
Abstract
Benzo[a]pyrene (B[a]P) exposure has been associated with the alteration in epigenetic marks that are involved in cancer development. Biotinidase (BTD) and holocarboxylase synthetase (HCS) are 2 major enzymes involved in maintaining the homeostasis of biotinylation, and the deregulation of this pathway has been associated with a number of cancers. However, the link between B[a]P exposure and the dysregulation of BTD/HCS in B[a]P-associated tumorigenesis is unknown. Here we showed that the expression of both BTD and HCS was significantly decreased upon B[a]P treatment in human bronchial epithelial (16HBE) cells. Benzo[a]pyrene exposure led to the global loss of DNA methylation by immunofluorescence, which coincided with the reduction in acetylation levels on histones H3 and H4 in 16HBE cells. Consistent with decreased histone acetylation, histone deacetylases (HDACs) HDAC2 and HDAC3 were significantly upregulated in a dosage-dependent manner. When DNA methylation or HDAC activity was inhibited, we found that the reduction in BTD and HCS was separately regulated through distinct epigenetic mechanisms. Together, our results suggested the potential link between B[a]P toxicity and deregulation of biotin homeostasis pathway in B[a]P-associated cancer development.
Introduction
The increased levels of environmental pollutants from manufacturing processes and anthropogenic activities have resulted in the contamination of human habitats with polycyclic aromatic hydrocarbons (PAHs), thus posing as a major environmental health problem. Benzo[a]pyrene (B[a]P) is the most common PAH and ranks among the top 20 in the National Priority List of Hazardous Substances in the United States. 1 It is a procarcinogen and an environment toxicant present in car exhaust, incomplete fossil fuel combustion, municipal waste incineration, tobacco smoke, and certain foods. 2 For most people, the primary route of environmental exposure to B[a]P is inhalation. 3 It has been proven that B[a]P may cause abnormal expression of genes in the early stage of tumorigenesis in human cells, which in turn may cause uncontrollable growth or promote tumorigenesis by increasing resistance to programmed cell death in various cell lines. 4 -6
Recent investigations have shed light on the role of B[a]P in epigenetic modifications, suggesting another pathway for B[a]P-associated cancer development. Sadikovic and Rodenhiser confirmed that B[a]P may lead to dynamic, sequence-specific patterns of hypo- and hypermethylation in breast cancer cells. 7 In addition, 5 of 6 B[a]P-induced hypomethylation events directly overlapped with genomic repeat elements, and 4 of these were short interspersed nucleotide repetitive elements. 8 These observations are in line with the genome-wide DNA hypomethylation and locus-specific hypermethylation on target gene promoters in most of the cancer genome. 9 Moreover, changes in histone acetylation have also been observed in genes and gene networks of B[a]P-treated MCF7 breast cancer cells. 8 Importantly, increased histone deacetylase (HDAC) activity has been shown in a variety of cancers, and HDACs have been the promising therapeutic targets for treatment of cancer. 10
Biotin, a water-soluble vitamin that functions as an essential coenzyme for carboxylases in mammals, is covalently bound to a lysine residue of the carboxylase proteins in acetyl-CoA carboxylase, propionyl-CoA carboxylase, 3-methylcrotonyl-CoA carboxylase, and pyruvate carboxylase. 11 Biotin homeostasis is crucial for maintaining normal body functions and is associated with cell proliferation and the metabolism of fats and amino acids. 12,13 Biotin homeostasis requires 2 major proteins, biotinidase (BTD) and holocarboxylase synthetase (HCS). 14 The discovery that BTD has biotinyl-transferase activity, in addition to BTD- hydrolase activity, presents the possibility that biotin may directly participate in epigenetic changes, and the specific transfer of biotin to histones by BTD provides a possible explanation for the occurrence of biotin in the nucleus of eukaryotic cells as well as its role in the regulation of protein transcription. 15 In addition, recent study indicated that nuclear HCS mediates events of transcriptional repression by interacting with chromatin-modifying protein complexes such as HDACs. 16 Furthermore, the rate of biotinylation had increased in all classes of histones in G1, S, G2, and M phases of the cell cycle compared to that of the G0 phase, which suggests that histone biotinylation is a part of a general mechanism that occurs in the nucleus. 17 Importantly, downregulation of BTD protein expression has been associated with breast cancer and papillary thyroid cancer development and has been suggested as a potential biomarker. 18,19 However, the link between B[a]P exposure and the deregulation of biotin homeostasis in B[a]P-related tumorigenesis has not been described previously.
The aim of the present study was to investigate whether the expression of these biotin-related proteins was altered by exposure to B[a]P as well as its underlying mechanism. The normal human bronchial epithelial cell line (16HBE) was employed in this study as an in vitro model. Our results showed that B[a]P treatment reduced the expression of both BTD and HCS. In addition, the inhibition of BTD and HCS was due to B[a]P-induced epigenetic changes. Together, our studies presented in this report, for the first time, establish a connection between B[a]P and biotin-related proteins and provide some potential underlying epigenetic links.
