
Abstract
Clinical pathology testing is routinely performed in target animal safety studies in order to identify potential toxicity associated with administration of an investigational veterinary pharmaceutical product. Regulatory and other testing guidelines that address such studies provide recommendations for clinical pathology testing but occasionally contain outdated analytes and do not take into account interspecies physiologic differences that affect the practical selection of appropriate clinical pathology tests. Additionally, strong emphasis is often placed on statistical analysis and use of reference intervals for interpretation of test article–related clinical pathology changes, with limited attention given to the critical scientific review of clinically, toxicologically, or biologically relevant changes. The purpose of this communication from the Regulatory Affairs Committee of the American Society for Veterinary Clinical Pathology is to provide current recommendations for clinical pathology testing and data interpretation in target animal safety studies and thereby enhance the value of clinical pathology testing in these studies.
Introduction
International regulatory authorities require the safety of drug products intended for animal use to be evaluated in guidance-driven safety studies in the species of interest. As with nonclinical toxicity studies conducted in laboratory animal species in support of regulatory submissions of investigational new drugs for human use, regulatory guidelines for target animal safety (TAS) studies for investigational veterinary pharmaceutical product (IVPP) development recommend the use of clinical data (physical examination, food consumption, body weights, etc), clinical pathology data, gross necropsy findings, organ weights, and histopathology findings to identify potential dose-limiting toxicities and to confirm an adequate margin of safety in the target species. 1 –3 For food-producing animals, in addition to assessing margin of safety for the target animal, the sponsor is also required to characterize and mitigate any potential consumer health risk associated with the unintended daily ingestion of drug residues from the administration of the IVPP. 4 Although not the focus of this manuscript, studies akin to clinical trials in human drug development that follow applicable TAS studies include clinical field studies to determine that the product is safe and effective in the target population under the conditions of intended use. 5
The International Cooperation on Harmonisation of Technical Requirements for Registration of Veterinary Medicinal Products (VICH) guideline on Target Animal Safety for Veterinary Pharmaceutical Products (VICH GL43) provides recommendations regarding the design and conduct of TAS studies for regulatory submission of an IVPP intended for major domestic species (canine, feline, bovine, ovine, caprine, porcine, equine, and poultry) to the US Food and Drug Administration (FDA) and other regions in the European Union (EU) and Japan that participate in the VICH. 1 However, specific regulatory oversight and guidelines for the scope and conduct of animal safety studies depend on the type of product being investigated and the indications sought for registration. In the United States, the FDA Center for Veterinary Medicine (CVM) typically regulates systemically acting pharmaceutical products intended for animal use for both internal and external indications, including external pests, 4 whereas the US Environmental Protection Agency (EPA) Office of Prevention, Pesticides and Toxic Substances (OPPTS) typically regulates products that act directly on pests or upon its ingestion after some type of topical application of the IVPP. 2 The OPPTS 870.7200 guidelines address the safety assessment requirements for products such as collars, dips, sprays, shampoos, and spot-on treatments applied directly to dogs and cats to treat external pests. 2 Further, the US Department of Agriculture (USDA) Center for Veterinary Biologics is responsible for the regulatory evaluation of animal vaccines and biotherapeutics.
With the main exception of USDA guidelines for vaccines and biologics, these guidelines provide similar recommendations for the design of animal safety studies, although there are some differences among them. Target animal safety studies under VICH GL43 should be conducted under good laboratory practices in the species and age group for which use of the IVPP is intended, and using a batch of final formulated product that was produced under current good manufacturing practices and is representative of that which will be marketed. Similarly, the dosing frequency and route of administration should follow the proposed labeling conditions. Similar to nonclinical animal studies and clinical trials in people, TAS studies should include a placebo or untreated control group along with the treated groups to assess the margin of safety of the formulated active pharmaceutical ingredient.
The types of TAS studies that are required for an IVPP are based on its conditions of use and characteristics. The focus of this communication is the margin of safety study, which includes a full panel of clinical pathology analytes. However, the principles discussed are applicable to clinical pathology analyte selection and interpretation for any study in a target animal species that may include clinical pathology evaluation, such as clinical field studies, which are used to determine efficacy and safety.
To define the margin of safety, the highest recommended therapeutic dose levels (1×) and multiples of this dose should be administered, most commonly 3 times (3×) and 5 times (5×) the proposed therapeutic dose level. The duration of administration is recommended to be at least 3 times the proposed period of administration up to a maximum of 90 days, although longer studies may be necessary depending on the product being investigated and the intended duration of administration. 1
As with nonclinical studies conducted in laboratory animals and human clinical trials, clinical pathology data have traditionally been collected in TAS studies as recommended by VICH GL43 and other guidelines and are valuable elements in the identification of potential toxicities. However, the recommendations for clinical pathology testing provided in such guidelines are somewhat generic, including tests that are not applicable to all species. Contemporary knowledge of appropriate clinical pathology tests and physiologic species differences is of paramount importance for the generation of quality clinical pathology data and for the correct interpretation of these data, highlighting the need for a broader input from veterinary clinical pathologists. Additionally, current recommendations frequently promote a reliance on the use of reference intervals (RIs) and statistical analysis for data interpretation. This practice may lead to errors in data interpretation and is not a substitute for critical scientific assessment by individuals experienced in the interpretation of clinical pathology data.
