
Abstract
DE-71, a commercial mixture of polybrominated diphenyl ethers widely used in flame retardants, is a pervasive environmental contaminant due to its continuing release from waste material and its long half-life in humans. Although the genotoxic potential of DE-71 appears to be low based on bacterial mutagenicity, it remains a public health concern due to its reported involvement in tumor development. Molecular mechanisms by which DE-71 influences tumor incidence or progression remain understudied. We used liver carcinoma tissue from mice exposed to DE-71 to test the hypothesis that epigenetic alterations consistent with tumor development, specifically DNA methylation, result from long-term DE-71 exposure. We profiled DNA methylation status using the methylated-CpG island recovery assay coupled with microarray analysis of hepatocellular carcinoma DNA from animals exposed to DE-71. DE-71 exposure had little impact on global DNA methylation. However, we detected gene body–specific hypomethylation within the Tbx3 locus, a transcription factor important in liver tumorigenesis and in embryonic and cancer stem cell proliferation. This nonpromoter hypomethylation was accompanied by upregulation of Tbx3 mRNA and protein and by alterations in downstream cell cycle–associated marker expression. Thus, exposure to DE-71 may facilitate tumor development by inducing epigenetic programs that favor expansion of progenitor cell populations.
Introduction
Understanding the contribution of the environment to disease processes is important in developing disease prevention strategies. This includes ascertaining the effects of the environment on both genetic and epigenetic programs. Here we examined how epigenetic changes induced by a set of persistent organic pollutants may contribute to disease.
Polybrominated diphenyl ethers (PBDEs) are members of a class of low-molecular-weight organic contaminants that are widespread due to their previous use as flame retardants in many common products, including furniture and electronics. The lipid-soluble components of the pentaBDE mixture, DE-71, eg, BDE 47, BDE 99, and BDE 153, are found in water, food, waste sites, and homes, 1 leading to exposure of both wildlife and humans. The presence of PBDEs in the US population is evidenced by their detection in human serum samples analyzed as part of the Center for Disease Control National Health and Nutrition Examination Survey. 2
Based on evidence of long-range atmospheric transport, environmental persistence, and bioaccumulation in various species, including humans, PBDE congeners, primarily the commercial penta-brominated diphenyl ether mixtures, have been added to the United Nations Economic Commission for Europe list of persistent organic pollutants protocol. 3 The European Union banned the use and sale of certain PBDEs in 2004 and the US manufacturer of penta- and octa-PBDE mixtures voluntarily phased out production that same year. 4 Nevertheless, the potential for PBDE exposure remains a concern because of continued widespread occurrence of these chemicals in the environment and the long half-life (up to 6 years) of PBDEs in humans. 5
Polybrominated diphenyl ether exposure can occur through multiple pathways. Polybrominated diphenyl ethers (including BDE 47, 99, and 153) leach into the environment from materials deposited at waste sites (eg, polyester foams) 6 –10 and are present in various food products including meat, poultry, and fish 11 as well as household dust, 12 with the relative contribution of each source varying from country to country. Of particular concern is the fact that PBDE levels are higher in children than in adults, at least in part because of exposure through mother’s milk and the mouthing behaviors of infants and toddlers. 5 Finally, incineration of PBDE-containing material may result in the formation of brominated dioxins and furans, chemicals that are also a health concern. 13
Recently, the National Institute of Environmental Health Sciences/National Toxicology Program (NTP) conducted PBDE (DE-71) cancer hazard identification studies in rodents and found that DE-71 was a liver carcinogen in both male and female rats and mice. 14 The doses for this 2-year DE-71 study were based on findings from a prechronic study that found doses higher than 50 mg/kg in rats and 100 mg/kg in mice compromised survival. The incidence of total hepatocellular tumors in male rats was 3/49, 2/50, 4/50, and 11/50 and in female rats, 3/50, 2/49, 8/50, and 21/47 for the 0, 3, 15, or 50 mg/kg/d dose groups, respectively. In mice, the incidence of total hepatocellular tumors (including hepatoblastomas in male mice) was 31/50, 40/50, 49/50, and 47/50, and in female mice, 8/50, 8/49, 33/50, and 47/49 for the 0, 3, 30, or 100 mg/kg/d dose groups, respectively. The liver tumors in this 2-year study were diagnosed based on microscopic findings as well as gross findings. The study reported in this article took mouse hepatocellular carcinomas (HCCs) from the control and high-dose groups of male mice in the 2-year study and examined them for treatment-related epigenetic changes. In the 2-year DE-71 study, there were also statistically significant increased incidences of Ctnnb1 mutations in mouse hepatocellular carcinomas following chronic administration of DE-71. 14 Furthermore, BDE 47 has been shown to induce various CYP genes through activation of the nuclear receptors, the constitutive androstane receptor (CAR) and the pregnane X receptor, suggesting a role for inappropriate receptor-mediated gene expression. 15 However, the mechanisms by which PBDEs alter liver tumor development are not fully understood. Since regulation of gene expression depends strongly on specific epigenetic marks, we hypothesized that epigenetic signals important for tumor development and progression might be altered by exposure to PBDEs.
To test this hypothesis, we examined DNA methylation changes in murine hepatocellular carcinomas from the NTP DE-71 study in which liver samples from male mice exposed to DE-71 were available. DNA methylation has been thoroughly documented to be one of the most common epigenetic alterations detected in human cancers. 16 We applied methylated-CpG island recovery assay (MIRA)-chip to analyze hepatocellular carcinomas from DE-71-treated mice and found specific DNA hypomethylation at the Tbx3 locus in DE-71-exposed cells. TBx3 overexpression has been previously shown in various cancers, 17 including hepatocellular carcinoma, 18,19 and has been connected to the oncogenic process. 20 –22 The expression of Tbx3 was upregulated in our mouse liver cells exposed to DE-71, consistent with prior data as well as with the observed DNA hypomethylation in this study. Molecular events downstream of Tbx3 suggest possible molecular mechanisms by which DE-71 could influence hepatocellular carcinogenesis.
Materials and Methods
Animal Study
DE-71 (technical pentabromodiphenyl oxide; CAS No. 32534-81-9; Lot 2550OA30A) was obtained from Great Lakes Chemical Corporation (Middlebury, CT). Components of this mixture include primarily the tetra- through penta-PBDEs with a small percentage of hexabromodiphenyl ethers. 14 In the 2-year study, DE-71 was administered to B6C3F1/N male mice (Taconic Biosciences, Inc.; Rensselaer, NY) by oral gavage in corn oil 5 days per week at doses of 100 mg/kg/d for up to 18 months. 14 Gavage with corn oil alone was used as a control. Tap water and the NTP-2000 diet (Ziegler Brothers, Inc.; Gardners, PA) were available ad libitum. Male mice were housed individually, and liver tissue from 2 to 5 mice was used for analysis as indicated in the figure legends.