Materials and Methods
Chemicals, Materials, and Cell Lines
Benzo[a]pyrene was purchased from Sigma-Aldrich (St Louis, Missouri). Minimum Essential Medium (MEM), antibiotics (penicillin and streptomycin), fetal bovine serum, and trypsin–EDTA solution were all purchased from Gibco BRL-Life Technologies (Grand Island, New York). Dimethyl sulfoxide was purchased from Bio Basic, Inc (Toronto, Canada). The 16HBE cell line was a kind gift from Professor Gruenert D.C. (University of California, San Francisco, California). Trichostatin A (TSA) and 5-aza-2′-deoxycytidine (Aza) were from Sigma. Trichostatin A was stored at −20°C, whereas Aza was dissolved in sterile deionized water at a concentration of 50 μmol/L and stored at −20°C.
Cell Culture and Chemical Treatment
Cells were cultured in MEM medium supplemented with 10% (vol/vol [v/v]) heat-inactivated fetal calf serum and antibiotic supplement (penicillin 100 U/mL and streptomycin 100 μg/mL) at 37°C in a humidified incubator with 5% CO2. When the cultured cells had grown to about 80% confluency, these were treated with different concentrations of B[a]P, ranging from 2.5 to 40 μmol/L (<1/2 IC50) for 24 hours, with 0.1% sterile deionized water as solvent control. The 16HBE cells were also treated either in the absence or in the presence of B[a]P (40 μmol/L) for 24 hours followed by the treatment with either TSA (500 nmol/L) or Aza (10 μmol/L) for another 24 hours. Three independent experiments were performed.
Confocal Microscopy Analysis
Fluorescence staining experiments were conducted as described elsewhere. 20 After treatment with B[a]P for 24 hours, 16HBE cells, grown on coverslips, were fixed in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (PBS) for 5 minutes, followed by washing 3 times with PBS prior to permeabilization by 0.3% Triton-X in PBS for 5 minutes. The fixed cells were then incubated with primary antibodies diluted 1:1000 in 1% BSA-phosphate-buffered saline with Tween 20 (PBST) for about 16 hours at 4°C, washed with PBS, and incubated with fluorescein-labeled secondary antibody diluted 1:400 in PBST at 37°C for 30 minutes in the dark and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 5 minutes. All immunofluorescence staining was observed using confocal laser scanning microscopy (Leica Tcs sp5, Leica Microsystems, Wetzlar, Germany) under dark field, and the images were analyzed using the Image-Pro Plus software. Three independent experiments were performed. The following antibodies were used in this study: anti-5-mC antibody (NA81; Calbiochem, Darmstadt, Germany) and immunoglobulin G fluorescein isothiocyanate (IgG-FITC) secondary antibodies (Santa Cruz, California,).
Western Blot Analysis
After the indicated treatments, total protein of 16HBE cells was extracted with lysis buffer (7 mol/L urea, 2 mol/L thiourea, 4% (3-[(3-Cholamidopropyl)dimethylammonio]propanesulfonate CHAPS), and 10 mmol/L Tris). The lysates were then centrifuged, and the insoluble debris was discarded. After boiling at 99°C for 5 minutes, the proteins were separated on 10% polyacrylamide gels and transferred onto polyvinylidene fluoride membranes. Membranes were blocked with Tris-buffered saline containing 0.1% (v/v) Tween-20 and 5% fat-free milk for 2 hours at room temperature, then incubated with primary antibodies overnight at 4°C, and then with secondary antibodies for 1 hour at room temperature. Glyceraldehyde-3-phosphate dehydrogenase was used for normalization of protein levels. Antibody signals were detected using the ImageJ quantification software (National Institutes of Health, Bethesda, MD, USA). Three independent experiments were performed. The following antibodies were used in this study: HDAC2 (C-8) antibody (sc-9959; Santa Cruz), HDAC3 (N-19) antibody (sc-8138; Santa Cruz), DNA (cytosine-5)-methyltransferase 1 (DNMT1) (H-12) antibody (sc-271729; Santa Cruz), DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3b) (52A1018) antibody (sc-52922; Santa Cruz), Methyl-CpG-binding domain protein 2 (MBD2) (H-70) antibody (sc-10752; Santa Cruz), BTD (K-17) antibody (sc-48432; Santa Cruz), HCS (N-19) antibody (sc-23732; Santa Cruz), anti-acetyl-histone H3 antibody (06-599; Upstate, Darmstadt, Germany), antihistone H3 antibody (ab137760; Abcam, Cambridge, United Kingdom), anti-acetyl-histone H4 antibody (06-598; Upstate), and antihistone H4 antibody (ab7311; Abcam).