Veterinary clinical pathologists have become increasingly involved in the development of human pharmaceuticals but have been less commonly engaged in veterinary drug development. 6 The scientific value of utilizing knowledge from veterinary clinical pathologists will be highlighted in the following sections on the selection of appropriate tests and analytes for specific target animal species and the scientific interpretation and reporting of clinical pathology data.
Regulatory Guidelines for Clinical Pathology in TAS Studies
Current regulatory guidelines for the conduct of animal safety studies generally contain recommendations for clinical pathology testing of study participants, although the scope of these recommendations is variable. The most comprehensive guideline for clinical pathology testing is included in VICH GL43 for TAS studies, which recommends evaluation of hematology (including coagulation), clinical chemistry, and urinalysis at multiple time points during the study. 1 While this guideline indicates that the types of observations, examinations, and tests for safety should depend on the target animal, the list provided for clinical pathology tests is generic and does not provide specific guidance for appropriate clinical pathology tests in the various animal species. Additionally, the guideline does not provide specific recommendations for clinical pathology sampling time points. The EPA’s OPPTS 870.7200 guideline for studies of topical pesticides contains similar recommendations but is more limited with respect to specific tests and timing of sample collection. 2 Other guidelines, however, such as EU Directive 2001/82/EC, amended by Directive 2009/9/EC, that is cited by the Committee for Medicinal Products for Veterinary Use of the European Medicines Agency, contains only very limited recommendations (eg, evaluation of hematology) with no specific clinical pathology tests listed. 3 Similarly, the European Food Safety Authority technical guidance on tolerance and efficacy studies for food additives in target animals recommends the evaluation of hematology and blood chemistry but does not provide guidance for specific tests to be performed and only recommends clinical pathology testing for animals administered the additive at several fold the highest recommended dose. 7 The lack of detailed information in these guidelines regarding species-specific considerations, supplementary testing, and data interpretation presents limitations for the sponsor looking to design an animal safety study.
Considerations and Recommendations for Species-Specific Clinical Pathology Test Selection
Although physiologic differences among target animal species necessitate the consideration of appropriate clinical pathology tests for the individual species being investigated, the majority of standard clinical pathology tests are appropriate for use in most species. A standard panel of clinical pathology tests was recently proposed for nonclinical toxicity studies in laboratory animal species by Tomlinson et al. 6 This list includes some tests that are not included in the VICH guideline, such as the hematocrit as an equivalent measurement to the packed cell volume (PCV) and the concentrations of total bilirubin and triglycerides. The addition of these tests provides a more complete assessment of red blood cell mass (hematocrit), liver function (bilirubin), and lipid metabolism (triglycerides), and these biomarkers are easily evaluated on currently available hematology and clinical chemistry analyzers. Similarly, no recommendation for collection of blood smears or bone marrow smears is included in the current VICH guidelines; however, microscopic evaluation of blood and bone marrow smears is highly relevant for assessment of potential treatment-related hematologic toxicity and are therefore recommended by the authors. Finally, there is no recommendation for the use of serum or plasma for chemistry profiles. This remains a matter of preference, but, importantly, serum values should not be directly compared with values measured in plasma. 6
Some of the tests listed in the guidelines are generally not recommended by the authors as part of the routine panel for any of the target animal species. They include whole blood clotting time (WBCT), buccal mucosal bleeding time (BMBT), concentration of total bile acids, and activity of lactate dehydrogenase and amylase. These analytes are not recommended for a variety of reasons. Some of these tests have been substituted by more sensitive and/or specific tests (eg, WBCT is less sensitive than activated partial thromboplastin time [APTT]), whereas other tests are second tier analytes not generally required for routine evaluation, such as amylase and bile acids. Further, some tests are functional evaluations for which accurate assessment depends on careful control of experimental conditions which may not always be practical or feasible (eg, BMBT, preprandial and postprandial bile acid assessment). For other profiles, such as urinalysis, reagent test strips designed for evaluating human urine are cost-effective and convenient to use. However, the tests for urine-specific gravity, urobilinogen, nitrites and/or leukocytes provided on some reagent strips are not useful or recommended in some target animal species. 6
Specific considerations for appropriate clinical pathology tests in the most prevalent companion and food animal species are discussed in the following sections. A comprehensive list of recommended tests for routine animal safety studies in each species is presented in Table 1.
