
Abstract
There are many reasons that molecules fail to progress to market and various principles of risk–benefit decisions that can help drive the molecule through development. This symposium included discussions on global strategies involved in pushing promising molecules to market, what to do when a molecule stalls in its progress to market, and options for rescuing the molecule and pushing it forward again. Innovative partnerships that bring stalled drugs back into clinical development were also addressed. A regulatory perspective on common reasons for a molecule to fail in its forward progress was presented. In addition, situations arise when a third-party advisory committee can provide input to help overcome issues identified by a regulatory agency. Using examples from the private and public domain, presentations centered on how to repurpose a molecule and when more science is needed.
How Can a Toxicologist Overcome Obstacles in Drug Development? (Krishna Allamneni, Jazz Pharmaceuticals)
Krishna Allamneni, PhD, introduced the topic of “How can a toxicologist overcome obstacles in drug development?” Toxicologists, at some point in their careers, will likely come across adverse findings in their toxicology studies that pose a serious threat to the advancement of the molecule into further development. The conventional approach for drug development is a standard linear pathway starting with target-based drug discovery using target identification, cell-based screening, lead optimization, and nonclinical, animal-based testing followed by clinical trials. 1 This approach is hypothesis-driven, based around target identification and validation, and thus focuses on a predicted mechanism of action. Target-based drug screening casts a wide net, usually incorporating high-throughput experimental testing of large libraries of structurally diverse compounds to identify new drug candidates. However, because of the broad net cast during high-throughput experimental testing, there is a high level of attrition present at each step of drug development. In fact, only 9.6% of drugs successfully make it to the market. 2
Analysis of compounds from 4 different companies revealed a few important reasons for drug failure. 3 The major cause of failure for terminated compounds at all development phases is nonclinical toxicology, and a significant fraction of these fail at candidate nomination. Furthermore, the majority of the failures are due to intrinsic factors such as nonclinical toxicity, clinical safety, efficacy, pharmacokinetics/bioavailability (PK/BA), and issues with formulation that affect any of the previous end points. Failures due to extrinsic factors are mainly driven by the company portfolio review and account for about one-third of terminations. As molecules progress into late stage clinical development, there is markedly reduced risk that a drug will fail due to lack of safety or appropriate PK. In phase 2 and 3, lack of efficacy is the primary reason for failure. Efficacy failures can account for 51% to 66% of all failures at phase 2 and 3. 4 –6
A primary strategy for overcoming attrition is to address toxicity issues early. Thus, is it important to find a primary indication with a favorable risk–benefit ratio. However, changing therapeutic indication is a sound strategy depending on the severity of the disease, the patient population, and tolerance for toxicity in such a population. Unforeseen toxicities in one population may actually be beneficial in a new indication, thereby justifying repurposing. Furthermore, since nonclinical findings may correlate with pharmacokinetic/toxicokinetic (PK/TK) parameters or specific target organs, it is possible to change the route of administration to address toxicities. By utilizing a route of administration that targets the tissue of interest (eg, dermal), systemic exposure can be decreased, resulting in lower systemic toxicities with higher, more effective, local concentrations. Nonclinical findings may also be addressed by showing that they are species-specific and not relevant to humans. In some cases, toxicities that are sex-specific, or subpopulation-specific, may require a change in the target population and development of a relevant biomarker for monitoring.
There are several case examples whereby changing indication made it possible for a candidate to be developed. For example, sildenafil had an initial indication for the treatment of coronary artery disease. However, it was only weakly effective as an antihypertensive/angina drug but had the unexpected side effect of causing penile erections. A link between the identified mechanism of action as a PDE5 inhibitor and establishment of PDE5 as a key enzyme in the biochemical pathway mediating erections led to the new indication of treating erectile dysfunction. Sildenafil was later approved for pulmonary arterial hypertension and also recently for the ultra-rare orphan disease of lymphatic malformation. 7 –9
A second example is thalidomide, which was marketed in Germany as a sedative and to treat morning sickness. A devastating side effect of this compound was induction of apoptosis during early embryonic development, leading to profound phocomelia, which resulted in removal of this teratogenic drug from the market. A later, serendipitous discovery showed that this compound was effective in treating erythema nodosum leprosum. Further investigation found that thalidomide’s antiangiogenic properties were also effective against multiple myeloma. 9,10
A third case example is minoxidil, a nitric oxide agonist originally developed as an oral drug for hypertension. An interesting adverse side effect was hypertrichosis. This occurred by prolongation of the anagen phase of hair cells, which promotes their growth, mediated by prolongation of ATP sensitive K+ channel opening and stimulation of prostaglandin synthase 1. Currently, the drug is approved for topical application in the treatment of androgenetic alopecia to prevent hair loss and promote regrowth. 11,12
Another repurposing strategy involves taking drugs that have been approved in nononcology indications and applying them to oncology indications when a strong mechanistic basis exists. Many drugs with a variety of mechanisms of action have been effective in oncology settings. 13
Changing the route of administration is another strategy to avoid nonclinical toxicities. This strategy necessitates a clear understanding of the PK of the drug and how PK drives toxicity. An example is cytarabine which is used to treat leukemias and lymphomas by targeting inhibition of cell cycle phase-specific progression (S-phase). Although this drug has a fast clearance, it also has high systemic toxicities. Intrathecal administration allowed a higher systemic tolerability and slower metabolism by cytidine deaminase which has lower activity in the central nervous system tissues and cerebrospinal fluid, allowing its use in an orphan indication of malignant lymphomatous meningitis, especially as a sustained release liposomal formulation. 14
A clear understanding of the underlying mechanism of toxicities in the animal species may sometimes help comment on the relevance of these findings in humans. If species-specific findings not relevant to human can be demonstrated, this information can be used to advance molecules into the clinic. As an example, mitogen activated protein kinase kinase (MEK) inhibitors have been indicated for use in tumors associated with ras/raf mutations such as B-Raf proto-oncogene serine/threonine kinase (BRAF)-mutated melanoma and Kirsten rat sarcoma viral oncogene homolog (KRAS)/BRAF-mutated colorectal cancer. Unfortunately, adverse findings in repeat dose rat toxicity studies with MEK inhibitors found increased serum calcium (Ca) and inorganic phosphate (iP) with ectopic/soft tissue mineralization. This was a species-specific finding in rat, mediated via fibroblast growth factor 23 (FGF23)/klotho regulation, which was not observed in nonrodents. 15 A mitigative approach was to institute clinical monitoring for Ca and iP and set thresholds. The clinical experience to date has shown unremarkable mineralization. There are numerous other drugs in which clinical trials were halted and then allowed to continue after the potential mechanism of toxicity was identified to be species-specific. 9
In summary, examples of cases where adverse findings posed serious threat to the advancement of the molecule in the original indication were highlighted. These challenges can be negotiated to bring them to patients at a new dose, different indication, and route or different patient population. Such approaches can be taken in a wide variety of therapeutic areas.