Statistical Analysis
Statistical analysis was conducted using 1-way analysis of variance with post hoc Bonferroni correction. The data were presented as the mean ± standard deviation. All experiments were repeated at least 3 times independently. Results were considered to be statistically significant at P ≤ 0.05.
Results
Exposure to B[a]P Leads to Decreased BTD and HCS in 16HBE Cells
Benzo[a]pyrene exposure has been associated with the increased cancer formation through both genetic and epigenetic changes. 21 Consistent with these notions, benzo[a]pyrene diolepoxide (BPDE), the major B[a]P metabolite, has been previously linked to the development of lung cancer in normal 16HBE cells through the formation of BPDE–DNA adducts at mutational hot spots at gene body of tumor suppressor P53. 22 Interestingly, the reduction in BTD expression has also been associated with cancer development, 18,19 and we hypothesized whether there was a link between B[a]P exposure and the change in expression of BTD as well as another key protein in the same biotin homeostasis pathway—HCS. To test our hypothesis, we selected 16HBE cells and treated with B[a]P for 24 hours followed by measurement for BTD and HCS using both Real Time (RT) polymerase chain reaction and Western blotting. In this study, we chose to use normal 16HBE, as exposure to B[a]P compounds is a well-documented cause of respiratory cancers. 23,24 In our previous study, we treated 16HBE cells with different doses of B[a]P for 24 hours and found that cell viability decreased by 14.6%, 16.5%, 40.7%, and 64.4% at 10, 20, 50, and 100 μmol/L BaP, respectively. 25 Therefore, in our study, we chose 24 hours as the incubation time, and 40 μmol/L (<1/2 LC50) was chosen as the highest concentration. To support our hypothesis, we found the mRNA (data not shown) and corresponding protein levels of both BTD and HCS were decreased in a dosage-dependent manner when cells were treated with B[a]P (Figure 1). Specifically, the levels of BTD and HCS were increased by 9.33% and 2.67% at 2.5 μmol/L BaP, respectively; then, the levels of BTD were decreased by 3.33%, 11.32%, 16.97%, and 22.21% at 5, 10, 20, and 40 μmol/L BaP; and the decreased percentage of HCS became 5.26%, 9.67%, 18.82%, and 32.54% at the same exposure dose. These results suggest a link between B[a]P exposure and the reduction in BTD/HCS in B[a]P-associated cancer development.