Recommended Clinical Pathology Tests for Common Companion Animal and Food Animal Species in Routine Animal Safety Studies for Veterinary Pharmaceutical Development.a
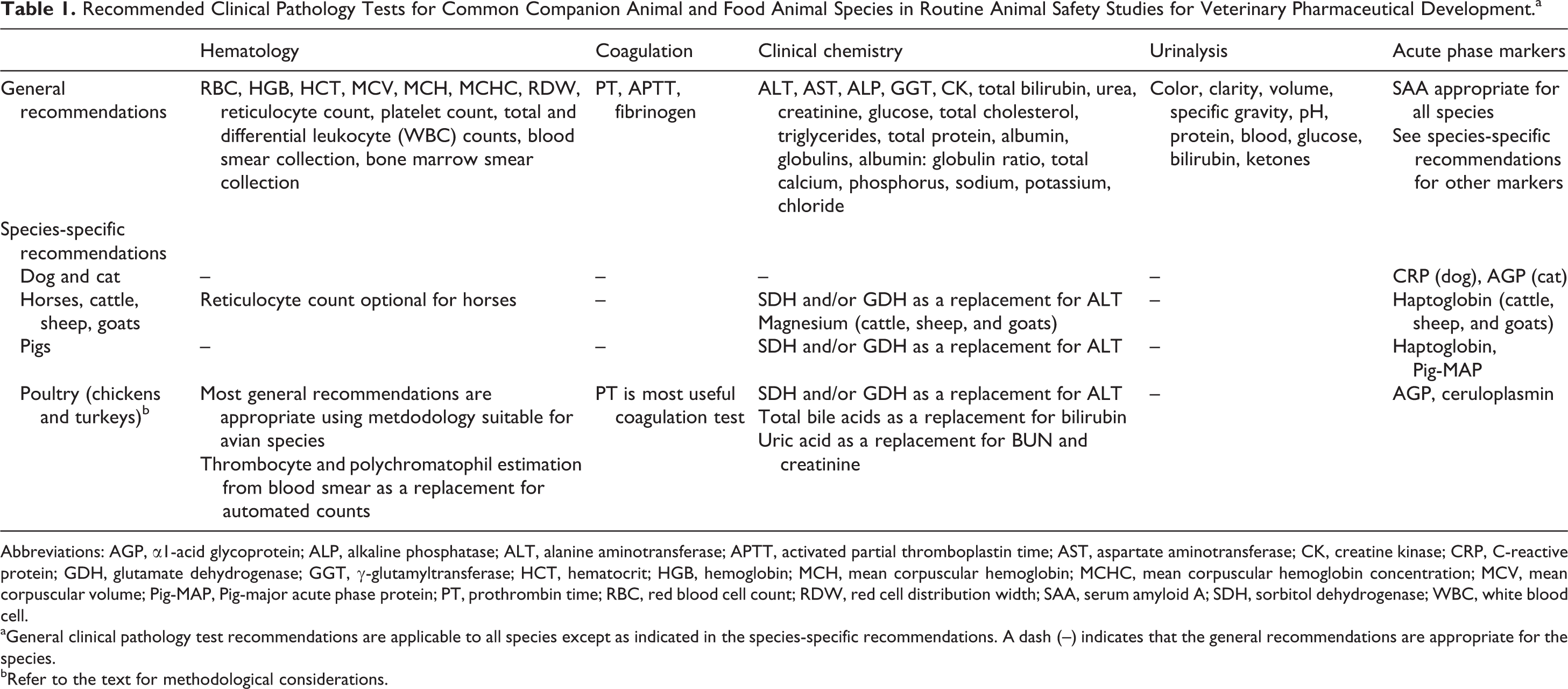
Abbreviations: AGP, α1-acid glycoprotein; ALP, alkaline phosphatase; ALT, alanine aminotransferase; APTT, activated partial thromboplastin time; AST, aspartate aminotransferase; CK, creatine kinase; CRP, C-reactive protein; GDH, glutamate dehydrogenase; GGT, γ-glutamyltransferase; HCT, hematocrit; HGB, hemoglobin; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; MCV, mean corpuscular volume; Pig-MAP, Pig-major acute phase protein; PT, prothrombin time; RBC, red blood cell count; RDW, red cell distribution width; SAA, serum amyloid A; SDH, sorbitol dehydrogenase; WBC, white blood cell.
aGeneral clinical pathology test recommendations are applicable to all species except as indicated in the species-specific recommendations. A dash (--) indicates that the general recommendations are appropriate for the species.
bRefer to the text for methodological considerations.
Dogs and Cats
All standard clinical pathology tests recommended by Tomlinson et al 6 are commonly utilized for TAS studies in dogs and cats. Activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are routinely used and considered liver-specific and reliable markers of hepatocellular injury in dogs and cats, even though borderline or mild elevations can occur with muscle injury. Due to high creatine kinase (CK) activity in skeletal muscle of dogs and cats, marked elevations of CK activity with muscle injury can be used to discriminate muscle from hepatocellular injury–related mild elevations in AST and ALT activity, and CK is therefore considered to be a useful marker in this species. For hepatobiliary toxicity, alkaline phosphatase (ALP) activity is generally considered more sensitive than γ-glutamyltransferase (GGT) activity, but correlative increases in the activity of both enzymes are highly predictive of hepatobiliary injury. Dogs have a corticosteroid or stress-induced ALP isoenzyme which is unique to dogs and can result in elevation of total ALP activity. Therefore, ALP should be cautiously assessed with respect to hepatobiliary toxicity when correlative increases in GGT activity or bilirubin concentration are not present.
Amylase and lipase activities are not recommended for routine evaluation but should be considered when exocrine pancreatic injury/pancreatitis is a concern in dogs; these analytes are of limited to no utility in cats. However, in cases of suspected exocrine pancreas dysfunction, testing for pancreas-specific lipase or pancreatic-lipase immunoreactivity may provide better sensitivity and specificity for detection of canine pancreatitis.