Mice were euthanized with carbon dioxide and, as part of the chronic DE-71 study, sections of grossly observed liver masses were minced and stored at −20°C for later RNA/DNA extraction. For histology, sections of the livers were fixed in 10% neutral buffer formalin, embedded in paraffin, sectioned at approximately 5 microns, and mounted on glass slides. Hematoxylin and eosin–stained tissues were evaluated microscopically by a board-certified veterinary pathologist. Hepatocellular carcinomas were diagnosed by the presence of a large mass, disrupting the normal architecture of the liver, composed of proliferations of hepatocytes that formed trabeculae at least 3 cells thick. Blunt-ended trabeculae were present in some sections and gave the appearance of nests of hepatocytes that were unattached to the rest of the tumor. Cells in identified tumors were typically large, with moderate to abundant amounts of eosinophilic cytoplasm, although there were areas in which the cells were small and close together and contained small, round, basophilic nuclei. There was moderate to marked cellular and nuclear pleomorphism, characterized by differences in size, shape, and staining characteristics of the cells and their nuclei. Occasional mitotic figures were present. For this current study, frozen tissues from lesions diagnosed as hepatocellular carcinomas (as shown in Figure 1B) were used for DNA/RNA extraction. An attempt was made to select tumors that did not have large areas of hemorrhage or necrosis, which are not uncommon in hepatocellular carcinomas in mice.
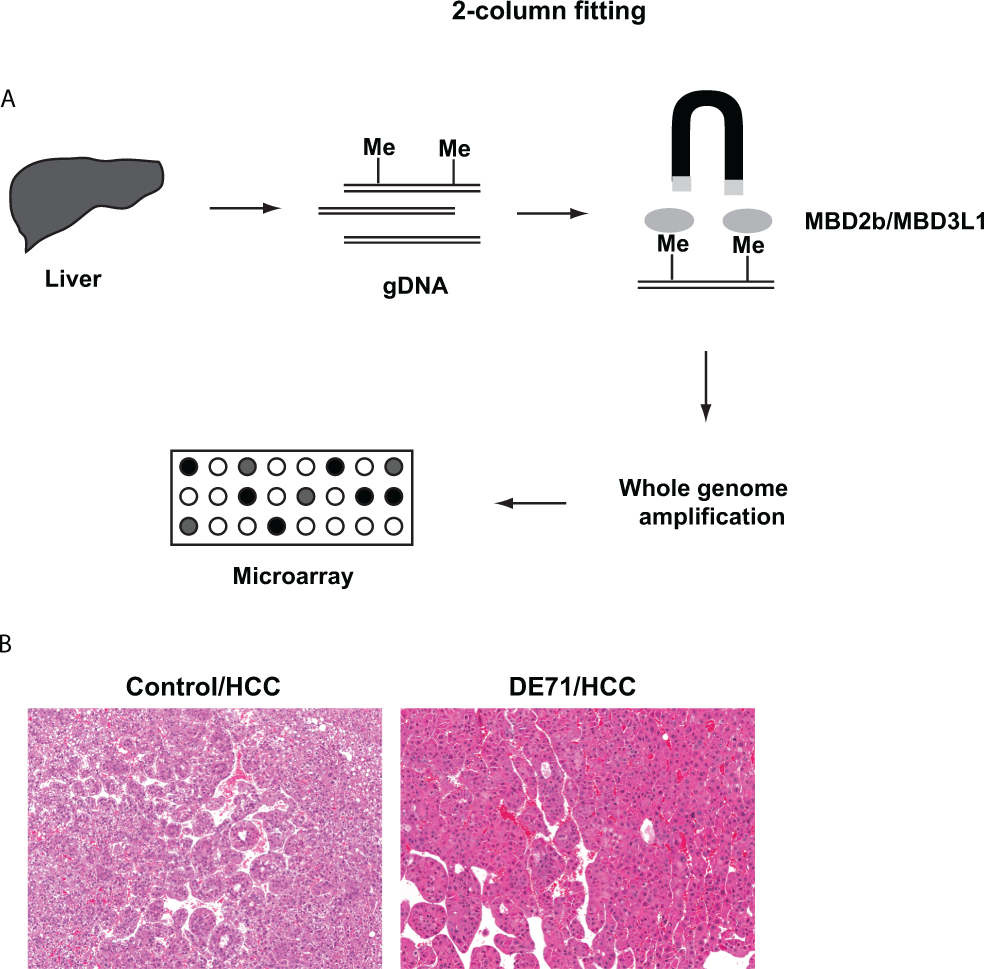
A, Graphical summary of the study. B, Photograph of HCCs from a control mouse and a mouse exposed to 100 mg/kg DE-71 for 528 days. Thick trabeculae are composed of pleomorphic hepatocytes. The H&E images displayed are representative examples of multiple tissues examined from different animals in each group (original magnification = 10×). H&E indicates hematoxylin and eosin; HCCs, hepatocellular carcinomas.
The care of animals on this study was according to NIH procedures as described in the “The US Public Health Service Policy on Humane Care and Use of Laboratory Animals,” available from the Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, Maryland, or at http://grants.nih.gov/grants/olaw/olaw.htm#pol. Study protocols were approved by the local institutional animal care and use committee.
Methylated-CpG Island Recovery Assay-Chip Analysis
The MIRA-chip analysis was performed as previously described. 23 Briefly, genomic DNA was purified and fragmented to 200 to 500 bp. Methylated DNA fragments were enriched using a MethylCollector Ultra kit (Active Motif) according to the manufacturer’s protocol. Input and enriched, methylated DNA were amplified, labeled, and hybridized onto a Nimblegen 2.1M Deluxe mouse promoter array (Roche) according to Nimblegen’s protocol (NimbleGen Arrays User’s Guide, version 6.2). The microarray slide was scanned using a DNA microarray scanner (G2565BA, Agilent Technologies; Santa Clara, CA), and the image was processed using the Nimblescan software.
Microarray Data Normalization
A 2-step normalization approach was used: the first step is designed to correct for GC bias and dye bias within a chip (intrachip correction) and the second step corrects for variations across chips (interchip correction). For intrachip correction, all probes were binned according to their GC content, which was computed as a ratio of C and G nucleotides to the total number of nucleotides in the probe sequence. The overall variability in GC content values was used to compute bin width according to zero-stage rule. 24 These bin widths are proven to be approximately L2 optimal; ie, they minimize mean integrated square error. The bins with fewer probes were then merged so that each bin contained at least 500 probes. Within each bin, Lowess regression was used to predict log-transformed cy5 values as a smooth function of log-transformed cy3 values. 25,26 The scaled (median of absolute residuals was used for scaling) difference between observed and predicted log(cy5) values was used as normalized signal.