Why Drugs Fail and When/How to Resurrect Them (Craig Wegner, AstraZeneca)
Dr. Craig D. Wegner then spoke on “Why Drugs Fail and When/How to Resurrect Them.” Despite advances in basic science, human genetics, medical research, enabling technologies, and project management and informatics, pharmaceutical research and development (R&D) productivity, defined as success divided by costs of a new drug coming to market, has plummeted over the last several decades. 3,16,17 The number of new drugs reaching patients (new drug application [NDA] approval) has decreased from approximately 50 per billion dollars of R&D spent in 1950 to less than 1 per billion dollars spent in 2010. 17 This astounding decline has contributed to escalating health-care costs and a “crisis” in patient care for both the depth and the breadth of therapeutic options available. To reverse this outcome, many companies and consultants have conducted investigations into the root causes and/or proposed revised pharmaceutical R&D strategies. 3,18–22 The following is a brief synopsis of some of the most intriguing findings, proposed solutions, and interim results.
New drug attrition occurs at alarmingly increasing rates at virtually all stages of drug discovery and clinical development. Attrition after candidate selection, during clinical development, is particularly both most frustrating and costly. 3 It is frustrating since the project/company has often focused down to betting on a single agent (compound); attrition typically means that project is terminated or at least drastically delayed. It is costly since the expenses prior to the attrition have accumulated to a substantial sum that is now wasted. Hence, while attrition at any stage is disappointing, the later the failure occurs, the greater its impact on overall R&D efficiency.
Safety and efficacy are the most common causes of new drug attrition. 3,22 Post candidate selection, unacceptable safety (eg, toxicology) and/or tolerability represent ∼80% of the failures prior to phase I, ∼60% during phase I, ∼35% during phase II, and still 10% to 20% during phase 3 and beyond. Efficacy, including improvement over existing standard of care, becomes the most frequent cause in phase 2 (∼60%) and beyond (up to 90%). While less frequent, inadequate pharmacokinetics or changes in company R&D focus (ie, strategy transformations) are other important causes. An internal Pfizer analysis of causes of attrition concluded that in up to almost 50% of phase II clinical efficacy failures, lack of sufficient coverage (engagement) of the primary molecular target (mechanism of action) of the compound could not be excluded. 21 Hence, testing of the primary compound/project hypothesis could not be confirmed.
A primary question is “What Can Be Done?” Several algorithms have been proposed to curb attrition. In 1997, Chris Lipinski authored the “Rule of Five” for medicinal chemists designing novel new chemical entities. 23 These focused on structural atomic features to reduce the risk of pharmacokinetic, both absorption and metabolism, as well as safety (eg, genetic and liver toxicity) attrition. Since then multiple improvements have been implemented in both the more predictive and earlier use (prior to candidate selection) of in vitro mutagenicity testing, 2-species in vivo 2-week dose escalation to clear toxicokinetic margins, safety pharmacology across the major organ systems, and exploratory toxicology to provide explanations for findings of questionable translation. To ensure target coverage (engagement) in clinical proof-of-principle efficacy studies, the “Three Pillars of Survival” criteria were advanced in a publication from Pfizer. 21 This was extended to additional criteria for molecular target selection, safety margin, optimal patient population, and commercial opportunity (return on investment) in the “5R’s Framework” analysis and recommendations from AstraZeneca. 22
Implementation of these principles has impressively enhanced survival rates. A reanalysis of success rates after applying the “5R’s Framework” has been invited for publication later this year. Yet, the failure of novel high-quality, optimized compounds is still present, significant, costly, and frustrating. Consequently, determining what, when, and how to resurrect such compounds has gained growing interest and focus.
The next question, “What to Resurrect?,” is crucial and completely dependent on the stage and known properties of the compound. Given the high rate of attrition in phase II clinical trials, most commonly due to efficacy insufficiently better than existing standard of care, a compound that is phase II ready is strongly preferred. These compounds have the properties and data set to support clinical testing straightaway in a novel indication. Of course, this is in part indication-specific such that a compound with some adverse effects may be acceptable for a life-threatening (eg, oncology) indication but not for a non-life-threatening “nuisance” (eg, allergic rhinitis) or chronic lifelong (eg, diabetes) indication. In general, the key features needed are evidence of: Target coverage (engagement) achievable in humans (in the target tissue/organ); Sufficient safety/tolerability margin; Novel mechanism of action (ie, not duplicative of another compound already approved or at a later stage of development); General drug-like features: potency, selectivity, attractive pharmacokinetics, low/no or manageable drug-drug interactions, and so on; and Confidence in the above.