A small proportion of beagle dogs are homozygous or heterozygous for a factor VII gene mutation. This mutation can result in prolongation of prothrombin time (PT) and an altered thromboelastography/thromboelastometry profile. 8 Nevertheless, carriers can be used for studies as long as they do not have to undergo any surgical procedures. Finally, in addition to the standard clinical chemistry profile, valuable markers for the assessment of an acute phase response/inflammation include serum amyloid A (SAA), C-reactive protein (dogs), and α1-acid glycoprotein (cats). 9,10
Horses, Cattle, Sheep, and Goats
Special considerations for horses, cattle, sheep, and goats are primarily limited to activity of serum enzymes. Sorbitol dehydrogenase (SDH) and AST increase during acute hepatocellular damage in these species and are, therefore, recommended tests for hepatocellular injury, whereas hepatocellular ALT activity is low in horses and ruminants and therefore is not a sensitive marker of hepatocellular injury. 11 Because skeletal muscle is another major source of AST in food animals and horses, increased AST should be interpreted in context with CK activity, a more specific marker of muscle injury. Glutamate dehydrogenase (GDH) activity also increases during hepatocellular injury in these farm animal species. Finally, GGT has better diagnostic utility than ALP for detection of biliary disorders in horses 12 and is also recommended for cattle, sheep, and goats. Major acute phase proteins in these species include SAA and, for ruminants, haptoglobin. 9
Although not recommended as a routine test for monogastric species, such as dogs, cats, pigs, and horses, serum magnesium concentration is a useful analyte in cases of prolonged anorexia, ruminal and intestinal disorders, renal disease, and milk fever in ruminants, especially in cattle. 13
Finally, while absolute reticulocyte counts are recommended for most species as part of the hematologic assessment and are often included as part of the complete hematologic profile generated by many automated instruments, horses do not commonly release reticulocytes from the bone marrow into the circulation during an erythropoietic response. Therefore, absolute reticulocyte counts are generally of limited value in this species and are considered to be optional for routine studies.
Pigs
The majority of the standard clinical pathology panel (Tomlinson et al) 6 is appropriate for domestic pigs and for minipigs used in biomedical research. For the detection of hepatocellular injury, SDH and/or GDH are recommended in addition to AST and ALT. Serum ALT has low specificity for hepatocellular injury in pigs (similar to horses, cattle, sheep, and goats) due to higher activity in cardiac and skeletal muscle and relatively low expression in hepatocytes. 14,15
The major acute phase proteins in the pig include haptoglobin, SAA, and pig major acute phase protein (Pig-MAP). These are considered more sensitive than fibrinogen and can be used for assessment of herd health status and stress levels in pigs. 9
Poultry (Chickens and Turkeys)
In general, hematology tests recommended for mammalian species are also recommended for poultry, although the methodology differs from that for mammalian species. Hematologic assessment of avian blood samples is more challenging due to the need to perform manual (as opposed to automated) red blood cell, thrombocyte, and total and differential white blood cell counts. Manual blood cell enumeration is required because the nucleated red blood cells of avian species may confound accurate enumeration of white blood cells and, to a lesser degree, red blood cells by most hematology analyzers. An estimation of red and white blood cell counts from peripheral blood smears is not recommended as this method lacks sufficient precision to be useful for animal safety studies but often needs to be performed for thrombocytes as there are limited alternatives. Similarly, polychromasia can be evaluated on peripheral blood smears in lieu of automated reticulocyte counts. Hemoglobin can be measured using automated methods available with most modern hematology analyzers, but the sample must be centrifuged a few times after the cell lysis step and prior to optical analysis to avoid interference from free nuclei. 16 Packed cell volume must be performed manually by the microhematocrit tube method rather than using automated instruments, and red blood cell indices (mean corpuscular volume, mean corpuscular hemoglobin, and mean corpuscular hemoglobin concentration) can be calculated using the red blood cell count, hemoglobin concentration, and PCV. Differential leukocyte counts should be performed manually by counting 100 to 200 leukocytes on a good quality blood smear.
Of the routine coagulation tests, only the PT is recommended for poultry and other birds, but routine analysis may be hindered by the need for species-specific reagents. 17 The APTT is generally not included in the standard panel as the presence and/or role of the intrinsic coagulation pathway in most avian species is questionable. 17 Fibrinogen concentration is difficult to accurately evaluate under routine conditions. Electrophoretic methods quantify only protein bands that often contain multiple proteins with similar size and charge (therefore fibrinogen is not quantified directly) and the alternative heat precipitation method for fibrinogen measurement lacks precision.
Many of the standard clinical chemistry analytes used in mammalian species are appropriate for poultry, with the main exceptions being serum urea (or urea nitrogen) and creatinine. These analytes have not traditionally been considered to be useful markers of renal function in avian species because of low plasma concentrations and are generally not recommended for routine assessment of renal pathology, although they may be useful in assessing dehydration. 18,19 If feasible, serum uric acid concentration should be measured, as it is the primary nitrogenous waste product in birds. 18 Additionally, ALT and ALP are of limited value due to the lack of liver specificity and low-measurable activity in liver tissue, respectively. 20,21 Inclusion of GDH in the clinical chemistry panel is recommended due to its higher liver specificity, although its sensitivity is lower and its serum half-life shorter than that of some other hepatocellular enzymes such as ALT and AST. 20,21 Although bilirubin concentration is of limited value in avian species due to a low level of production, bile acids may be a more useful indicator of hepatic function. 22 Either fasting or postprandial bile acids can be measured; however, as feeding is expected to increase bile acid concentrations, the feeding status for all animals should be consistent for each sample collection throughout the study to allow for appropriate interpretation. Collection of a fasted sample for bile acid measurement may help to reduce variability associated with differences in feeding and gastrointestinal transit time. Total protein determination in avian species should ideally be performed using the biuret method that is available on many automated instruments, as opposed to refractometric estimation, and albumin and globulin fractions should be determined using electrophoretic methods when possible as the traditional dye-binding method for albumin measurement is not accurate in avian species. 17,18 Useful positive acute phase proteins for poultry species include α1-acid glycoprotein, ceruloplasmin, and SAA. 9
Urine can be collected for chickens and turkeys from a fresh dropping using a syringe or capillary tube; however, the sample should be obtained from a clean surface and care should be taken to avoid fecal contamination as much as possible during sample collection. Urine reagent test strip (dipstick) assays can be used to semiquantitatively assess biochemical constituents in uncontaminated avian urine samples. In contrast, urine sediment examination may be impeded by the high numbers of urate crystals excreted in health, although sodium hydroxide can be added to the sample to dissolve these crystals and more easily identify other urine sediment constituents. 18 Nevertheless, microscopic sediment examination is generally not recommended for routine TAS studies unless there is an expected test article effect on the urine sediment.
