 10-fold higher dose (3 mg). B, Erosion and ulceration, mucosal and submucosal neutrophilic infiltrates, and degeneration and regeneration of the respiratory epithelium were produced by the PTP1B inhibitor and pharmacologically inactive analog to a similar extent. No changes were produced by ketorolac tromethamine (0.3 mg/dose), with minimal changes produced at a 10-fold higher dose (3 mg). C, Acute necrosis and neutrophilic exudate of the olfactory epithelium were produced by the PTP1B inhibitor and pharmacologically inactive analog to a similar extent. No changes were produced by ketorolac tromethamine (0.3 mg/dose), with minimal changes produced at a 10-fold higher dose (3 mg).
In Vitro Cytotoxicity Assay
An in vitro study using the CRL1213 rat skin fibroblast cell line was performed to assess the cytotoxicity of various PTP1B inhibitors to further characterize SARs and correlate with in vivo data. Measures of cytotoxicity included overall cell metabolism (CellTiter-Blue) and LDH leakage from cells into the culture medium. Compounds were ranked by their TC50 values. Paraquat, a herbicide known to induce respiratory toxicity in humans 17,18 and the detergent NDS were used as positive controls. The latter was selected because the PTP1B inhibitors evaluated have amphiphilic structures that may have detergent-like properties. Ketorolac tromethamine, a marketed drug, was included as a comparator compound.
Sodium dodecylbenzene sulfonate was the most cytotoxic of the compounds tested, with TC50 values of 0.3 mM (Table 3). All of the PTP1B inhibitors evaluated produced cytotoxicity, with a range of TC50 values of 0.3 to 3.2 mM. Compounds G, B, and C produced similar results as NDS. Compounds E, D, and A were less cytotoxic and had similar TC50 values as paraquat (3 mM for CellTiter-Blue). Ketorolac tromethamine was the least cytotoxic compound tested with TC50 values of 9.9 mM (CellTiter-Blue) and 6.2 mM (LDH). At test compound concentrations that produced changes of ≥50% in CellTiter-Blue and/or LDH, the integrity of the cell monolayers was compromised and cell morphology was impacted, including condensed nuclei. Although the cytotoxicity range was fairly narrow (within 10-fold), the in vitro cytotoxicity potency of the PTP1B inhibitors was similar to that observed in vivo in which compounds B and G were the most toxic in both experimental systems. Ketorolac tromethamine was the least toxic compound tested in vivo and in vitro.
In Vitro Cytotoxicity Assay in Rat Fibroblast Cells.a
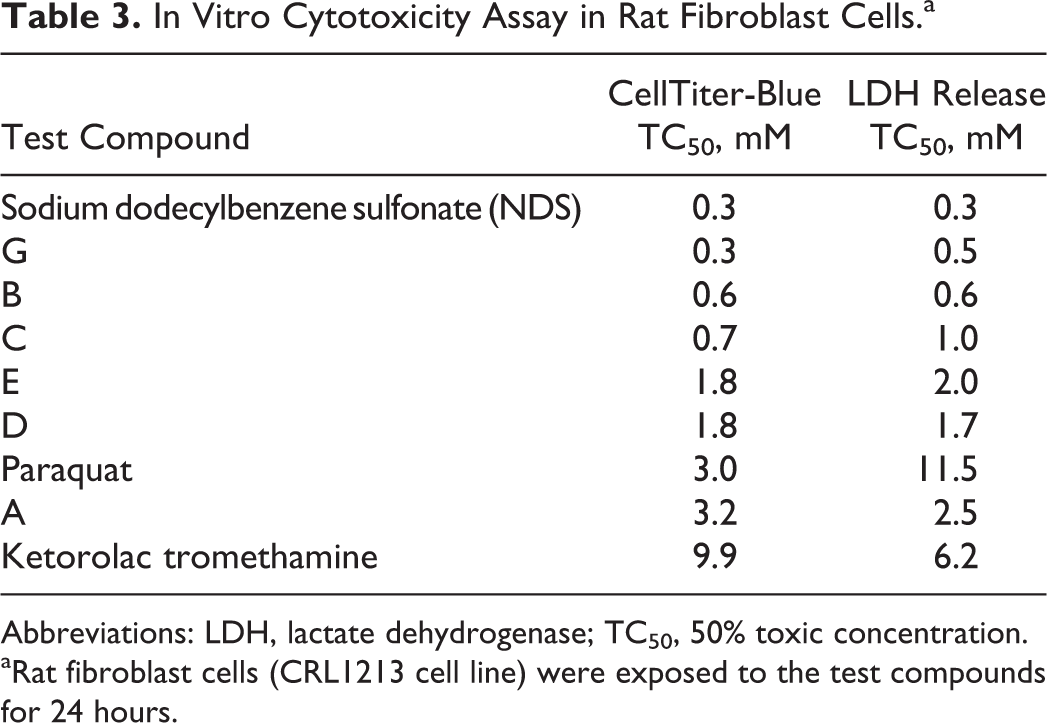
Abbreviations: LDH, lactate dehydrogenase; TC50, 50% toxic concentration.
aRat fibroblast cells (CRL1213 cell line) were exposed to the test compounds for 24 hours.