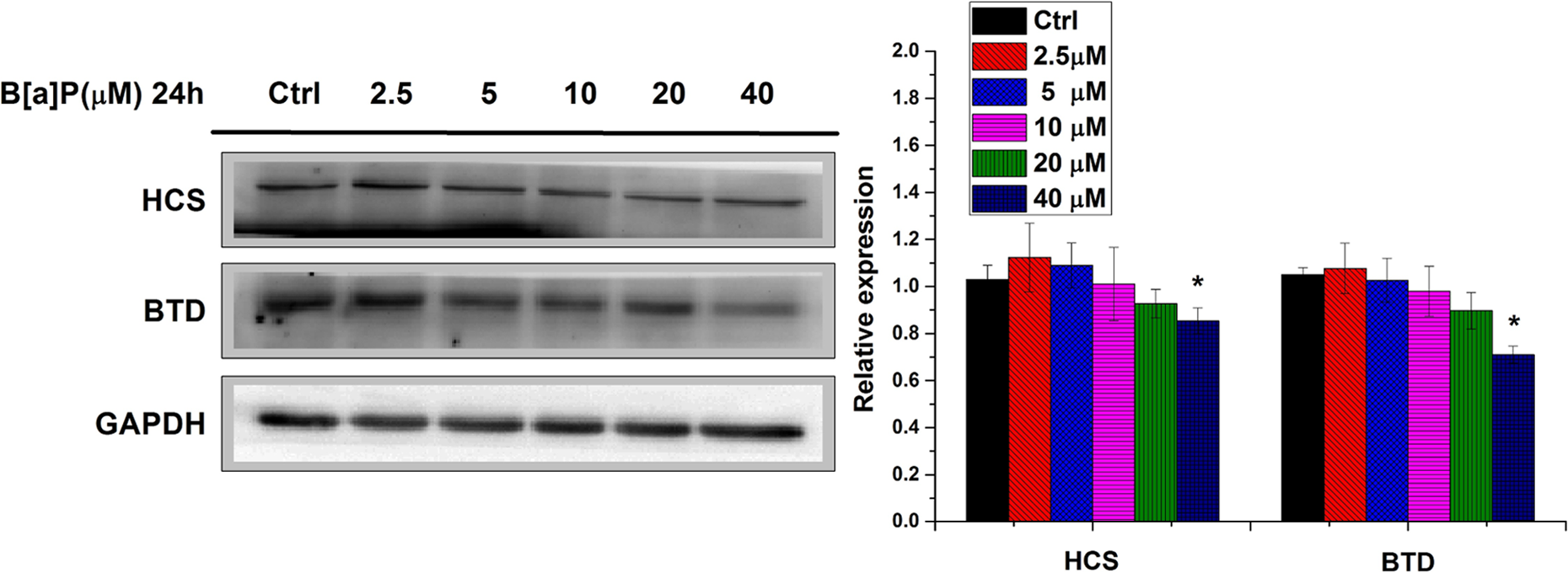
B[a]P exposure leads to decreased BTD and HCS protein levels in 16HBE cells. After indicated treatment, total protein of 16HBE cells was subjected to SDS-PAGE and probed with BTD or HCS-specific antibodies. Data are presented in terms of percentage versus the results using non-B[a]P-treated controls, which were assigned a value of 100% and as means ± standard deviations. Significant difference versus non-B[a]P-treated control, *P < 0.05. B[a]P indicates benzo[a]pyrene; BTD, biotinidase; HCS, holocarboxylase synthetase; 16HBE, human bronchial epithelial cells; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Benzo[a]pyrene Treatment Results in Global Loss of DNA Methylation in 16HBE Cells
One of the hallmarks of cancer is the global level of hypomethylation in cancer genome. 9 Given that B[a]P exposure has been associated with the increased rate of various cancers during epidemiology studies, 26 we reason that B[a]P exposure may associate with genome-wide loss of DNA methylation. To test this, we treated 16HBE cells with B[a]P for 24 hours, followed by detecting the global level of DNA methylation by immunofluorescence staining using antibody against 5-methyl Cytosine. As shown in Figure 2A and B, the global level of DNA methylation in B[a]P-treated cells was decreased in a B[a]P concentration-dependent manner. Specifically, global DNA methylation decreased by 3.43%, 9.27%, 23.76%, 32.55%, and 43.15% at 2.5, 5, 10, 20, and 40μmol/L BaP, respectively. Compared to the control group, DNA methylation of 16HBE cells was decreased significantly from 20 μmol/L of BaP treatment (P < 0.05).
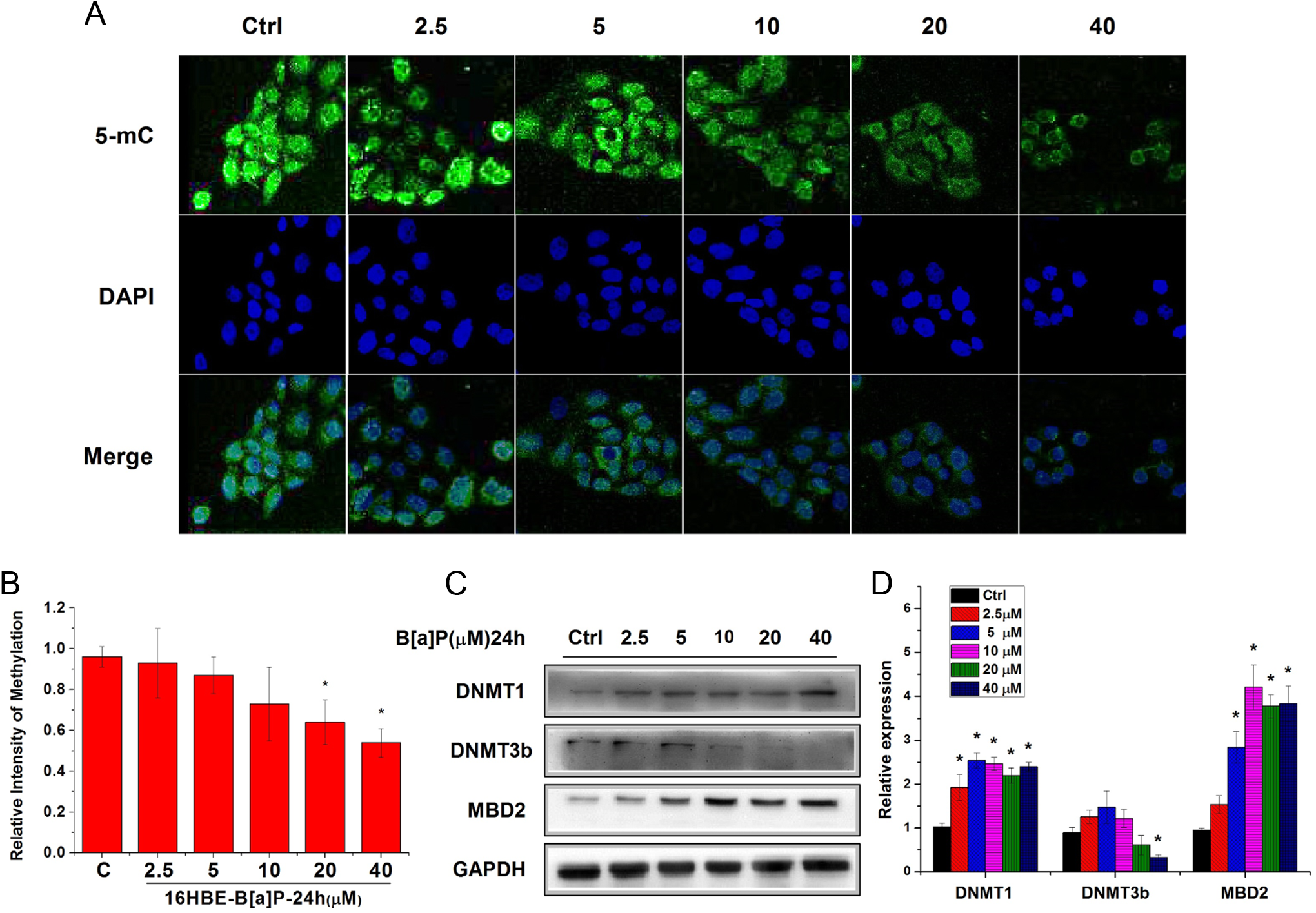
B[a]P treatment results in global loss of DNA methylation in 16HBE cells. A and B, Confocal microscopy analysis of 5-mC-positive cells using a 5-mC-specific antibody; DAPI was used as control. C and D, Total protein of 16HBE cells was subjected to SDS-PAGE and probed with DNMT1, DNMT3b, or MBD2-specific antibodies. Data are presented in terms of percentage versus the results using non-B[a]P-treated controls, which was assigned a value of 100%, and as means ± standard deviations. Significant difference versus non-B[a]P-treated control, *P < 0.05. B[a]P indicates benzo[a]pyrene; 16HBE, human bronchial epithelial cells; DAPI, 4′,6-diamidino-2-phenylindole; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; DNMT, DNA methyltransferases; MBD2, methyl-CpG-binding protein 2.