Considerations and Recommendations for Clinical Pathology Data Interpretation in TAS Studies
Timing of Clinical Pathology Sample Collection
The time points at which clinical pathology samples are collected can be critical to the identification of a test article–related effect; however, specific guidelines for the timing of sample collection are not available for TAS studies. The most comprehensive recommendations for clinical pathology sampling time points for animal toxicity studies are provided in the FDA Redbook 23 2000 and vary slightly depending on the length of the proposed study. We propose that these recommendations can be extrapolated to TAS studies and advise that samples for clinical pathology assessment should be collected at least once prior to the initiation of dosing, during the first 2 weeks after dosing begins, at least monthly thereafter, and at the end of the study. However, consideration must be given to the size and, by extension, blood volume of the species being evaluated, in that, collection of baseline samples prior to the initiation of dosing may not be possible for smaller species such as some birds or pocket pets (hamsters, guinea pigs, etc).
Despite these general recommendations, consideration must also be given to the dosing regimen, and any expected clinical pathology or organ system effects and the timing of sample collection should be carefully planned in order to increase the likelihood of capturing any such effects. For example, clinical pathology sample collection after 2 weeks and subsequently at monthly intervals may be appropriate for a 3-month study in which a small molecule is administered via daily oral gavage; however, this sample collection schedule may fail to accurately identify effects associated with an immunomodulatory large molecule administered via weekly intravenous injection that exhibits rapid and transient effects on peripheral leukocyte counts. In the latter case, the inclusion of time points in close proximity to dose administration (eg, 24 hours postdose) may be appropriate. Accordingly, it is incumbent on investigators to carefully consider all available information in designing a clinical pathology collection schedule that maximizes the likelihood of identifying pharmacodynamic and toxiologic effects.
Use of Reference Intervals
Reference intervals, sometimes referred to as reference ranges or historical control ranges, may be useful for the diagnostic evaluation of individual animals or to provide additional support to assessment of adversity (ie, a change is less likely to be adverse if within the RI). However, RIs are not necessarily considered appropriate for identification of test article–related effects in TAS studies for the following reasons: Meaningful RI must be established with reference populations of animals that are defined by breed, sex, and age, in addition to strain, supplier, management practices, and fasting status, to name the most important properties. Specifically, for a scientifically sound application of RI in the context of a TAS study, the animals used for the RI calculation and the study animals in question should be comparable in terms of breed; age; sex; strain; supplier; duration, type, and frequency of handling and restraint; blood collection site; number of previous phlebotomies; and any administered substances, such as a vehicle or control article. In practice, generating a robust data set that meets each of these criteria for a particular study design is generally not feasible. Meaningful RI are valid for the laboratory equipment in a particular laboratory including preanalytic/analytic factors defined by standard operating procedures and quality control practices. Unless equipment and quality control practices in different sites/laboratories have been harmonized and analyzers calibrated to provide comparable results, RI should not be used interchangeably for data generated in different laboratories under different conditions.
24
The calculation of RI requires the application of recommended statistical procedures as proposed by the Clinical and Laboratory Standards Institute (CLSI) or the Recommendations by the American Society for Veterinary Clinical Pathology (ASVCP), which is often not the case with available RIs.
24
In clinical pathology reference laboratories, leftover samples from clinically healthy animals such as study controls or health check populations can be used. However, since such animals usually do not undergo the same procedures as the study animals (dosing, blood sampling, handling, etc) and may not be of a uniform strain or age, the use of RI is discouraged for identifying test article–related effects in TAS studies. Although the strain and age of animals used for RI generation are often more representative of typical study animals for facilities that routinely conduct animal safety studies due to the common practice of using control animals for this purpose, even under these circumstances, there may be differences in the amount or type of restraint and handling, number of previous venipunctures, and other procedures that may impact clinical pathology data. Therefore, comparison of clinical pathology data to concurrent control animals is considered to be more appropriate for distinguishing procedure-related effects from test article–related effects. 25 Finally, comparison of each individual animal’s data to its baseline (pretreatment) data is a valuable component of data interpretation as it takes the potential biologic variation between individual animals into consideration. This is particularly relevant if the study population includes animals of different breeds (as may occur with field studies in dogs, cats, horses, cattle, etc), housed in different facilities or maintained under different management practices (horses and cattle). 26 Nevertheless, it is important to note that comparison to baseline data should be done judiciously as some analytes exhibit changes due to skeletal growth (such as ALP and phosphorus), pregnancy (such as red blood cell mass and albumin), or other physiologic processes such as stress (leukocyte and glucose concentrations), and therefore, changes relative to both control and baseline data should be assessed when possible.