For interchip normalization, once the data were corrected for dye and GC bias as described in the first step, quantile normalization was used to correct for between-sample variations. The resulting data set was referred to as normalized data and was used for further investigations. The methylation data from this study have been submitted to the NCBI Gene Expression Omnibus (GSE73326).
Identification of Differentially Methylated Regions or DMRs
A modified algorithm for contacts in a multiphysics environment (ACME algorithm 27 ) was used along with probe-wise difference in average (across all replicates) methylation signal in treated and control samples. This algorithm utilized three user-specified parameters: window size (w), signal threshold (s), and P value threshold (p). The up (or down)-regulated peaks, the number (x) of signal values within a window of size w (centered at probe) that are greater than 100sth percentile (or less than 100[1 − s]th percentile) was first computed across all replicates. Next, the enrichment P value for probe was computed using hypergeometric distribution as following:
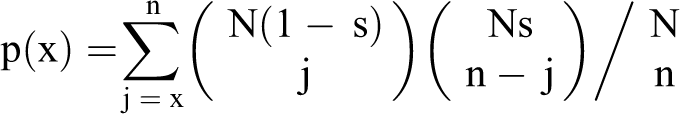
where N denotes total number of probes and n denotes number of probes in the window.
Finally, peaks were identified as runs of enrichment P values that are less than the P value threshold (p). The analysis presented here corresponds to a signal threshold of s = 0.95, a window size of w = 500, and a P value threshold of P = 0.001, with peaks containing less than 4 probes excluded.
Quantitative Real-Time PCR
Total RNA was purified from hepatocellular carcinoma tissue from untreated and DE-71-treated mice using the AllPrep DNA/RNA/Protein Mini Kit (Qiagen; Germantown, MD) and converted to cDNA using an iScrip cDNA synthesis kit (Bio-Rad; Hercules, CA) according to the manufacturer’s protocols. Real-time polymerase chain reaction (PCR) was conducted using iQ SYBR Green Supermix (Bio-Rad) and iCycler iQ (Bio-Rad). All data were normalized to 18S ribosomal RNA (Qiagen, QuantiTect Primer Assay, QT02448075). All data are presented as the mean with standard deviation and analyzed by the 1-way analysis of variance. P value <0.05 was considered as statistically significant.
Primer sequences are described below. Ms_Tbx3_F TTGCAAAGGGTTTTCGAGAC Ms_Tbx3_R TGCAGTGTGAGCTGCTTTCT Ms_p16INK4a_F ATCTGGAGCAGCATGGAGTC Ms_p16INK4a_R TCGAATCTGCACCGTAGTTG Ms_p19ARF_F CCCACTCCAAGAGAGGGTTT Ms_p19ARF_R TCTGCACCGTAGTTGAGCAG
Methylation-Specific PCR
Genomic DNA was purified from mouse hepatocellular carcinomas from either control (corn oil) or DE-71-treated animals. For some, but not all, experiments, matching tissue was available from both carcinoma and an adjacent nontumor liver tissue. In either case, DNA was prepared using the AllPrep DNA/RNA/Protein mini kit (Qiagen). Genomic DNA was treated with sodium bisulfite (EpiTect Bisulfite kit, Qiagen) according to the manufacturer’s protocol. The primers for methylated and unmethylated DNA for CpG 22 region were designed using Meth Primer. 28 Polymerase chain reaction was performed using Amplitaq (Invitrogen/ThermoFisher Scientific; Waltham, MA), and PCR products were separated on an agarose gel.
Primer sequences are described below: MSP_mTbx3_CpG22_MF (methylation-specific) TTTATTATTAATGGTTTTCGTCGC MSP_mTbx3_CpG22_MR (methylation-specific) ACCGCTAAATACTCTACAATTCG MSP_mTbx3_CpG22_UF (unmethylation-specific) TTATTATTAATGGTTTTTGTTGTGG MSP_mTbx3_CpG22_UR (unmethylation-specific) ACCACTAAATACTCTACAATTCACT
Immunoblot
Hepatocellular carcinoma samples from control and DE-71-treated mice were crushed on dry ice and lysed in 1× sample buffer [50 mM Tris-HCl, pH 6.8, 2% (w/v) sodium dodecyl sulfate (SDS), 6% (v/v) glycerol, 1% (v/v) β-mercaptoethanol, 0.004% (w/v) bromophenol blue]. The lysates were sonicated using a Bioruptor for 5 minutes (5 cycles of 30 seconds on and 30 seconds off) at “HIGH” setting and then boiled for 5 minutes. Primary antibodies used were anti-Tbx3 (Invitrogen/ThermoFisher Scientific, #42-4800) and anti-β-actin (ab8226, Abcam; Cambridge, MA).
Results
Previous analysis has shown that after 13 weeks of exposure, DE-71 caused treatment-related liver lesions in mice, including liver hepatocyte hypertrophy, hepatocyte necrosis, and/or hepatocyte cytoplasmic vacuolization. 29 This spectrum of lesions is often associated with the development of liver cancer. 30 To elucidate potential molecular mechanisms by which DE-71 exposure may facilitate tumorigenesis, we investigated the impact of chronic DE-71 exposure on global DNA methylation status in nontumor liver and HCC tissues by MIRA-chip (Figure 1). After purifying genomic DNA from nontumor liver and HCC from DE-71-treated mice, methylated DNA fragments were enriched, amplified, labeled, and hybridized as described in the Nimblegen Mouse ChIP-chip Promoter Array, which covers −8 kb to +3 kb with respect to transcription start sites and about 16,000 CpG islands across the mouse genome. DNA methylation status in all samples showed high correlation values (Table 1), with Spearman correlation coefficient ranging from 0.695 to 0.779 across conditions. These values were quite similar to the correlation coefficients observed between biological replicates. This high correlation indicated that DE-71 had only a subtle impact on global DNA methylation status in the normal and cancerous tissues analyzed (Figure 2A, Supplemental Figure 1).