DNA methylation is directly maintained by DNA methyltransferases (DNMTs), including DNMT1, DNMT3a, and DNMT3b, and recognized by methyl-binding proteins, such as methyl-CpG-binding protein 2 (MBD2). 27,28 While DNMT1 is required to maintain methylation patterns following DNA replication, 29 DNMT3a and DNMT3b are essential for de novo methylation and mammalian development. 30 Methyl-CpG-binding protein 2 is the only member of the methyl-binding protein family that has been reported to be both a DNA demethylase and a transcriptional repressor. 31 Therefore, we set out to determine the protein expression levels of DNMTs and MBD2 after treatment of cells with B[a]P for 24 hours. Benzo[a]pyrene showed no effect on DNMT3a (data not shown), but the protein expression levels of DNMT1 and MBD2 (Figure 2C and 2D) were significantly increased from 10 μmol/L of BaP treatment (P < 0.05). The result seems in contradictory, however, to those of a previous study, which indicated that although B[a]P increased the protein expression level of DNMT1, it sharply reduced its enzymatic activity level. 32 In addition, overexpression of MBD2 may have a direct role in the genome-wide DNA hypomethylation. 33 On the other hand, the level of DNMT3b, a DNA methyltransferase that is thought to function in de novo methylation, was reduced by up to 32% at 40 μmol/L, relative to untreated controls. Thus, these data suggest that the alteration of DNMT3b and MBD2 may be involved in the B[a]P-induced reduction in DNA methylation.
Benzo[a]pyrene Reduces Histone H3 and H4 Acetylation Levels in 16HBE Cells
Methyl-CpG-binding protein 2 has been previously demonstrated to interact with HDAC repression complex for gene repression. 34 In line with the increased MBD2 protein level, when treated with B[a]P, Western blot analysis showed that B[a]P increased both HDAC2 and HDAC3 levels, and this effect of B[a]P was concentration dependent (Figure 3B) and significant from 10 μmol/L of BaP treatment (P < 0.05). Consistent with the globally increased protein expression of HDAC2 and HDAC3, Western blot revealed that high concentrations (20 and 40μmol/L) of BaP caused a significant decrease (P < .05) in H3 and H4 acetylation, and a dose-dependent manner was shown (Figure 3A).