Because of individual variation and breed- and sex-related differences, RIs that are not partitioned for different cohorts and include a large reference population tend to exhibit a relatively wide range. Renal biomarkers 27 and tightly regulated analytes (such as electrolytes) are examples where wide range RIs present the risk of failing to identify small, yet real, test article–related changes. Comparison with concurrent control (and with baseline values for larger animal species) and corroborative evidence in other data (other clinical pathology tests, histopathology, clinical observations, body weight, food consumption, etc) usually helps with identification of minor but significant individual trends which can be missed or dismissed if compared against an RI covering a varied group of animals.
On the other hand, the comparison to an RI can occasionally result in “flagging” a value, falsely indicating a potential treatment-related effect for a value that may in reality be within the realm of a normal but varied study population. Importantly, the statistical method used for calculating RI depends on the number of reference animals used and the normal distribution of data. Ideally, the reference population should include at least 120 animals; however, such a large group may not be available in some environments. Alternatively, a minimum of 40 animals may be acceptable, and as long as the data are normally distributed, RI can be calculated using the group mean (2 standard deviations). 24 If the data are not normally distributed, the central 95% is calculated using a nonparametric evaluation of rank percentiles excluding bottom and top 2.5% of the values. In either case, approximately 5% of clinically healthy animals are eliminated from the RI by the statistical evaluation. As a consequence, supposed normal healthy animals may be excluded as outliers before the study start when RIs are calculated using data from control animals from previous studies, although often the values interpreted as outliers or outside of the expected range represent biologic or physiologic variation in the healthy population (eg, leukocyte counts in dogs).
Unfortunately, it appears to be relatively common practice for many smaller laboratories to rely on RI published in text books or journal articles. Published RIs are frequently constructed based on data collected from different lots, strains, ages, or origins of animals; using different instrumentation; and under different environmental conditions than the institutions using this information for interpretive purposes, and therefore, these values do not necessarily provide a valid comparator for animals enrolled in TAS studies. Thus, this approach is not considered appropriate and is highly discouraged, as it may result in erroneous data interpretation.
An additional consideration is that for some studies, such as field studies designated to evaluate efficacy and safety in the target species, data may be generated from multiple institutions using different equipment under varying conditions. In the majority of cases, it is unlikely that the practices and equipment at each participating institution have been harmonized to provide equivalent results, resulting in analytic variability in the data generated among institutions and variable laboratory RI. Therefore, the comparison of individual values to their respective baseline values (presumably analyzed at the same laboratory) and/or to control values at the same laboratory will provide the greatest accuracy in identifying test article–related effects and interpreting their importance under the conditions of the study.
Considerations for Statistical Analysis
Statistical analyses in TAS studies generally include descriptive statistics (number of subjects, mean, standard deviation, etc) and inferential statistical analyses, usually by applying a repeated measures analysis of covariance (ANCOVA). A recent detailed guidance published by the FDA CVM regarding TAS data presentation and statistical analysis (CVM Guidance for Industry #226) 28 recommends that statistical analysis of continuous variables measured over time, including most clinical pathology data, should start with investigation of potential interactions (sex, time, treatment). The first step in the analysis determines if the treatment-by-sex-by-time interaction is significant, followed by separate evaluation of the treatment-by-sex and/or treatment-by-time interactions, if required. The results of these analyses determine if there is a need to compare treatment group mean data by sex and/or time point. If the previously described interactions are not significant and the effect of dose (treatment group) is not significant, the guidance recommends concluding that there is no difference between treatment group means.
Although statistical analysis is a helpful adjunct to identify differences between treatment groups in TAS studies that warrant further attention, 1 the utilization of statistical methods should be complemented with the scientific interpretation of clinical pathology data. 1,6,25,29 The scientific interpretation adds the assessment of magnitude, incidence, and direction of a particular change within the context of other in-life and postmortem observations. The final assessment will, therefore, focus on the biologic relevance and potential risk to the target animal species. Importantly, statistically significant changes may not be related to test article administration. In fact, statistically significant differences can be seen relatively often among baseline values, before application of a test article. These are either incidental or reflect other study-related parameters. Likewise, there may be subtle but toxicologically meaningful treatment-related changes that will not result in statistically significant differences between treated groups and the control group due to the biologic variation previously discussed. Specifically, statistical analyses comparing the treated groups relative to a negative control group do not take into account changes in relation to individual baseline values. Noteworthy changes in individual animals can be missed in animal safety studies when group sizes are small and interindividual variation may be considerable. Therefore, the interpretation of the biologic and toxicologic relevance of a clinical pathology change and its relationship to test article administration should be based on the comparison of individual and group mean values to control and baseline values, taking into consideration expected biologic variability and experience-driven scientific judgment. 6,25,29
Case Study Examples
In order to illustrate the principles described above with respect to the use of RIs and statistical analysis for data interpretation, 2 case examples are presented.
Example 1
Dogs were administered “Compound X” via once daily oral gavage for 30 days at dose levels of 0 (control), 5, 15, or 25 mg/kg/d (5 animals/sex/group). Blood and urine samples were collected for clinical pathology evaluation (hematology, coagulation, clinical chemistry, and urinalysis) twice prior to the initiation and weekly after the start of dose administration. Group mean data for serum albumin concentration are provided in Table 2. The laboratory-specific RI for albumin in dogs is 2.4 to 3.4 g/dL.