Overall Correlation Among Normal and Tumor Samples From Control and DE-71-Treated Mice.a
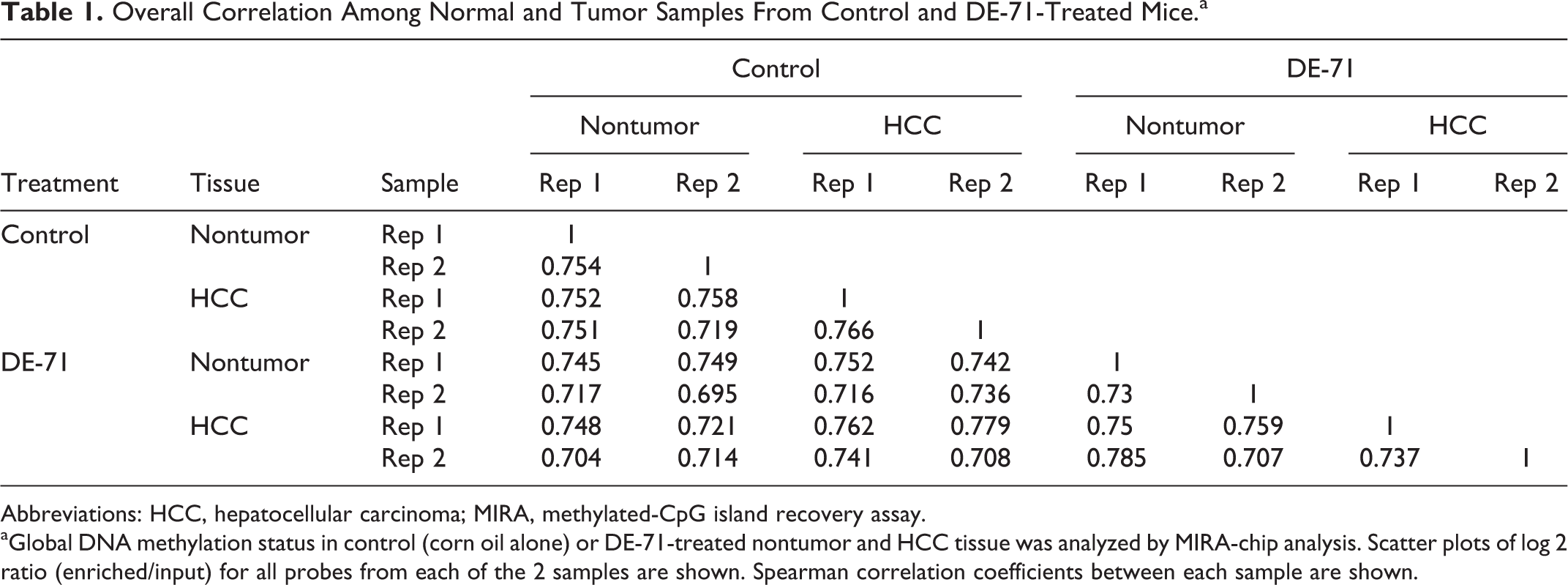
Abbreviations: HCC, hepatocellular carcinoma; MIRA, methylated-CpG island recovery assay.
aGlobal DNA methylation status in control (corn oil alone) or DE-71-treated nontumor and HCC tissue was analyzed by MIRA-chip analysis. Scatter plots of log 2 ratio (enriched/input) for all probes from each of the 2 samples are shown. Spearman correlation coefficients between each sample are shown.
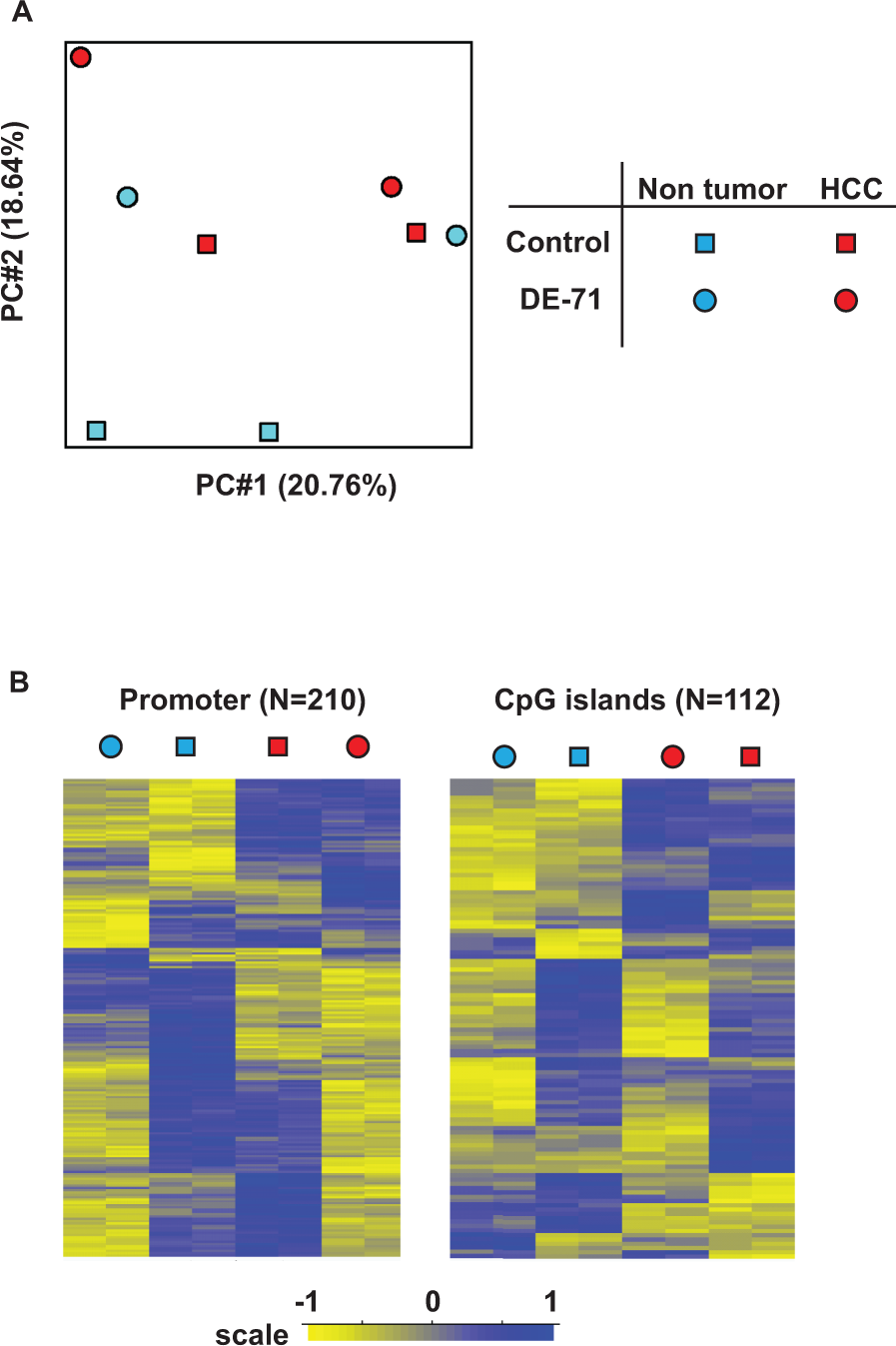
DE-71 induces local loss of methylation but not changes in the global DNA methylation pattern. A, PCA plot of the DNA methylation status in nontumor liver and HCC upon DE-71 treatment. Two animals from each group were analyzed. B, Differentially methylated regions that distinguish each group (same tissues as in panel A were used, with 2 biological replicates from each of the nontumor control, nontumor DE-71-treated, HCC control, and HCC DE-71-treated samples) were identified and presented in a heat map. Specific probe sets both within promoter regions (± 1kb) and across CpG islands were used for analysis. HCC indicates hepatocellular carcinoma; PCA, Principal component analysis.