Compounds that fulfill these features are not as frequent as many perceive. Of >450 compounds advanced to candidate stage within AstraZeneca and “discontinued” in their original indication, only 22 met these criteria. 24
The next question to arise is “When to Resurrect?” One may quickly notice, and perhaps be surprised, that a novel “composition of matter” patent status is not a crucial feature in the abovementioned list. While certainly not unimportant, resurrection of a previously “discontinued” compound often must rely on data exclusivity rules, a method of use patent in the new indication, or orphan status to protect its postlaunch commercial return on development investment. For the typical new chemical entity, composition of matter is filed at candidate nomination. With the average development durations of 1 to1.5 years in nonclinical development, 1 year in phase 1, 1.5 to 2 years in phase 2a, another 1.5 to 2 years in phase 2b, 3 years in phase 3, and 1 to 1.5 years in NDA filing to launch, a total of 9 to 11 years of the patent life is consumed, leaving perhaps 8 to 10 years of intellectual property exclusivity at launch. Data exclusivity rules in Europe (10 years) and Japan (8 years) provide essentially equivalent protection. Only for new chemical entities in the United States (5 years) is this duration substantially lower. Existence of a granted formulation or method of use patent for the new indication would likely extend the exclusivity an additional ≥1.5 years in the United States, as such patents cannot be challenged until the data exclusivity period has expired. Therefore, it is never too late to resurrect a high-quality previously “discontinued” clinical compound.
Several methodologies exist to address the question of “How to Resurrect?” each to various degrees of sophistication. These have been employed to identify and prioritize new uses for existing high-quality compounds. At the outset, it is important to note that >75%, and perhaps as high as 90%, of all drug approvals are for indications in addition to or distinct from those for which the compound was initially designed.
25
Hence, drug repositioning/repurposing has not only been successful but is actually the norm. How to do it deliberately, however, has been a constant debate with multiple approaches advanced. These include the following: Reformulation to fix a poor pharmacokinetic profile, increase the safety margin, and/or optimize and focus penetration to the target organ.
26
While leveraging an existing high-quality compound, this approach requires generation of a new formulation, PK/pharmacodynamics (PD) understanding (ie, data package) in nonclinical models, and typically the generation of a new nonclinical safety/toxicology package. Hence, this is neither a quick nor an inexpensive path to clinical hypothesis testing. Retrospective analysis, reanalysis or meta-analysis, of existing clinical data to identify a patient subpopulation or adjacent indication where an efficacy (or efficacy compared to safety/tolerability) signal is more favorable.
27,28
This personalized medicine approach is extremely attractive in principle, but often difficult to discover in practice, due to the finite size of clinical data sets available, including parameters measured, and the inherent bias of retrospective criteria election. Literature mining via sophisticated connection and machine learning algorithms that interrogate the vast amounts of published “observations.”
29,30
The challenge here is publication bias, variation in experimental design, and recording indiscrimination. Phenotypic screening in “disease-relevant” in vitro assays and/or nonclinical in vivo models.
31,32
This is only limited by the translatable predictivity to human disease of the assay/model system and the number of end points, biomarkers, and disease-relevant pathways that can be practically measured. Chemogenomic connectivity mapping via matching the transcriptomic changes induced by a compound to all disease transcriptomic, genetic, and phenotypic data.
33,34
This approach is quite attractive in being raw data-driven but limited by the distinct disease and subpopulation data available and bandwidth to confirm experimentally. System/network mapping which encompasses a combination of all of the above.
35,36
Extensive manual curation and application of best guess (open to investigational bias) weighting of data, pathways, and connections are the major challenges with this approach. Crowdsourcing of ideas and prioritization via effectively engaging the intellect of the worldwide medical science community. This requires a skillful and mutual rewarding outreach, clear statement of the problem/request, open iterative deliberations, and an effective peer review prioritization of ideas.
24,37
AstraZeneca has utilized each of the abovementioned approaches to varying degrees of success. Yet, it is the last in which efforts have been both the most unique and arguably the most successful, hence, expounded upon in this communication.
The rationale for drug rescue via crowdsourcing is to connect and leverage the collective wisdom of parties with similar objectives but distinct knowledge sets. The AstraZeneca Open Innovation platform (https://openinnovation.astrazeneca.com/) offers access to clinically enabled high-quality compounds to investigators with an understanding and insight to a novel indication. Both parties are motivated by the desire to improve human health and/or minimally advance medical science, but neither alone has the collective tools (compound, biomarker assays, etc.), insight (breadth of specific disease understand, including personalized subpopulations), or resources (access to funding, clinical facilities, etc.) to move forward without the other.
After participating in pilot pioneering partnership programs sponsored by the Medical Research Council (MRC) in the United Kingdom and National Center for Advancing Translational Sciences (NCATS) in the United States, 24 AstraZeneca launched a worldwide “Open Innovation” platform in March 2014. To date, 30 proof-of-principle clinical studies in new indications have been completed, are ongoing, or approved. Of the 6 that have been completed, 4 yielded positive efficacy results that have triggered follow-on proposals and/or now ongoing studies. Of the 30 studies, 26 have been funded by public grants from, for example, the MRC, NCATS, Taiwan National Research Program for Biopharmaceuticals (NRPB), other divisions of the National Institutes of Health (NIH), and disease foundations. In all cases, AstraZeneca provides the formulated clinical supplies (active and matched placebo), compound background knowledge, cross-reference to regulatory documents, cross-study pharmacovigilance, and drug development/clinical study design advice for these investigator-sponsored studies. The diversity of novel indications being tested is impressive, spanning common to ultra-orphan diseases, including acute myeloid leukemia, alcohol abuse, Alzheimer’s disease, autism, bronchiolitis obliterans syndrome, cancer bone pain, chronic cough, chronic idiopathic urticaria, chronic sinusitis, diabetic wound healing, dystonia, glioblastoma, high-risk atherosclerosis, menopausal hot flush, iatrogenic Cushing’s syndrome, idiopathic intracranial hypertension, lymphangioleiomyomatosis, nonalcoholic steatohepatitis, neuropathy, osteopenia, peripheral artery disease/intermittent claudication, post-traumatic stress disorder, psychosis, renal scleroderma, refractory gastroesophageal reflex disease, and type 2 diabetes. It is now difficult, and even disturbing, to recognize that none of these would be happening if the parties had not joined forces and, in most cases, received public funding.
In summary, novel drugs fail frequently, at all stages and for multiple reasons with safety/tolerability and insufficient efficacy being the major two. Through careful retrospective analyses, criteria (increased rigor) to minimize this attrition have been implemented and are leading to improvements. However, attrition still exists and is costly and frustrating. Resurrection of those high-quality clinical compounds that fail is warranted and has yielded early signals of success. Figure 1 provides a summary of an emerging pharmaceutical R&D strategy to curb attrition as well as enhance success through drug rescue or repositioning (drug repo).