Example 1: Group Means of Albumin Concentration (g/dL) in Dogs Administered “Compound X” via Daily Oral Gavage for 30 days at Doses of 0 (control), 5, 15, or 25 mg/kg/d.a
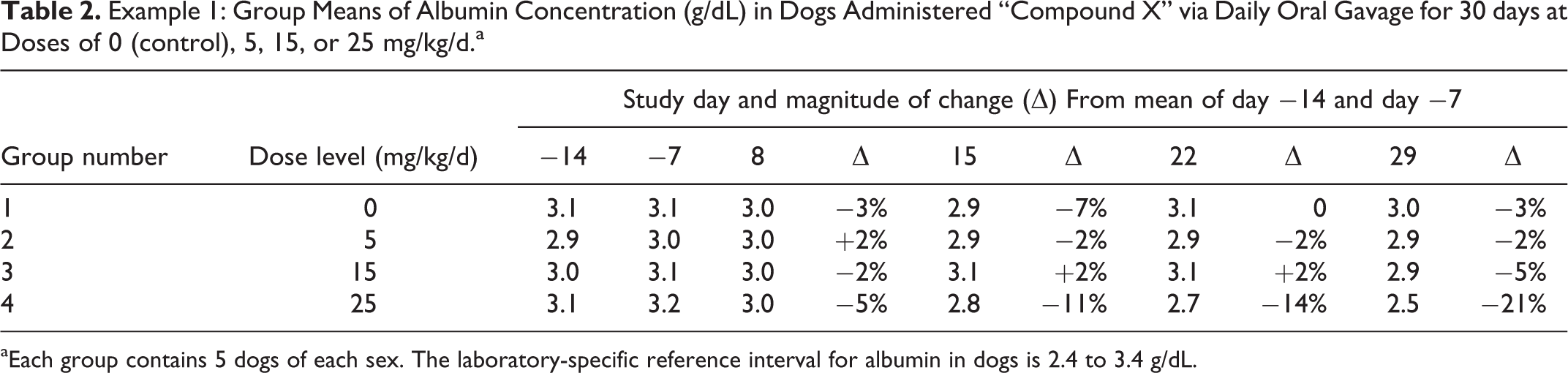
aEach group contains 5 dogs of each sex. The laboratory-specific reference interval for albumin in dogs is 2.4 to 3.4 g/dL.
In this example, all group mean values for albumin are within the laboratory-specific RI. There is minimal variability over time for the control group, and comparable variability for groups 2 and 3 (low- and mid-dose groups). For group 4 (high-dose group), however, there is a gradual trend for decreasing albumin values beginning on day 8. Although all mean values are within the RI, the magnitude of the decrease in group 4 (approximately 20% relative to the baseline values), the occurrence only at the highest dose, and the progressive decrease with continued dosing suggest a test article–related effect. Furthermore, while there was a large overlap among the individual control group and group 4 values prior to the initiation of dosing, nearly all group 4 animals had values lower than the range of individual control values by day 22 (data not shown), adding to the weight of evidence in support of a test article–related effect. In this study, dogs in group 4 also had mildly increased neutrophils, fibrinogen, and globulins (data not shown); therefore, the decreasing albumin, a negative acute phase protein, was likely part of an acute phase response. This example illustrates the importance of evaluating changes relative to control and baseline values on their own merit and not discounting a possible test article–related effect solely on the basis of RI.
Example 2
“Compound Y” was administered to cattle via subcutaneous injection once weekly for 4 weeks at doses of 0 (control), 1, 3, or 5 mg/kg (3 animals/sex/group). Blood samples for clinical pathology evaluation (hematology, coagulation, and clinical chemistry) were collected once prior to the initiation of dosing and twice during the dosing period. Selected clinical pathology data are presented in Table 3. Laboratory-specific RIs were 55 to 132 U/L for AST and 20 to 297 U/L for CK. Statistical analysis was conducted using ANCOVA. Pairwise comparisons of group means between groups 2 to 4 and the control group mean for each time point were performed, with statistical significance set at α ≤ .05. Mean values include both males and females.