Discussion
Previous nonclinical research has suggested that the intracellular enzyme PTP1B functions as a negative regulator for both insulin and leptin signaling pathways. 12 Inhibition of PTP1B in the hypothalamus may increase leptin sensitivity, thereby providing a pharmacological means for eliciting an anorectic response. 9 A research program was initiated at Novartis to evaluate whether small molecule inhibitors of PTP1B could be delivered via the IN route as a strategy for the treatment of obesity. Molecules with a phosphate mimetic were identified that inhibited PTP1B in a biochemical assay with IC50 values in the nanomolar range. The IN route of administration was investigated as a means for drug delivery to the CNS and hypothalamus via an extracellular route along the olfactory- or trigeminal-associated neuronal pathways. 13
Initial studies in DIO mice appeared to support the hypothesis that IN delivery of a PTP1B inhibitor could decrease food consumption through a centrally mediated pharmacological response. However, subsequent data indicated that acute injury to the nasal epithelium was the more likely mechanism of action. Multiple studies in DIO mice demonstrated that IN administration of various PTP1B inhibitors produced significant injury to the major types of nasal epithelium, which included the squamous, transitional, respiratory, and olfactory epithelium. 19 Injury was observed shortly after dosing, with damage to diverse epithelial cell populations in a nonspecific manner. Single-cell necrosis of the olfactory epithelium was observed, which could impact olfaction leading to decreased food consumption. Similarly, IN administration of zinc oxide nanoparticles in rats or zinc gluconate in mice resulted in olfactory epithelial injury and impaired olfaction. 20,21 Injury to olfactory epithelium occurred as early as 2 hours following IN administration of zinc gluconate in mice, with detachment of the epithelium from the basement membrane by 4 hours. 21 These data provide further support that injury to olfactory epithelium can occur rapidly upon IN administration of a toxicant. Degeneration resulting in atrophy or complete necrosis of the olfactory epithelium is a common change after administration of an olfactory toxin. 22 Respiratory metaplasia of the olfactory epithelium and squamous metaplasia in the respiratory and transitional epithelium were observed following repeat dosing of the PTP1B inhibitors over a 2-week period. These metaplastic changes represented a regenerative response to epithelial cell necrosis. 22,23 Although a limited number of PTP1B inhibitors were studied, SAR data suggested that compounds with a cLogP >3.0 were the most toxic. Toxicity to the nasal epithelium occurred to a similar extent with a pharmacologically inactive analog of a PTP1B inhibitor, demonstrating that the mechanism of action was independent from the inhibition of PTP1B. These results support the hypothesis that these compounds produce local cytotoxicity to epithelium, possibly due to the disruption of cell membranes. Protein tyrosine phosphatase 1B inhibitors studied are comprised of a lipophilic region with a phosphate mimetic negatively charged at physiologic pH and therefore have an amphiphilic structure that may act to disrupt lipid membranes (Figure 1). Although the initial studies were confounded to a limited extent with the use of water as a vehicle for dosing (due to hypotonic stress on the nasal epithelium), subsequent studies utilized PBS as the vehicle to minimize local irritancy due to pH and/or osmolality. 15,16 A correlation was observed between increased cLogP of PTP1B inhibitors and decreased food consumption in the DIO mouse model (Figure 6), lending further support for a mechanism of action due to nasal airway toxicity.
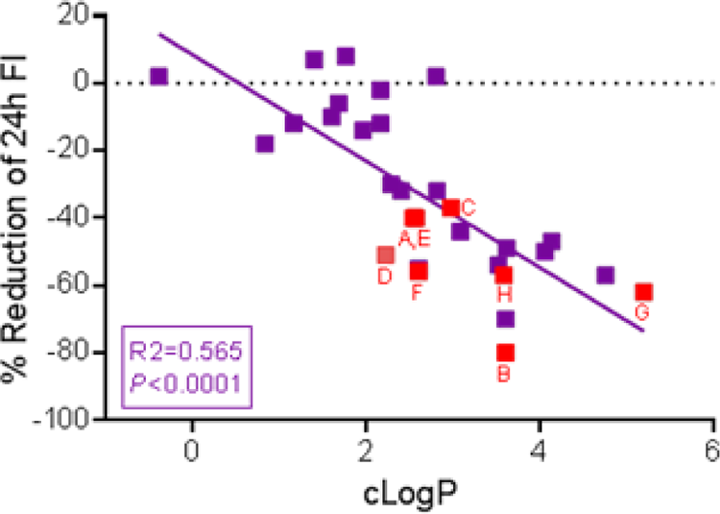
The percentage reduction in 24-hour food consumption in diet-induced obese (DIO) mice was plotted against calculated log octanol; water partition coefficient (cLogP) of various protein tyrosine phosphatase 1B (PTP1B) inhibitors, including those compounds (in red) studied for nasal toxicity. A correlation exists between food intake reduction produced by intranasal (IN) administration of PTP1B inhibitors and cLogP.