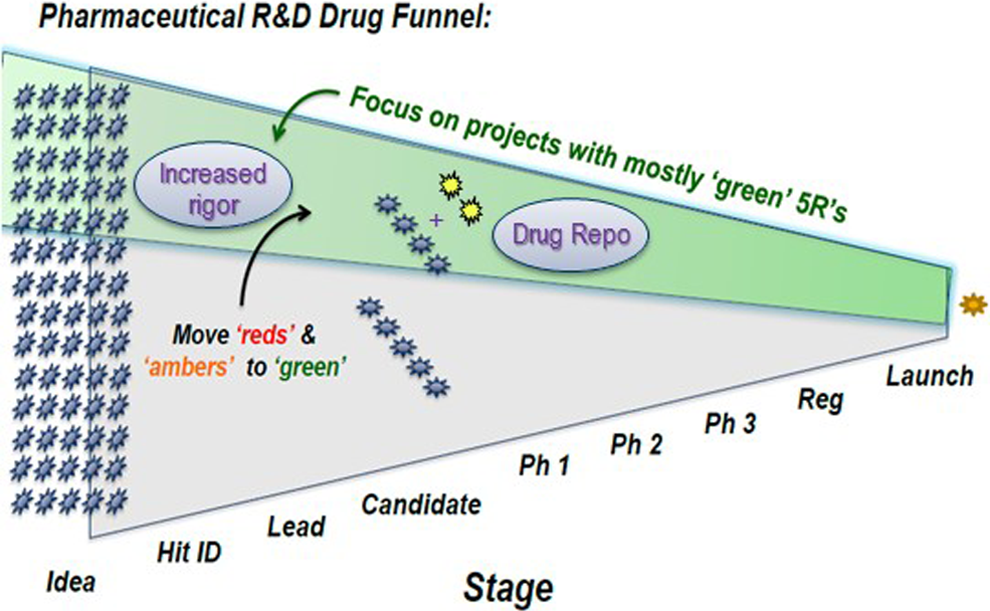
Representation of the funnel (attrition) of compounds across stages of pharmaceutical R&D. Criteria (increased rigor) learned from analyses of past attrition, such as the 5R’s Framework, are being applied to prioritize and focus on those compounds/projects with a higher probability of success (green shaded area). Drug repositioning (Drug Repo) is being employed to identify, prioritize, and test high-quality compounds in additional indications (yellow stars) to further enhance productivity (success/costs).
National Center for Advancing Translational Sciences (NCATS) Action Plan for Repurposing Existing Molecules (Christine Colvis, National Institutes of Health)
Dr. Christine Colvis then presented “National Center for Advancing Translational Sciences (NCATS) Action Plan for Repurposing Existing Molecules,” which highlighted 3 initiatives: the Biomedical Data Translator initiative, the initiative for Bench to Clinic Testing of drug/indication pairing strategies, and the New Therapeutic Uses (NTU) partnership with pharmaceutical companies.
The mission of NCATS is to catalyze the development of methods, technologies, and processes that enhance the development, testing, and implementation of diagnostics and therapeutics across a wide range of human diseases and conditions. The Center’s programs and initiatives are designed to address particular barriers or challenges to diagnostic and therapeutic development. Each of the initiatives achieves this goal in a different way. The first initiative, the Biomedical Data Translator, is intended to leverage clinical and nonclinical data from several different domains. The challenge is that those data currently exist not only in different locations but also in different formats, which presents a significant barrier to data integration. This barrier is seen by the Center as not only a technical issue but one that runs much deeper into the biomedical community, in that clinical practitioners and biomedical researchers think of disease in different ways and speak different languages. Physicians focus on signs and symptoms, while researchers tend to focus on molecular changes in pathways or cells. Due to the complexity of the problem, the program was launched with a 2-year feasibility assessment initiative, which began in the fall of 2016.
The second initiative, Bench Testing Therapeutic/Indication Pairing Strategies, is designed to support nonclinical studies to repurpose existing drugs or biologics (therapeutics) that have already begun or completed at least a phase I trial. The initiative is structured such that therapeutics with a positive outcome in nonclinical studies will be eligible for an award for clinical trial planning without the need to submit another application to the NIH. While the program is open to repurposing marketed therapeutics as well as investigational therapies, the repurposing hypothesis has to originate from the use of a published or publicly available method (computational algorithm) for identifying new indications for existing therapeutics or from using a crowdsourcing strategy in the case of investigational therapies.
The third initiative, the NTU initiative, forms partnerships between pharmaceutical companies and academic researchers. In this initiative, pharmaceutical companies offer investigational assets that have a known mechanism of action and that have completed at least a phase I trial. In offering the asset, the company agrees to provide drug and matched placebo for trials that are supported by the NIH. The Center uses this initiative to identify ideas from the broader research community for new uses of existing investigational therapeutics from industry. The new partnerships between the pharmaceutical company and the NIH awardee (generally an academic institution) are formed using template collaborative research agreements drafted by the NIH and the companies. The utility of these template agreements is the catalyst in therapeutic development that the Center is demonstrating through this initiative.
FDA Perspectives on Overcoming Obstacles for Drugs (Andrew Goodwin and Jane Sohn, US Food and Drug Administration)
Dr. Andrew Goodwin (US Food and Drug Administration, Silver Spring, MD) presented a talk entitled “FDA Perspectives on Overcoming Obstacles for Drugs.” The first portion of the presentation provided an overview of the process guiding nonclinical safety assessment at the Food and Drug Administration (FDA). Nonclinical data supporting the proposed clinical protocol are evaluated, considering the dose, duration, route of administration, and risk–benefit balance for the intended study subject population. The primary pharmacology–toxicology reviewer evaluates the study reports submitted by the sponsor to identify drug-related toxicities and assess safety margins between the no observed adverse effect level (NOAEL) in the pivotal animal studies and the proposed clinical dose level. During the review process, the nonclinical reviewer consults with his or her team leader/supervisor and, as needed with FDA Center for Drug Evaluation and Research Pharmacology–Toxicology leadership, subject matter experts, and/or other members of the interdisciplinary review team (eg, medical officer, chemist, clinical pharmacologist, or regulatory project manager). If necessary, the sponsor may be asked to answer questions or provide additional information. The nonclinical team then provides a summary and recommendations to division management as to whether the proposed clinical protocol is safe to proceed.