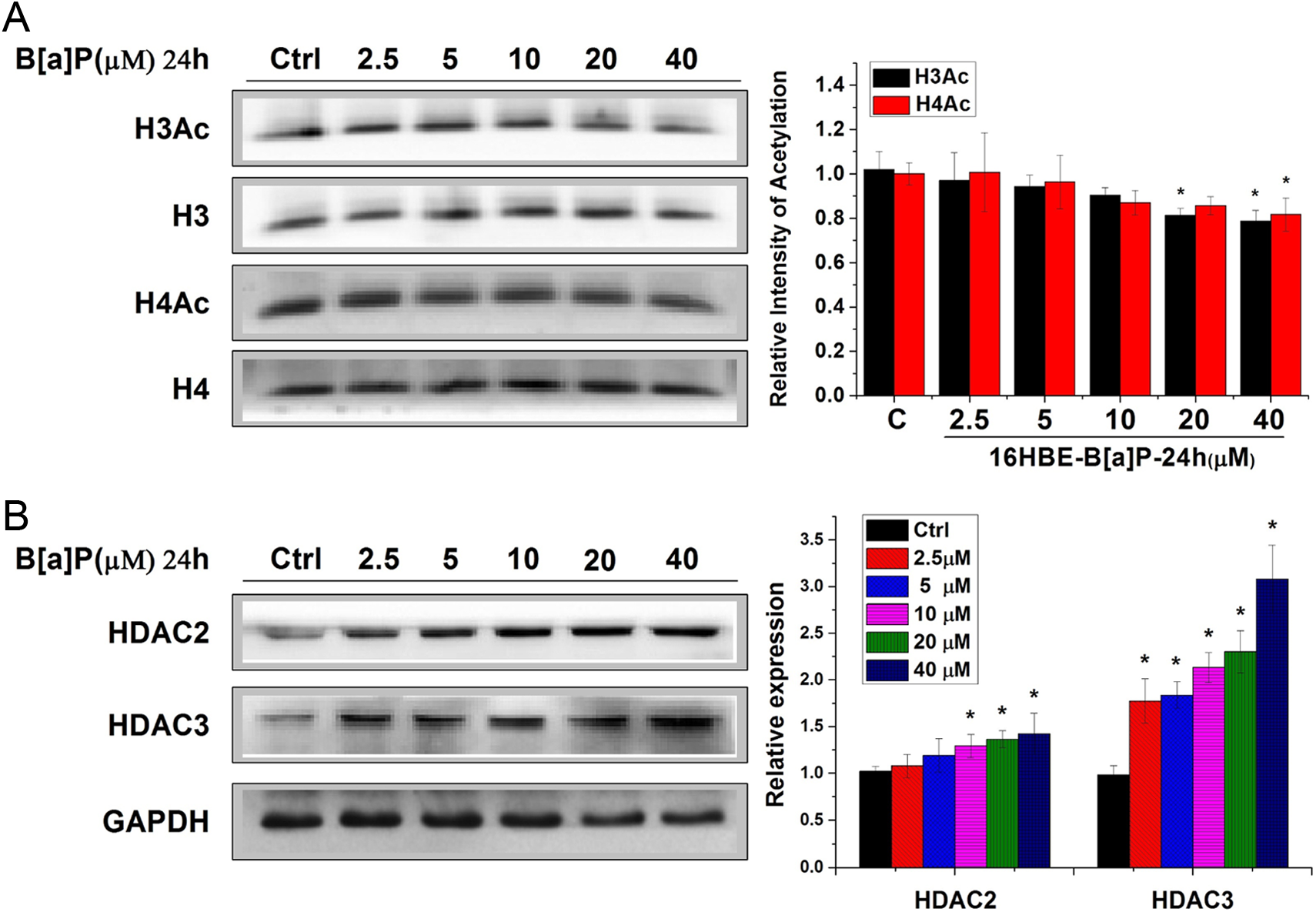
B[a]P reduces histone H3 and H4 acetylation levels in 16HBE cells. A, Total protein of 16HBE cells was subjected to SDS-PAGE and probed with H3 acetyl- or H4 acetyl-specific antibodies. B, Total protein of 16HBE cells was subjected to SDS-PAGE and probed with HDAC2- or HDAC3-specific antibodies. Data are presented in terms of percentage versus the results using non-B[a]P-treated controls, which were assigned a value of 100%, and as means ± standard deviations. Significant difference versus non-B[a]P-treated control, *P < .05. B[a]P indicates benzo[a]pyrene; 16HBE, human bronchial epithelial cells; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; HDAC, histone deacetylase.
Holocarboxylase Synthetase and BTD may be Regulated Through Distinct Epigenetic Mechanisms in 16HBE Cells
To further explore the relationship between loss of DNA methylation, increased histone deacetylation, and changes in biotin-related proteins in 16HBE cells, we examined the effect of B[a]P, TSA, which is a Streptomyces metabolite that specifically inhibits mammalian HDAC and causes accumulation of highly acetylated histone molecules in cells, and Aza, which is an epigenetic modifier that inhibits DNMT activity and thus causes DNA demethylation or hemidemethylation, on HCS and BTD. In order to dissect what epigenetic changes may be mainly responsible for the change in the expression of BTD and/or HCS, 16HBE cells were treated either in the absence or in the presence of B[a]P (40 μmol/L) for 24 hours followed by the treatment with either TSA (500 nM) or Aza (10 μmol/L) for another 24 hours. In the absence of B[a]P treatment, the treatment of Aza resulted in the increased protein expression of HCS but did not change that of BTD. In contrast, TSA treatment led to the increased protein expression of BTD but did not alter that of HCS (Figure 4). These results suggested that HCS and BTD may be mainly regulated through DNA methylation and histone acetylation, respectively. In line with these notions, TSA or Aza treatment had no significant further effect on either HCS or BTD expression level in B[a]P-treated cells, respectively (Figure 4). Furthermore, TSA and Aza treatments seemed to rescue B[a]P-induced reduction in HCS and BTD expression, respectively. These results suggest that HCS and BTD expression might be regulated through distinct epigenetic mechanisms.