To further examine the effect of DE-71 on global DNA methylation status, we performed peak overlap analysis. In this analysis, significant local enrichment of methylation signal was defined as a peak (see Methods section), and the percent overlap between each sample was calculated (Table 2). Peak overlap analysis also indicated that DE-71 had only a small impact on global DNA methylation patterns as measured in this assay.
Peak Overlap Analysis of MIRA-Chip Data.
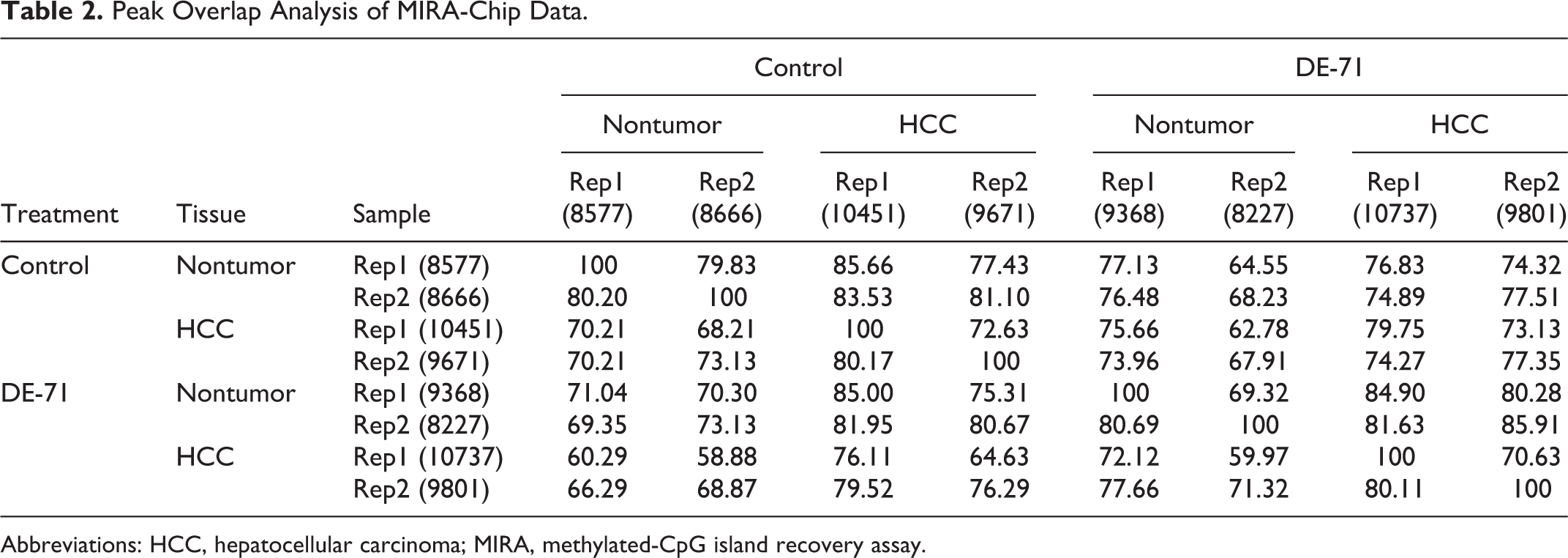
Abbreviations: HCC, hepatocellular carcinoma; MIRA, methylated-CpG island recovery assay.
It is well known that both global and gene-specific DNA methylation changes are observed in hepatocellular carcinogenesis. 31 To gain insight into specific genomic features where DE-71 exposure might have an impact on epigenetic status, we identified differentially methylated regions in promoters and CpG islands using a mixed-effect model (see Methods section). From this analysis, we detected 210 promoters and 122 CpG islands that were differentially methylated (Figure 2B) in tumor and normal liver tissue from control and DE-71-treated mice. We observed that gene promoters displaying differential methylation generally tended to exhibit decreased methylation upon DE-71 exposure (circles vs squares in Figure 2B). Ingenuity pathway analysis of these promoters indicated significant enrichment for genes involved in cell cycle, cell-to-cell signaling, cell growth, and cell movement pathways (data not shown).
A similar trend was observed in CpG islands that showed differential methylation; that is, DE-71 exposure led to decreased methylation at this subset of sites in the genome. We then compared tumor tissue to nontumor tissue and found generally increased levels of methylation at CpG islands from the tumor samples (Figure 2B), but an insufficient number of CpG islands displaying this characteristic were identified to permit a more robust enrichment analysis for this genomic feature.
Though the DE-71 effects on global methylation were subtle, we were aware that it is possible that DE-71 may impact some methylation sites but not others. Careful examination of MIRA-chip data across the genome revealed an interesting differentially methylated region at the Tbx3 locus on the chromosome 5. This locus was consistently hypomethylated in HCC from DE-71-treated animals when compared to HCC from untreated animals (Figure 3A). The DE-71-specific hypomethylation was detected around a CpG island (CpG22) roughly coincident with the final exon of the Tbx3 gene, thus, in a nonpromoter region of the gene. No differential methylation was detected elsewhere at the Tbx3 locus. To validate the hypomethylation detected by microarray, we analyzed the methylation status of CpG22 by methylation-specific PCR (see Methods section). First, we analyzed control and DE-71-treated HCC tissue and found that CpG22 was unmethylated in all DE-71-exposed HCC samples whereas the methylation pattern in tumors from control animals tended to show hypermethylation at this CpG island (Figure 3B) relative to tumors from DE-71-exposed animals. To determine whether DE-71 treatment also changed the methylation status in nontumor tissue, we performed methylation-specific PCR with matched sets of HCC and adjacent nontumor liver tissue from treated animals. In all 4 cases, we observed that only tumors from DE-71-exposed animals exhibited hypomethylation at this region of the Tbx3 locus (Figure 3C). These data suggest that DE-71 influences local DNA methylation status within the Tbx3 gene body preferentially in tumor tissue.