Dr. Goodwin provided an overview of potential factors leading the development of certain drugs to “stall” during the FDA review process. In some cases, pivotal nonclinical data are not provided by the sponsor. To avoid or address these deficiencies, sponsors are encouraged to refer to regulatory guidance documents and to seek advice from the review division in the form of meetings or written feedback. Alternatively, FDA pharmacology–toxicology review may identify adverse findings in nonclinical studies with NOAELs that do not adequately support the proposed clinical protocol. One possible path forward in these cases is to modify the clinical program to achieve a more favorable risk–benefit profile. In other cases, the sponsor may be able to provide additional data or analyses to support a conclusion that the nonclinical findings are (1) not adverse, (2) monitorable in the clinical setting, or (3) not relevant to humans. Three case studies were presented to illustrate circumstances which led drugs to stall, as well as how they were resolved in order to facilitate continued development.
The first example was a small molecule drug that elicited testicular toxicity, including degeneration and necrosis, in both rodents and nonrodents. In longer studies, there was no NOAEL for male reproductive toxicity, and the findings were not considered clinically monitorable. Therefore, there was inadequate nonclinical support for the sponsor’s intended development in a large indication with effective FDA-approved therapies available. However, a path forward could be development in a different patient population such as refractory patients or a serious disease with unmet medical need. In such a scenario, the clinical team would assess whether the potential toxicity represents an acceptable risk for that particular patient population; the prospect of benefit based on available clinical data could also be considered. As development proceeds, the nonclinical toxicities and potential risk to humans would be detailed in the Informed Consent and other clinical protocol documents.
In the second case study, a small molecule drug produced severe toxicity and mortality in rodents but exhibited a benign profile in nonrodents. Studies demonstrated that a reactive epoxide metabolite was formed in the rodent species but not in the nonrodent species or in humans. Coumarin, an analogous example from the public domain, was presented for illustrative purposes. 38 The sponsor asserted that this species-specific difference in metabolism may account for the variable toxicologic findings and tested the hypothesis in supplemental rodent studies. Rodent toxicology studies were conducted with 2 additional test articles: the synthesized epoxide metabolite and a pharmacologically active analog that cannot be converted to the reactive metabolite. Administration of the epoxide recapitulated the target organ toxicity and lethality while the analog was not toxic even at a large multiple of the clinical exposure. Based on the additional data provided, the rodent toxicity was not considered relevant to the human safety assessment, allowing a path forward for clinical development. Corresponding adjustments to other aspects of the nonclinical development program (eg, carcinogenicity and developmental and reproductive toxicology) were implemented, based on the conclusion that further rodent studies were not feasible.
The final case study involved a small molecule drug in development for a serious disease indication with unmet medical need. Investigational New Drug (IND) application-enabling toxicology studies in rodents and nonrodents were unremarkable and supported the initial proposed clinical evaluation. However, drug-related mortality was observed at all doses in the nonrodent chronic toxicology studies, leading to a full clinical hold. The sponsor subsequently proposed to restrict the clinical development program to a high-risk subset of the patient population and to limit the duration of dosing. The FDA concluded that it was acceptable to proceed with the revised clinical development program based on (1) an acceptable clinical safety profile at the proposed dose and duration, (2) prospect of benefit based on a novel mechanism of action with supportive in vitro and in vivo efficacy data, and (3) potential benefits to patients in terms of safety and convenience compared to other available therapies.
Dr. Goodwin concluded his remarks by reiterating that protection of the safety of subjects in clinical trials is the top consideration in the nonclinical safety assessment process. This evaluation is conducted on an individual basis, and final regulatory decisions are informed by risk–benefit considerations. As shown in the case study examples, it is sometimes possible to overcome nonclinical obstacles with a sound scientific approach. A hypothesis as to why a particular finding is not a safety concern for humans is not sufficient; additional nonclinical data may be required. The most productive strategy to avoid drugs stalling due to nonclinical safety concerns, and to resolve these issues should they occur, is proactive communication with the appropriate FDA review division.
Disclaimer: The preceding section of this article reflects the views of Dr. Goodwin and Dr. Sohn and should not be construed to represent FDA’s views or policies. Dr. Goodwin, Dr. Sohn, and the FDA did not contribute to and do not endorse the views expressed by the coauthors in other sections of the article.
Preclinical Findings Resulting in a Clinical Hold and the Journey to Gather More Scientific Data: Case Studies Including Pregabalin-Induced Hemangiosarcoma in Mice and Its Human Relevance (Jon Cook, Pfizer)
Finally, Dr. Jon C. Cook spoke about the “Mode-of-Action Associated With Development of Hemangiosarcoma in Mice Given Pregabalin, and Assessment of Human Relevance.” It is extremely rare that a nonclinical finding results in a clinical hold, especially findings from carcinogenicity studies that are generally reported during phase III and can be addressed in the label with a short discussion of human relevance. However, this was not the case with pregabalin. In 1997, Pfizer initiated phase II trials for the development of pregabalin for the treatment of neuropathic pain, as adjunctive therapy in treatment of partial seizures, and in treatment of generalized anxiety disorders and fibromyalgia. In late 2000, the carcinogenicity study results demonstrated that pregabalin produced a dose-dependent increase in hemangiosarcoma in male and female mice. 39 These tumors were located predominantly in liver, spleen, and bone marrow, which are hematopoietic tissues in mice. There were no other treatment-related tumors seen in mice, and there was no increase in tumors of any type in rats. In February 2001, the FDA placed a clinical hold on the clinical trials because of these findings. There was a numerical increase in hemangiosarcoma in the female low dose and while this was within historical control incidence, it occurred at the highest therapeutic dose. The FDA expressed concern about this tumor for the following reasons: (1) genotoxic compounds such as arsenic, Thorotrast, and vinyl chloride are known human carcinogens 40 ; (2) the mode of action (MOA) for hemangiosarcoma was not known for nongenotoxic compounds, and therefore biomarkers could not be used in the clinical trials to monitor for this risk; and (3) the human relevance of this tumor type could not be evaluated with any certainty without an MOA. This resulted in a nearly 10-year research effort by Pfizer scientists to understand the human relevance of these tumors. There were 2 periods of intense research, 2001 to 2004 where a pregabalin MOA was initially described that led to the removal of a clinical hold, and 2008 to 2010 where end points identified in the Society of Toxicology Contemporary Concepts in Toxicology (CCT) workshop were assessed with pregabalin to support the generalized anxiety disorder (GAD) indication in the United States.