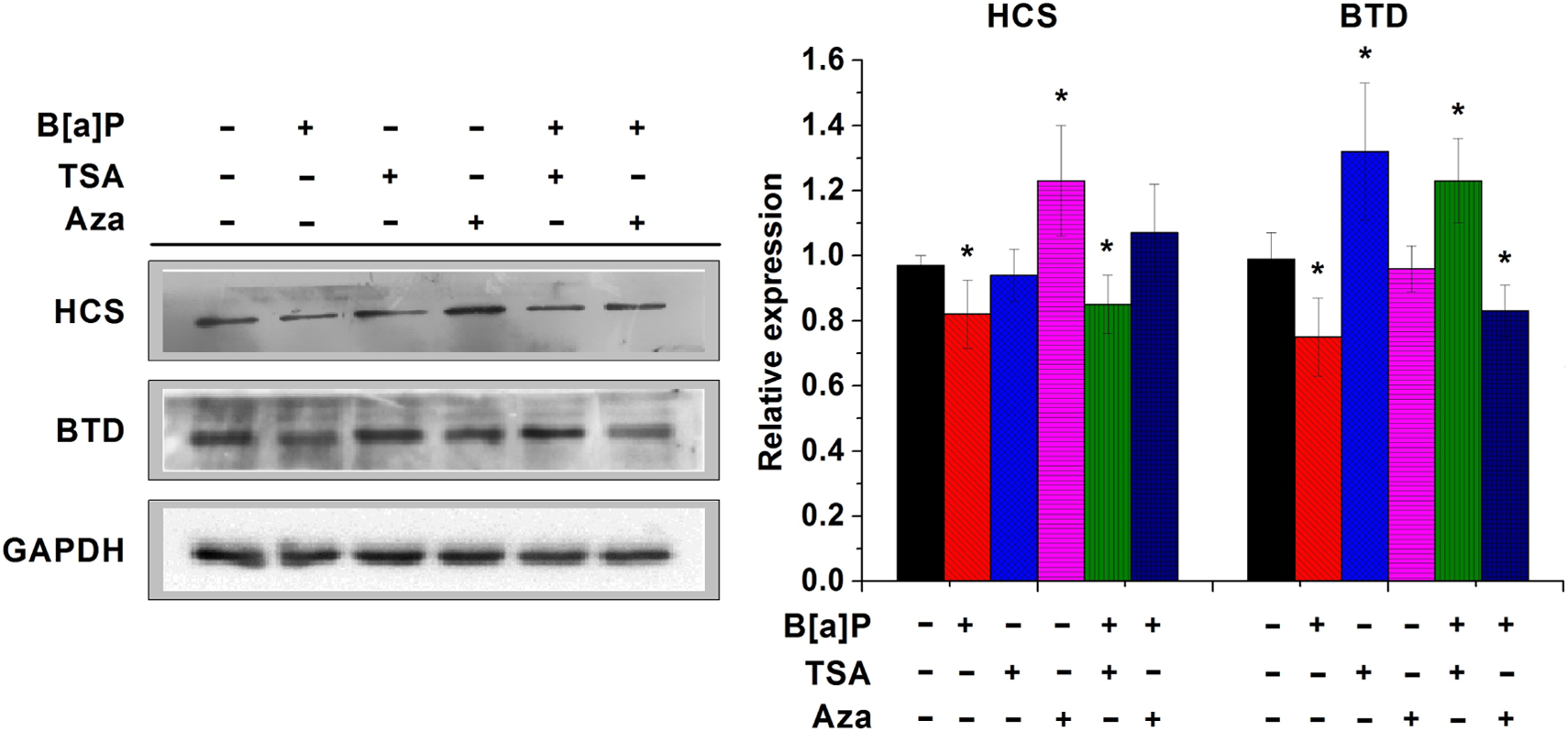
HCS and BTD are separately regulated through distinct epigenetic mechanisms in 16HBE cells. Comparative effect of B[a]P, Aza, and TSA on the protein expression levels of HCS and BTD in 16HBE cells was determined using Western blot analysis after treating the cells with B[a]P for 24 hours. Data are presented in terms of relative density of control as means ± standard deviations in different treatment groups. Significant difference versus control, *P < .05. HCS indicates holocarboxylase synthetase; BTD, biotinidase; 16HBE, human bronchial epithelial cells; B[a]P, benzo[a]pyrene; Aza, 5-aza-2′-deoxycytidine; TSA, trichostatin A.