Ketorolac tromethamine is a FDA-approved nasal drug indicated for short-term management of moderate to severe pain that requires analgesia at the opioid level and delivered at high daily doses (with respect to other IN drugs). The approved dose is 31.5 mg for every 6 to 8 hours, up to a maximum of 126 mg/d, using an active pharmaceutical ingredient formulation concentration of 157.5 mg/mL (418 mM). Ketorolac is negatively charged with a cLogP of 1.6 and has a similar molecular weight to the PTP1B inhibitors. The most common adverse events for IN ketorolac tromethamine are nasal discomfort and nasal pain. 24 Although 0.3 mg/dose of any of the PTP1B inhibitors studied produced significant injury to the nasal epithelium, ketorolac tromethamine was nontoxic at 0.3 mg/dose and produced significantly less injury to the epithelium at the 3 mg/dose in comparison. These results support the use of ketorolac tromethamine as a benchmark or comparator compound when evaluating for potential effects to the nasal epithelium.
An in vitro study assessed the cytotoxicity of various PTP1B inhibitors to better characterize SAR and correlate with in vivo data. Sodium dodecylbenzene sulfonate, which can act as a detergent due to hydrophobic and charged regions on the molecule, and the herbicide paraquat were included as positive controls, whereas ketorolac tromethamine was included as a comparator compound. Sodium dodecylbenzene sulfonate and ketorolac tromethamine were the most and least cytotoxic compounds, respectively, to the fibroblast cell line. The rank order for in vitro cytotoxicity produced by the various PTP1B inhibitors was similar to that observed in vivo. These results support the use of in vitro cytotoxicity testing as a means for screening molecules prior to IN administration in animal studies. It is recommended that assays incorporate both positive (highly cytotoxic) and negative controls, along with a range of test compound concentrations which bracket the concentration to be used in vivo. A similar in vitro screen (using L6 rat myotube cells) was recently proposed to identify compounds with reduced risk of producing irritation and injection site reactions upon parenteral administration. 25
Although pharmacologic activity of the PTP1B inhibitors was not evident, a risk assessment for IN administration in man can be conducted as a case example for evaluating IN drugs. 26 The lowest IN dose that reduced food consumption in DIO mice was 0.1 mg. In a 0.040-kg mouse, this equates to 2.5 mg/kg or 7.5 mg/m2. The human-equivalent dose (by body weight) is 0.20 mg/kg, which equates to 12 mg in a 60-kg adult. 27 A standard dose volume for nasal drug products is 0.2 mL (0.1 mL/nostril); therefore, a 60 mg/mL dosage formulation would be necessary to deliver a 12-mg dose. A similar assessment can be conducted based on nasal cavity surface area. 28 In mice, this results in 0.1 mg/2.8 cm2 equating to 0.036 mg/cm2. The total nasal surface area in man is considered to be 160 cm2; therefore, the 0.036 mg/cm2 dose equates to a nasal dose of approximately 6 mg. This dose requires a 30 mg/mL dosage formulation. Because nasal toxicity was observed in the animal studies with PTP1B inhibitor concentrations ≥1.2 mg/mL, no safety margin exists with respect to projected human doses when considering body weight or nasal cavity surface area for cross-species comparisons.
In conclusion, various small molecule PTP1B inhibitors produced toxicity to the major cell types of the nasal epithelium upon IN administration. Injury to the nasal epithelium was not due to PTP1B inhibition, and compounds with a cLogP >3.0 were the most toxic. Although the mechanism for toxicity is not known with certainty, it was likely due to nonspecific cytotoxicity such as disruption of cell membranes. Results from an in vitro cytotoxicity assay corresponded to those obtained following IN administration. Therefore, in vitro cytotoxicity screens should be used as an early assessment prior to IN administration in animals. Evaluation of local tolerance for drug delivery by nonoral routes of administration should be conducted as early as possible during drug discovery activities.
Footnotes
Author Contributions
Alan P. Brown contributed to conception and design, data analysis, and interpretation of the studies and prepared the manuscript. David Barnes-Seeman, Jiaping Gao, and Ken Yamada contributed to conception and design of the PTP1B inhibitor research program, along with data analysis and critical review of the manuscript. Chandrassegar Saravanan conducted all pathologic evaluations and provided analysis and interpretation of the in vivo studies. Patrick Devine and Maria Magnifico contributed to design, conduct, analysis, and interpretation of the in vitro studies. Valerie Beaulieu contributed to design and conduct of the in vivo studies. Fupeng Ma and Kayo Yasoshima contributed to the medicinal chemistry design and synthesis of the PTP1B inhibitors. All authors gave final approval and agree to be accountable for all aspects of the work ensuring integrity and accuracy.
Acknowledgments
The authors acknowledge the following individuals at the Novartis Institutes for BioMedical Research for their contributions to the PTP1B inhibitor research program: Micah Hollis-Symynkywicz, Lac Lee, Monish Jain, Jeyaseelan Raju, Geeti Gangal, and Sunita Yadav.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: When these data were generated, all authors were employed by the Novartis Institutes for BioMedical Research.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Novartis Institutes for BioMedical Research funded this research.