Pregabalin binds with high affinity to the α2δ subunit of voltage-gated calcium channels, and the analgesic, antiseizure, and anxiolytic activities of pregabalin are related to binding affinity at that site, although the precise mechanism of efficacy is not known.
41
–44
Structurally, pregabalin is related to the endogenous amino acid
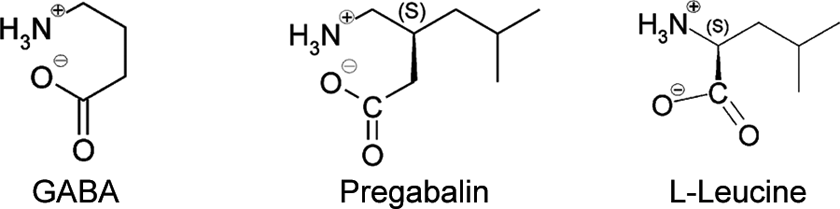
Pregabalin (S-3-(aminoethyl)-5-methylhexanoic acid; CAS 148553-50-8) and the structurally related, endogenous molecules GABA and l-leucine.
Hemangiosarcomas are endothelial cell (EC)-derived tumors that are comprised of poorly differentiated, proliferating ECs. 46 These tumors form spontaneously and in response to many different compounds in mice but are rare in humans. To our knowledge, there are only 2 examples of hemangiosarcoma induction by a chemical stimulus that occur in both humans and rodents (ie, mouse and rat). Both examples are genotoxic carcinogens (vinyl halides and Thorotrast), and both produce primarily liver hemangiosarcomas. 47,48 In contrast, numerous commercial pharmaceutical products and chemicals that produce hemangiosarcomas solely in rodents act by nongenotoxic, proliferative mechanisms. 40
In 2009, a unified MOA framework for induction of hemangiosarcoma by nongenotoxic compounds was proposed based on the output of a CCT workshop. This workshop evaluated the MOA for several nongenotoxic compounds that induced hemangiosarcoma. From this analysis, it became apparent that all of these compounds shared similarities in the induction of hemangiosarcoma: induction of hypoxia as an initiating event, dysregulated angiogenesis and/or erythropoiesis, macrophage activation, increased angiogenic growth factors, and increased EC proliferation. 40 Specifically, agents that induce hemangiosarcoma appear to have initiating events that lead to local tissue hypoxia and macrophage activation. These 2 changes increase angiogenic growth factors. This process is analogous to physiologic angiogenesis, but it is hypothesized that dysregulated angiogenesis occurs because these responses are sustained many times by dysregulated erythropoiesis. Dysregulated angiogenesis also leads to EC proliferation and contributes toward hemangiosarcoma formation.
Based on this information, a series of new investigative studies were conducted to determine the MOA for mouse-specific hemangiosarcomas by pregabalin. 41 –44 Table 1 summarizes the key events in the pregabalin MOA and contrasts the findings between those seen in mice versus rats, monkeys, and humans. Mice react inadequately to metabolic alkalosis following pregabalin administration, thus creating a cascade of events starting with an increase in serum bicarbonate and leading to sustained depression of respiratory rate and minute volume. 49 The sustained increase in blood pH hampers oxygen release from hemoglobin due to the Bohr effect, creating conditions conducive to hypoxia. Persistent perturbation of acid–base balance in mice is consistent with the identification of chronic tissue hypoxia in this species. 50 One method of compensating for hypoxia is an increased production of erythrocytes to provide greater oxygen carrying capacity. An appropriate circulating erythrocytosis is seen in rats but is lacking in mice, highlighting an additional key species-specific event that contributes to tissue hypoxia in mice alone. 49 The inability of the mouse to increase erythrocytes is due to dysregulated erythropoiesis that is apparent in bone marrow.
Species Specificity of Pregabalin-Induced Changes in Parameters Associated With Hemangiosarcoma in Mice.