Example 2: Group Means of Aspartate Aminotransferase (AST; U/L) and Creatine kinase (CK; U/L) Activity in Cattle Administered “Compound Y” Once Weekly for 4 Weeks at Doses of 0 (control), 1, 3, or 5 mg/kga
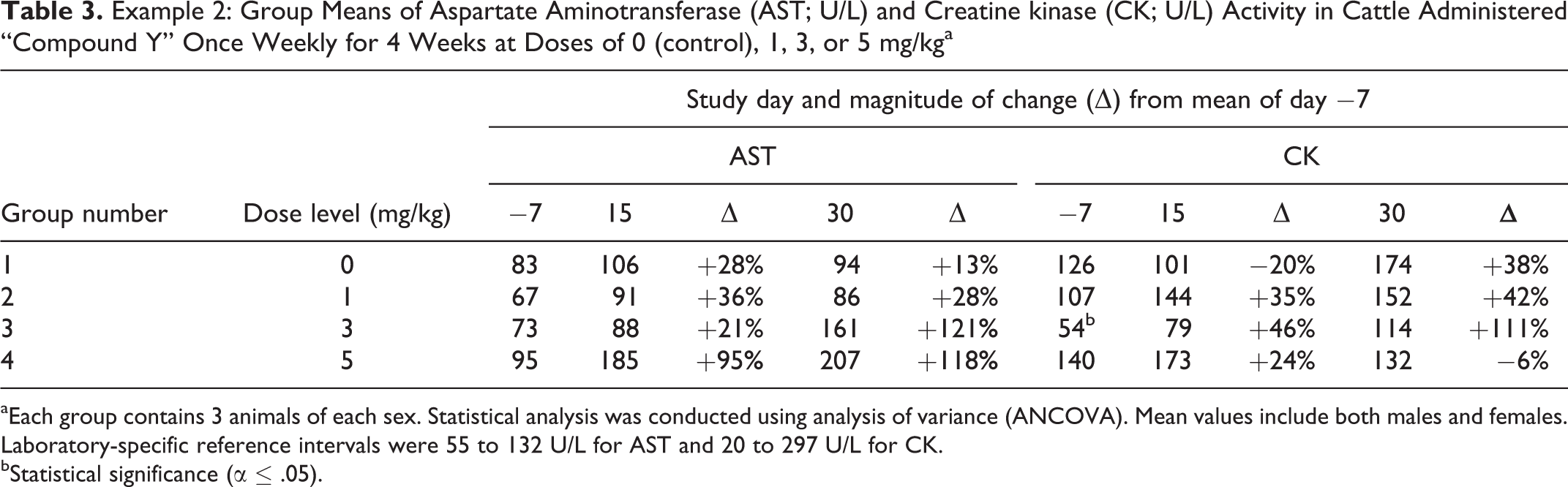
aEach group contains 3 animals of each sex. Statistical analysis was conducted using analysis of variance (ANCOVA). Mean values include both males and females. Laboratory-specific reference intervals were 55 to 132 U/L for AST and 20 to 297 U/L for CK.
bStatistical significance (α ≤ .05).
In this example, AST was mildly increased for animals in the high-dose group after 2 and 4 weekly doses and in animals in the mid-dose group after 4 weekly doses. Although none of the mean AST differences at any time points were statistically significant, there was a trend of increasing difference in comparison to the baseline and control values in this study, suggesting a treatment-related increase in AST activity in the high-dose group.
The lack of statistical significance for otherwise noteworthy changes in serum enzyme activity is frequently due to high interindividual variability. Although it is important to identify test article–related changes in the presence of high biologic variability, it is equally important to rule out a test article–related effect in the presence of high biologic variability. For CK activity, there is moderate variability over time in all groups including the control. The statistically significant lower value for group 3 on day 7 (prior to the initiation of dosing) is clearly not test article related and is therefore likely a reflection of biologic variability. Similarly, the lower CK values for this group at other time points, although not statistically significant, further suggest that lower serum CK activity for animals in this group is likely due to biologic variation. Finally, the magnitudes of change were very moderate for CK, as an increase due to muscle damage would often be 10× or higher. In this study, the lack of clearly increased mean CK activities in any group also provides useful information supporting possible test article–related hepatocellular injury as a cause for the increased AST, as opposed to procedure-related (eg, injection or venipuncture) or test article–related muscle injury.
Conclusions
Clinical pathology data collected as part of TAS studies are a valuable addition to the overall body of knowledge that is used to characterize the toxicologic profile of IVPPs. The assessment of clinical pathology data helps to identify target organs of toxicity, may aid in elucidating the pathophysiologic mechanism of that toxicity, and may also identify valuable biomarkers for clinical monitoring. Importantly, to take full advantage of such data, adequate species-specific test selection and an accurate assessment of clinical pathology data require an understanding of the diagnostic power of different analytes and interpretive nuances unique to a particular species, including a careful scientific assessment of study data based on a strong understanding of pathophysiology.
Although existing regulatory guidelines provide a useful framework from which to develop the clinical pathology component of an animal safety study, it is important to understand the limitations of a “one-size-fits-all” approach and to judiciously consider the utility of each test in the species for which the study is planned. Careful consideration of clinical pathology tests and appropriate timing of sample collection will ensure the highest likelihood of accurately identifying and characterizing a test article-related effect. Additionally, we recognize that data analysis tools such as inferential statistics and RI can be useful aids in identifying trends and differences warranting further attention; however, they should not replace critical scientific interpretation of data in assessing potential test article-related effects within the entire context of a particular study.
The recommendations provided in this communication are intended to add to the value of clinical pathology testing in animal safety studies by providing the perspective of veterinary clinical pathologists involved in the veterinary biopharmaceutical industry with respect to interspecies differences in commonly evaluated clinical pathology tests and considerations for study data analysis and interpretation. It is hoped that this manuscript might serve as a reference for regulatory authorities and leads to further discussion among sponsors, investigators, and veterinary clinical pathologists during TAS study protocol development and data interpretation.
This manuscript represents a consensus opinion among members of the ASVCP–Regulatory Affairs Committee. The ASVCP executive board reviewed and endorsed this manuscript.
Footnotes
Acknowledgments
The authors would like to acknowledge the substantial contributions of Dr Anne Provencher in the preparation and review of this manuscript. The authors also thank the members of the American Society for Veterinary Clinical Pathology’s Regulatory Affairs Committee for critical scientific review and valuable insight.
Authors’ Contribution
Siska, Gupta, Tomlinson, Tripathi, and von Beust contributed to conception and design, drafted manuscript, and critically revised manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.