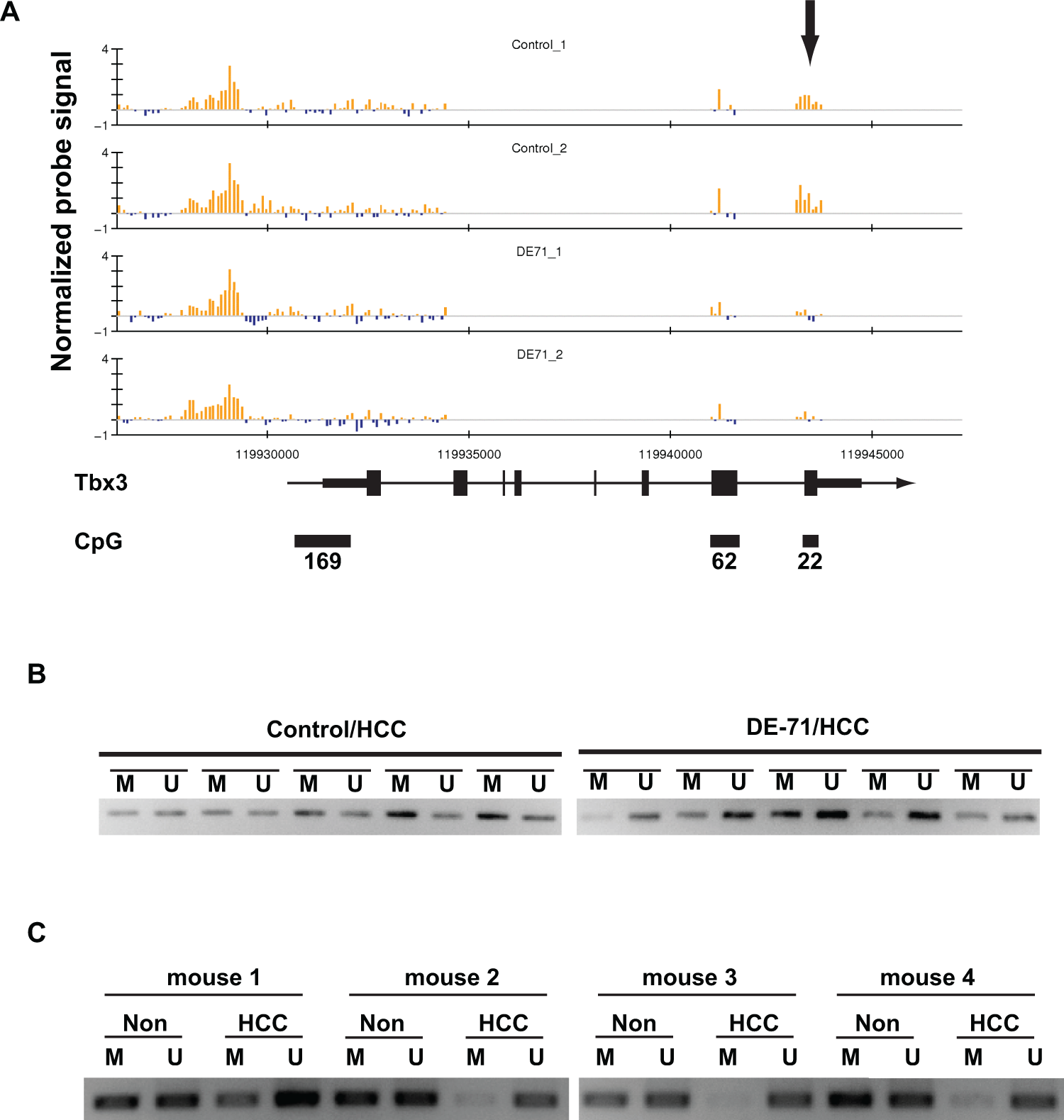
DE-71 treatment leads to hypomethylation at the Tbx3 locus. A, MIRA-chip analysis of control and DE-71-treated HCC across the Tbx3 locus on chromosome 5. Each vertical line indicates normalized log 2 ratio (enriched/input), and blue and yellow bars represent methylated and unmethylated regions, respectively. The direction of transcription, genomic structure of Tbx3 gene, and 3 CpG islands are also indicated. The differentially methylated region is indicated with an arrow. Data are generated from tissues from 2 different animals from each group. B, DNA methylation status at the CpG22 region within Tbx3 locus was analyzed by methylation-specific PCR. For this analysis, we used HCC tissue samples from 5 different control mice and HCC tissue samples from 5 different DE-71-treated mice. C, Methylation-specific PCR was performed for 4 sets of matched HCC and the adjacent nontumor regions in livers collected from the DE-71-treated group. HCC indicates hepatocellular carcinoma; M, methylation-specific primer set; MIRA, methylated-CpG island recovery assay; Non, nontumor region; PCR, polymerase chain reaction; U, unmethylation-specific primer set.
Tbx3 has a critical role in normal liver development; its depletion causes aberrant liver organogenesis. 32 The expression of Tbx3 is silenced in most adult tissues. Therefore, we asked whether hypomethylation in the Tbx3 gene body correlated with expression. We analyzed Tbx3 mRNA expression by qPCR in HCCs from control and DE-71-treated groups and observed significant upregulation of Tbx3 mRNA in HCC derived from DE-71-treated animals (Figure 4A). We also used immunoblotting to examine Tbx3 protein expression levels in animals from the same cohort. Tbx3 protein was readily detected in all 4 DE-71-treated HCC while only 1 HCC from the control group expressed Tbx3 (Figure 4B).