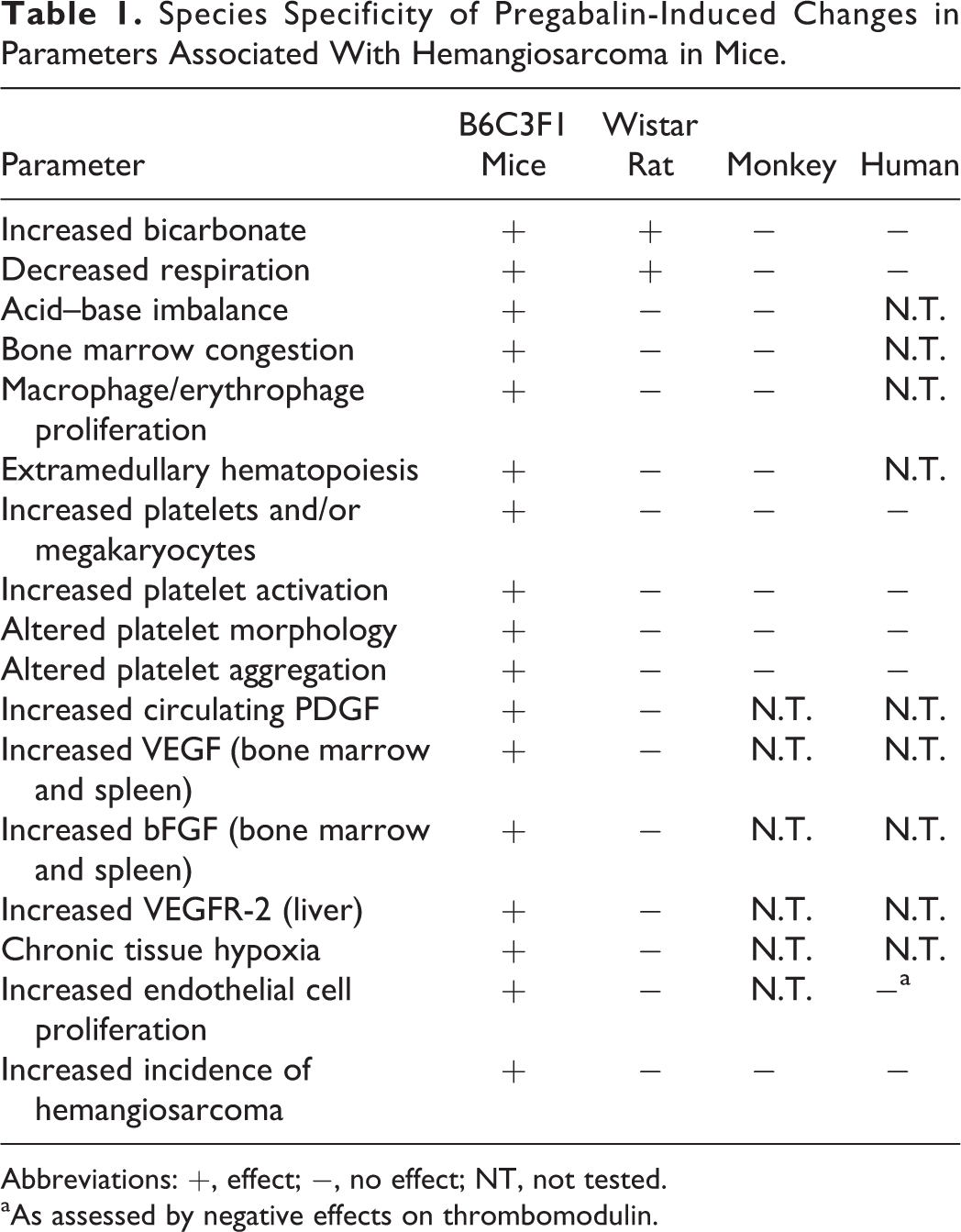
Abbreviations: +, effect; −, no effect; NT, not tested.
aAs assessed by negative effects on thrombomodulin.
In mice, pregabalin treatment increased platelet and megakaryocyte counts, decreased myeloid to erythroid ratio (M:E; up to 49%), increased the number of bone marrow macrophages/erythrophages, and was associated with congestion of bone marrow and spleen and extramedullary splenic hematopoiesis. 49 Bone marrow changes occurred early, within 1 to 3 months of drug in diet so were not the result of tumor formation 49 and are consistent with a temporal association. These findings demonstrate that dysregulated erythropoiesis is occurring in mouse bone marrow where enhanced red blood cell (RBC) production is occurring, but there is a failure of release. This failure of release explains the decreased RBC response of the mouse when compared to the rat and results in macrophage activation and increased hemosiderin production that increases oxidative stress. 49
In contrast to mice, pregabalin administration decreased overall bone marrow cellularity in rats, including megakaryocytes (up to a 24% decrease 49 ). Decreases in megakaryocytes in rats correspond with decreased peripheral platelet counts, which are in contrast to elevations in platelet counts, and highly proliferative bone marrow, in mice. There was no evidence of macrophage or erythrophage accumulation in rats, and M:E ratios were similar to controls. 49 The effects seen in mice and absent in rats, monkeys, and humans suggest an association between hematopoietic stimulus and selective endothelial tumor induction in mice. In addition, they provided sentinel biomarkers for assessing potential risk to humans.
An inflammatory component is also considered important in the MOA 40 and is evidenced by a dose- and time-dependent increase in macrophages in bone marrow, spleen, and liver of pregabalin-treated mice. 49 Macrophages can release reactive oxygen species that can damage ECs as well as release angiogenic cytokines such as IL-6 to stimulate EC proliferation. Using immunohistochemistry, pregabalin was shown to increase tissue angiogenic growth factors including vascular endothelial growth factor alpha and basic fibroblast growth factor (VEGFa and bFGF) 49 and increase EC proliferation as early as 2 weeks and sustained out to 12 months. 50 Therefore, mice treated with pregabalin demonstrate all of the components proposed by the unified MOA for nongenotoxic hemangiosarcoma formation. 40
Angiogenesis is an important physiologic process. It occurs monthly in the uterus of menstruating women, during pregnancy with the formation of the placenta, and in wound healing. Because angiogenesis is so important, it is tightly regulated with over 20 angiogenic growth factors and more than 300 angiogenic inhibitors (http://www.angio.org/understanding/fact.php). The production of hemangiosarcomas in pregabalin-treated mice is believed to occur due to pathologic angiogenesis that is characterized by dysregulated angiogenesis. Dysregulated angiogenesis refers to the condition where normal blood vessel formation does not occur in response to a hypoxia signal. Hence, there is a continued hypoxia signal leading to angiogenic growth factors exceeding inhibitors coupled with macrophage activation and release of reactive oxygen species that damage ECs, all of which promote increased EC activation, proliferation, and damage of EC DNA. Figure 3 illustrates a hemangiosarcoma in mouse liver where there is a pooling of blood from the blood vessels surrounded by the hemangiosarcoma, illustrating the concept of dysregulated angiogenesis.