Discussion
Epigenetic alterations are considered as a key mechanism involved in carcinogenicity. Changes in normal epigenetic patterns necessary for transcriptional regulation may cause serious disruptions. Previous evidences show that the activity of B[a]P is affected by the methylation status of CpG sequences of genes. 35 In addition, B[a]P affects epigenetic pathways not only through methylation-enhanced B[a]P binding by B[a]P metabolites that form DNA adducts 36,37 but also results in the disruption of DNMTs. 38 In the current study, decreased global DNA methylation was observed in B[a]P-treated 16HBE cells, which is consistent with previous studies. 39 In addition, high doses of B[a]P caused alterations of maintenance (DNMT1) and de novo DNMTs (DNMT3b) and methyl-binding protein MBD2. Combined with previous observations, 32,33 it is reasonable to deduce that B[a]P-induced demethylation in 16HBE cells is partly caused by DNMT3b and MBD2. Moreover, the results of our laboratory research also showed that the protein expression levels of DNMTs were altered when treated with B[a]P for 72 hours, and DNMT3a, another de novo DNA methyltransferase, should be responsible for B[a]P-induced demethylation. 40 Taken together, B[a]P-induced global hypomethylation may contribute to tumorigenesis induced by B[a]P exposure.
Histone modifications, on the other hand, are more complicated than DNA methylation. Posttranslational modifications of histones usually correlate with chromatin accessibility and transcriptional activity, and their presence potentially influences various fundamental biological processes. 41,42 Histone acetylation generally leads to an open chromatin structure that is associated with transcriptional activation, whereas deacetylation of histone results in a closed chromatin structure with transcriptional repression. 43 A previous study reported that specific histone acetylations such as H3K18ac and H3K27ac are involved in the regulation of the expression of AhR (Aryl hydrocarbon receptor), which in turn may contribute to BaP-induced toxicity. 44 The results of the present study demonstrated that B[a]P significantly increased the expression of HDAC2 and HDAC3 and, in turn, resulted in global deacetylation of histones H3 and H4. In line with B[a]P-associated tumorigenesis, B[a]P-induced HDAC activity is reminiscent of the similar feature in most cancer cells.
It has recently become apparent that epigenetic modifications such as DNA methylation and histone acetylation can work together for a common purpose, and this relationship is of physiological importance and has implications for understanding normal development as well as somatic cell reprogramming and tumorigenesis. 45 Both B[a]P exposure and deregulation of biotin homeostasis pathway have been associated with tumorigenesis. However, the link between B[a]P exposure and deregulation of biotin homeostasis pathway is missing. Our results showed that B[a]P treatment led to the reduction in BTD and HCS, suggesting a new pathway of tumorigenesis in B[a]P exposure. Given that B[a]P exposure results in loss of DNA methylation, increased HDAC activity, and reduction in BTD and HCS, we have further determined which epigenetic pathway is mainly responsible for the reduction in BTD and HCS by either inhibition of DNA methylation by Aza or repression of HDAC activity using TSA. Our results indicated that Aza significantly increased the protein expression level of HCS, whereas TSA significantly increased BTD. In addition, in B[a]P-induced cells, Aza and TSA reverse the effects of B[a]P on the protein expression level of BTD and HCS, respectively. These results suggest that HCS and BTD are inhibited through different epigenetic pathways in B[a]P-treated cells. Further studies of the effects of B[a]P on the dysregulation of biotin-dependent pathways are thus warranted. A better understanding of the role of B[a]P in biotin metabolism may shed new light on B[a]P toxicity and B[a]P-associated human cancer.
Footnotes
Author Contribution
Bo Xia contributed to conception and design and critically revised the manuscript. Lin-qing Yang and Hai-yan Huang contributed to conception and drafted the manuscript. Li Pang contributed to acquisition, analysis, interpretation and drafted the manuscript. Xi-fei Yang contributed to acquisition, analysis, interpretation and critically revised the manuscript. You-Jin Yi contributed to design and critically revised the manuscript. Xiao-hu Ren contributed to acquisition, analysis, interpretation and drafted the manuscript. Jie Li contributed to design and drafted the manuscript. Zhi-xiong Zhuang and Jian-jun Liu contributed to conception, design, and critically revised the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work ensuring integrity and accuracy.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by Natural Science Foundation of Guangdong (Contract No. 2014A030310508), Natural Science Foundation of China (Contract No. 81273127), the Future Industry Special Project of Shenzhen (Contract No. ZDSYS20140509101335476), Foundation for High-level Personnel of Hunan Agricultural University (Contract No. 14RCPT07), Science and technology projects of Shenzhen (Contract No. JCYJ20150402102135497) and Shenzhen Industrial Development Sponsored Research Project (Contract No. CXB201104220031A).