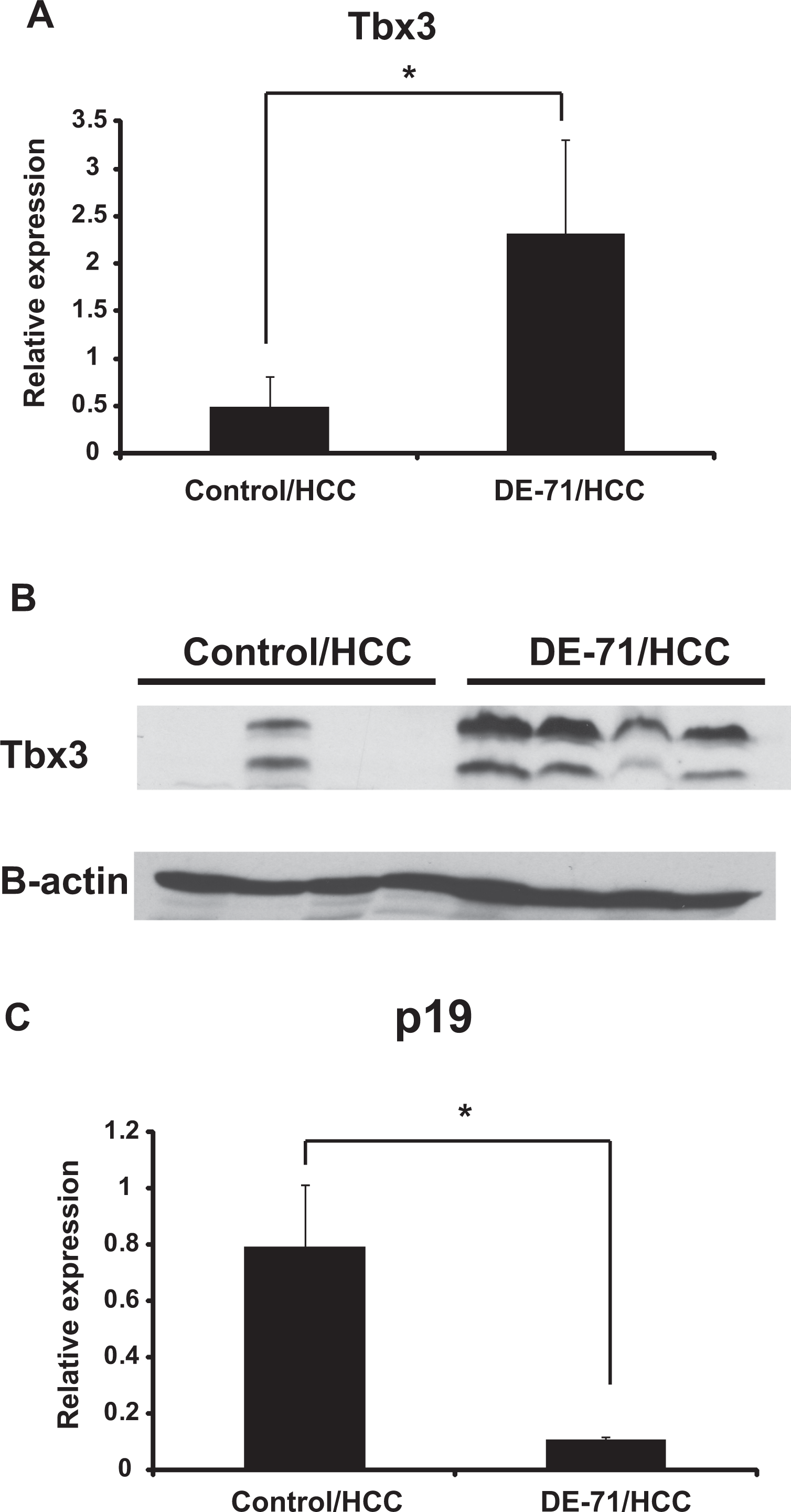
Tbx3 expression is upregulated in DE-71-treated HCC. A, Tbx3 mRNA levels in control and DE-71-treated HCC were analyzed by quantitative real-time PCR (n = 4 biological replicates for each group). The Tbx3 expression levels were normalized to 18S RNA expression; relative values are presented as mean (SD). B, Tbx3 protein expression levels in control and DE-71-treated HCC were analyzed by immunoblotting (n = 4 for each group). β-actin was also detected and used as a loading control. C, p19 mRNA levels in control and DE-71-treated HCC were analyzed by quantitative real-time PCR (n = 5 for each group). The p19 expression levels were normalized for 18S expression; relative values are presented as mean (SD). HCC, hepatocellular carcinoma; mRNA, messenger RNA; PCR, polymerase chain reaction; SD, standard deviation.
To clarify whether upregulation of Tbx3 impacts tumor development, we searched potential targets of Tbx3. Tbx3 is reported to maintain the proliferative state of hepatoblasts by suppressing the tumor suppressor p19ARF, a key regulator of cell cycle progression in normal cells. 32 We therefore tested whether upregulation of Tbx3 in HCC specimens from DE-71-exposed animals was associated with alterations in p19 status. We observed a significant reduction of p19 mRNA in carcinoma samples from DE-71-exposed animals when compared to controls, ie, HCC samples from animals not exposed to DE-71 (Figure 4C).
Discussion
Polybrominated diphenyl ethers are a class of persistent, lipophilic molecules that have been connected to liver toxicity, thyroid toxicity, neurodevelopmental defects, and endocrine disruption. 33 A mixture of PBDEs has been found to be carcinogenic in rats and mice. 14 Since it appears that these compounds are at best weakly genotoxic, their possible role in carcinogenesis through other mechanisms is of significant interest. Strong evidence exists for specific nuclear receptor activation by PBDEs, 15 which may be responsible for the disruption of multiple cell metabolic pathways. Another potential pathway of interest is the epigenetic program that controls gene expression. 16 In this study, we examined the DNA methylation status in murine hepatocellular carcinoma cells following DE-71 exposure. We show for the first time that long-term exposure to a mixture of PBDE’s leads to changes in DNA methylation at a specific gene, Tbx3, known to influence proliferative cell development. The increased expression of Tbx3 in murine hepatocellular carcinoma and the concomitant changes in downstream factors, including p19 and cell cycle markers, suggest a mechanism by which these nongenotoxic environmental contaminants may lead to an enhanced incidence of detectable cancers in exposed populations.
Tbx3 functions as a transcriptional repressor, and the mutation of Tbx3 is found in the human genetic developmental disorder, Ulnar-Mammary Syndrome. 34,35 Tbx3 is essential for liver organogenesis through the suppression of p19ARF, a known p53 pathway tumor suppressor gene; small and immature liver development is observed in a Tbx3 knockout mouse. 32 Although the expression of Tbx3 is suppressed in adult liver, the reactivation of Tbx3 is reported in human liver tumor samples, corresponding to Wnt/β-catenin activation. 19 It is interesting that connections exist between the activation of the nuclear xenobiotic receptor CAR and β-catenin and murine liver tumorigenesis. 36 Whether and how these specific receptor-mediated gene activations and epigenetic reprogramming are connected is an important but understudied aspect of the response of cells to these environmental pollutants.
The upregulated Tbx3 closely correlates to the high proliferation rate of these liver tumor cells, and hepatic tumor patients with high Tbx3 expression display a poor prognosis compared to patients with low liver Tbx3 expression. Some evidence has supported Tbx3 reactivation in tumor pathogenesis, but the detailed mechanism of the reactivation is not fully understood. Published reports indicate that Tbx3 may negatively effect tumor proliferation while supporting a more invasive phenotype. 19,37,38 Other studies find the Tbx3 may inhibit the tumor suppression activity of the p19ARF-Mdm2-p53-p21 pathway. 17 The correlation in our study between Tbx3 expression and indications of proliferation occurs in the presence of a specific microenvironment in which a particular PBDE toxicant is present, and thus, may be unique for those conditions. Our observations do not rule out a role for Tbx3 in the development of aggressive cancer phenotypes, which may in fact be selected for during the evolution of cancer genotypes that arise during the rapid proliferation of cancer cell clones.
Several studies have found that Tbx3 can be epigenetically silenced by methylation in glioblastoma, gastric, and bladder cancers. 39 There is also strong evidence that Tbx3 expression is linked to oncogenic processes. 20 –22 Our findings suggest that changes in methylation patterns induced by environmental toxicants, specifically hypomethylation in a localized nonpromoter region, may initiate changes in genetic programs, such as that involving Tbx3 regulation of p19ARF, that result in altered proliferative behaviors. Elucidating the precise details and patterns of altered methylation is critical for understanding the ultimate effects of toxicants on disease initiation and progression.