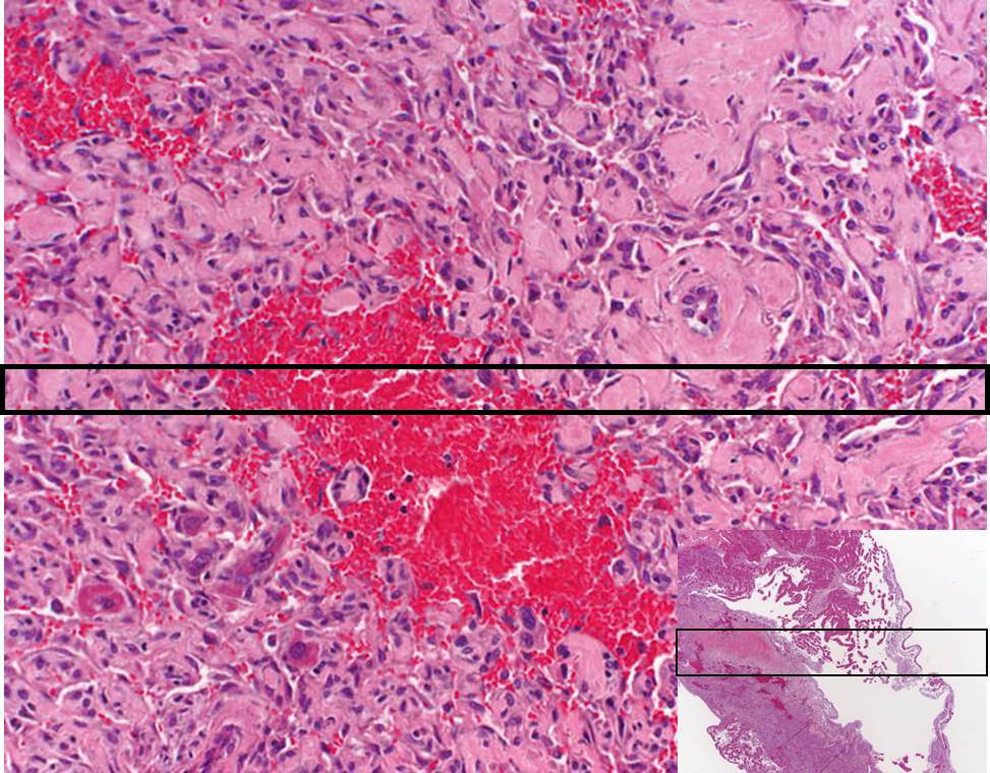
This slide illustrates a hemangiosarcoma in a B6C3F1 mouse liver. The red region is the pooling of blood that occurs when the endothelial cells do not form the normal tube-like blood vessel structure.
Hypoxia has been demonstrated in the liver of mice treated with pregabalin as well as time-related increases in multiple angiogenic growth factors. 49,50 Additionally, the discontinuous endothelium found in liver, spleen, and bone marrow is supported by numerous macrophages (Kupffer cells in the liver), an additional source of angiogenic growth factors. Angiogenesis is controlled by 2 different components, paracrine and autocrine. The paracrine component is driven by nonendothelial expression of angiogenic growth factors such as VEGF and bFGF. In the autocrine component, the ECs themselves are induced to express VEGF. 51 In pregabalin-treated mice, paracrine VEGF and bFGF were demonstrable in bone marrow and spleen and were particularly noteworthy in macrophages, megakaryocytes, and early erythroid cells. 49 In the liver of these animals, VEGFR2 expression was increased. This suggests that both paracrine and autocrine increases in angiogenic growth factors are involved in pregabalin-treated mice, which drive an increase in proliferation rate of ECs. 49,50 The increase in EC proliferation can lead to an enhanced potential to fix spontaneous mutations since more cells are undergoing mitosis, resulting in growth dysregulation, clonal expansion of altered cells, and ultimately hemangiosarcoma formation. 40 Based on that mentioned earlier, the MOA for pregabalin has been established in mice and is consistent with the proposed unified MOA for hemangiosarcoma by other nongenotoxic compounds. 40
Potential effects of pregabalin on key elements of the MOA established in mice were investigated in human clinical trials. It was not ethically possible to duplicate EC proliferation, and hypoxia assays in humans as current methods would have required obtaining liver sections. Therefore, selective biomarkers were evaluated in a clinical trial to assess whether pregabalin treatment at the maximum therapeutic dose altered key elements of the MOA established in mice. Bicarbonate was measured because this is hypothesized to be the initial event in the mouse for the induction of hemangiosarcoma. White blood cell (WBC) counts, platelet numbers, and platelet function were measured as biomarkers of dysregulated erythropoiesis. Since increased EC proliferation is paramount in the production of hemangiosarcomas by nongenotoxic mechanisms and direct assessment of EC proliferation in humans was not possible, thrombomodulin and the generation of Annexin V-labeled circulating endothelial microparticles, markers of EC activation or injury, were evaluated as alternate biomarkers of EC proliferation. Since EC activation is a prerequisite step for EC proliferation to occur in normal angiogenesis, it was considered a relevant biomarker for EC proliferation. There was no evidence that pregabalin-treated patients or volunteers had changes in bicarbonate (the first event in the mouse for hemangiosarcoma formation), dysregulated erythropoiesis (changes in WBC or platelet function), or activated ECs (thrombomodulin, Annexin V, or microparticles), surrogates for EC proliferation. These data support the conclusion that humans do not exhibit the specific responses to pregabalin treatment that were observed in the mouse and hence do not appear to be at increased risk for hemangiosarcoma at the maximum daily dosage of pregabalin (Table 1). Collectively, these data support the conclusion that humans do not exhibit the specific responses to pregabalin treatment that were observed in the mouse and hence are not at increased risk for hemangiosarcoma at the maximum daily dose of pregabalin.
In conclusion, there are a variety of avenues open to progress promising therapies when they have failed at some point in development. Further investigation of these therapies is often warranted, and significant investments have already been made in their advancement. Capitalizing on existing information can hopefully push forward therapies that merit it.
Footnotes
Author Contribution
C. D. Wegner contributed to design, drafted the manuscript, and critically revised the manuscript. A. Goodwin contributed to design and drafted and critically revised the manuscript. J. C. Cook contributed to design and drafted the manuscript. K. Allamneni contributed to conception and design. J. Sohn contributed to conception and design and critically revised the manuscript. M. McVean contributed to conception and design, drafted and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.