It is unlikely that DE-71 induces Tbx3 expression directly, because there was no Tbx3 upregulation in DE-71-treated normal liver. Given the changes in proliferative markers, one possibility is that the tumor microenvironment, generated by the combination of tumor cells and DE-71 treatment, selects for Tbx3-expressing tumor cells. It is well known that selective pressures can drive tumor cell mutagenesis, which then provides a growth advantage to cancer cells. 40 A more detailed and comprehensive study of gene expression in DE-71 HCC and nontumor tissue would be an interesting component of future studies that might shed light on patterns of gene expression in an environment of tumor formation and PBDE exposure.
We also showed that methylation changes occurred in localized, nonpromoter regions of the Tbx3 locus, in this case within the terminal exon of Tbx3, rather than throughout the gene, and correlated with the upregulation of Tbx3 in DE-71-treated HCC. We did not uncover significant changes in the global DNA methylation status of cells exposed to DE-71. However, previous studies showed that while changes in global DNA methylation patterns occur throughout hepatocellular carcinogenesis, regional, and gene-specific hypo- and hypermethylation occur primarily in the later stages of carcinogenesis. 31 This suggests that DE-71 may be involved in the progression, rather than the initiation, of hepatocellular carcinogenesis. As indicated above, however, there have been previously observed increased incidences of Ctnnb1 mutations in mouse hepatocellular carcinomas following chronic administration of DE-71. 14 The combination of these studies suggests that there may be multiple mechanisms at work for how exposure to PBDEs enhances cancer development: mutations that drive cancer initiation and late stage epigenetic alterations that, under appropriate conditions, can generate more aggressive cancer progression. Thus, reducing exposure to PBDEs may reduce both genetic and epigenetic changes regulating pathways that impact the formation of malignancies (Figure 5). Furthermore, this study addressed only one of the many epigenetic marks that control gene expression, and it would be of interest to understand the full epigenetic landscape in animals exposed to PBDEs.
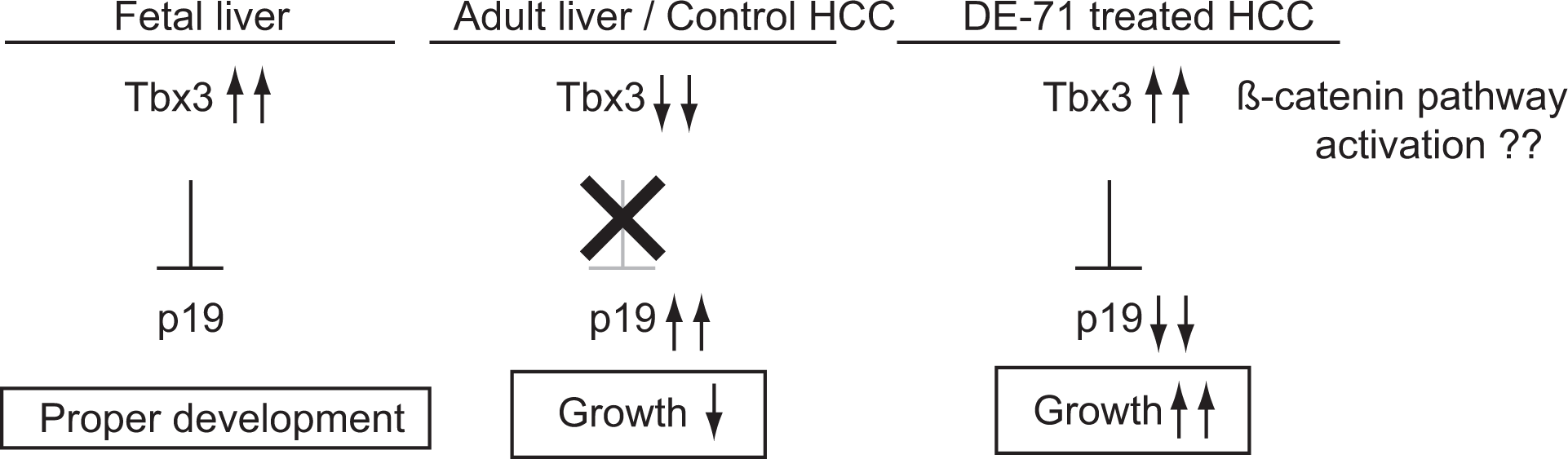
A model of the mechanism by which DE-71 induces hepatocellular carcinogenesis.
The ability to monitor the effects of environmental exposures is a critical component in identifying risks to human health. A recent cost-effectiveness study found that eliminating disease-causing environmental exposures is an important strategy in developing health-care policy. 41 The MIRA-chip approach used in this study successfully detected a methylation status change at the Tbx3 locus and suggested a potential mechanism for how DE-71 affects hepatocellular carcinogenesis. This method requires a minimal amount of genomic DNA (∼200 ng), allowing its use with limited materials such as those frequently obtained from human clinical samples. Thus, MIRA-chip provides researchers with a promising tool for understanding at least 1 type of epigenetic regulation and a connection to the influence of toxic compounds and/or environmental factors on human health.
Footnotes
Acknowledgments
The authors gratefully acknowledge the members of the Wade laboratory for many useful discussions and comments in the course of this work. The authors would also like to thank Dr Linda S. Birnbaum, NIEHS, for many helpful discussions throughout the study and careful readings of the manuscript during preparation. The authors also thank the NIEHS microarray facility for their assistance in MIRA-chip experiment. This work was supported by the intramural program of the National Institute of Environmental Health Sciences (NIEHS), Research Triangle Park, North Carolina. The statements, opinions, or conclusions contained therein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the US government. The in-life portion of this study was conducted under the supervision of Dr C. Hébert, Southern Research Institute, under NIEHS contract NO1-ES-45516.
Author Contributions
T. Shimbo contributed to conception, design, acquisition, analysis, interpretation, and drafted the manuscript. J. K. Dunnick contributed to conception, design, acquisition, analysis, interpretation, and critically revised the manuscript. A. Brix contributed to acquisition, analysis, and critically revised the manuscript. D. Mav and R. Shah contributed to analysis and critically revised the manuscript. J. D. Roberts contributed to analysis and interpretation, drafted the manuscript, and critically revised the manuscript. P. A. Wade contributed to conception and design, analysis, interpretation, drafted the manuscript, and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the Division of Intramural Research (DIR) of the National Institute of Environmental Health Sciences (NIEHS), and the Division of the National Toxicology Program (DNTP), NIEHS, National Institutes of Health, Research Triangle Park, North Carolina.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.