
Abstract
Although several studies have shown that chemically mediated epigenetic changes are an etiological factor in several human disease conditions, the utility of epigenetic data, such as DNA methylation, in the current human health risk assessment paradigm is unclear. The objective of this study is to investigate the relationship between the points of departure (PODs) for cancer incidence and DNA methylation changes in laboratory animals exposed to the following environmental toxicants: bromodichloromethane, dibromochloromethane, chloroform, hydrazine, trichloroethylene, benzidine, trichloroacetic acid, and di(2-ethylhexyl) phthalate (DEHP; a known reproductive toxicant). The results demonstrate that the PODs for cancer incidence and altered DNA methylation are similar. Furthermore, based on the available data, the POD for DNA methylation appeared more sensitive compared to that for cancer incidence following the administration of DEHP to rats during different life stages. The high degree of correlation between PODs for cancer incidence and DNA methylation (for both total DNA and individual genes) suggests that DNA methylation end points could potentially be used as a screening tool in predicting the potential toxicity/carcinogenicity and in prioritizing large numbers of chemicals with sparse toxicity databases. The life stage during which treatment occurs is also an important consideration when assessing the potential application of epigenetic end points as a screening tool.
Introduction
The human health risk assessment (HHRA) paradigm currently relies heavily on data from traditional toxicity testing (eg, chronic duration in vivo bioassays), which can be a time- and resource-intensive process that requires years to complete. Therefore, only a small fraction of commercial chemicals have any associated HHRA information. 1,2 Considering that thousands of environmental chemicals are in need of evaluation for their potential risk to human health, regulatory organizations have recognized the need to increase the efficiency of the current risk assessment process to include the potential use and integration of alternative toxicity testing data. 1,3 Molecular toxicology testing is an example of an alternative data source that could increase the efficiency of the current HHRA paradigm. Recently, Thomas et al 4,5 indicated that benchmark dose (BMD; note 1) values for transcriptional changes may be used to estimate points of departure (PODs; note 2) in both noncancer and cancer risk assessment. This was based on an observed high degree of correlation between transcriptional and apical BMD values for 10 chemicals in studies using rats and mice. Although there is an ongoing attempt to use alternative data streams in the HHRA process, the applicability of the epigenetics area of molecular toxicology for improving the efficiency of HHRA is understudied.
Epigenetics refers to heritable modifications of the genome in a cell that occur without a change in the primary DNA sequence. 6,7 The 3 most common types of epigenetic modifications are via DNA methylation, histone modification, and noncoding RNA. 8 –10 DNA methylation has been a subject of interest because of its recognized role in many disease processes, including cancer and cardiovascular injury. 11,12 Compared to the extensive DNA methylation data available, limited data exist with respect to the involvement of histone modification and noncoding RNA in environmental chemical-induced disease processes. Therefore, this article focuses only on DNA methylation epigenetic changes induced by environmental chemicals. DNA methylation occurs primarily at the 5′ carbon position of cytosine residues or at the 6′ nitrogen position of adenine residues in mammals. 13 The pattern of DNA methylation is finely controlled in a normal cell, whereas the control of DNA methylation is often altered in a cancer cell. 14 Total DNA, as well as the coding and promoter regions of individual genes, are susceptible to methylation changes. Hypermethylation of a gene is usually associated with gene silencing, which has been demonstrated to affect numerous tumor suppressor genes. 15 Cancer genomes are characterized by hypomethylation, which is usually associated with overexpression of oncogenes. 16 Recent investigations have identified a number of environmental toxicants (eg, arsenic, nickel, chromium, trichloroethylene, dichloroacetic acid, and pesticides) that cause DNA methylation alterations in genes of various tissues (eg, liver, lung, kidney, and testes) leading to both noncancer and cancer effects. 17 –19
Few studies have reviewed the current status of the application of epigenetics in the HHRA process. Goodman et al, 20 LeBaron et al, 21 and Rasoulpour et al 22 provide an overview of epigenetic mechanisms, experimental models with target end points to evaluate epigenetic changes, and regulatory perspectives on the potential for incorporating epigenetic data in HHRA. Evidence of epigenetic alterations in chemical-induced carcinogenesis and the advantage of using epigenetic biomarkers in cancer risk assessment was presented by Koturbash et al. 23 Alyea et al 24 compared PODs between epigenetic and apical end points and stated that the current HHRA practice of using apical end points for deriving reference values is also protective against epigenetic alterations and does not warrant a change in the prevailing methodology. The objective of the present study is to provide an estimation and validation of the correlation between the PODs for cancer incidence and DNA methylation for several environmental chemical carcinogens.
Materials and Methods
A data mining experimental approach was used for this analysis and is summarized in Figure 1. The US Environmental Protection Agency’s (US EPA’s) Integrated Risk Information System (IRIS) 25 database, and the National Toxicology Program (NTP) 26 database were searched for environmental chemical carcinogens (step 1). Specifically, the US EPA’s IRIS database (http://www.epa.gov/iris/) was searched using the term “oral slope factor.” The 13th Report on Carcinogens 26 was searched for environmental toxicants designated as probable or known human carcinogens. Both mutagenic and nonmutagenic carcinogens were included in this analysis because DNA methylation could influence mutagenesis 27 as well as contribute to tumorigenesis via nonmutagenic events. 28 Literature searches were then performed on these selected carcinogens in PubMed using the search term “DNA methylation” in conjunction with their chemical name or their Chemical Abstracts Service Registry Number. DNA methylation data from total DNA and individual genes were included in this analysis. Only chemicals with in vivo DNA methylation data in laboratory animals were considered for further analysis. For the DNA methylation studies, no exposure duration restriction was applied, and a chemical was removed from consideration if the respective study lacked a control group (step 2). The chemicals identified in step 2 were then mined for cancer incidence data. Because epigenetic changes, specifically DNA methylation alterations, do not correlate well across different strains (eg, in mice), 29 different outbred lines (eg, in rats), 30 and different sexes (eg, in mice and rats), the resultant cancer studies were cross-referenced with the DNA methylation studies identified for each chemical in step 2 to determine which studies possessed data on the identical species, strain, sex, target organ, and type of oral exposure (step 3). Based on those criteria, the principal DNA methylation and cancer studies for each chemical to be used in further analyses were identified.
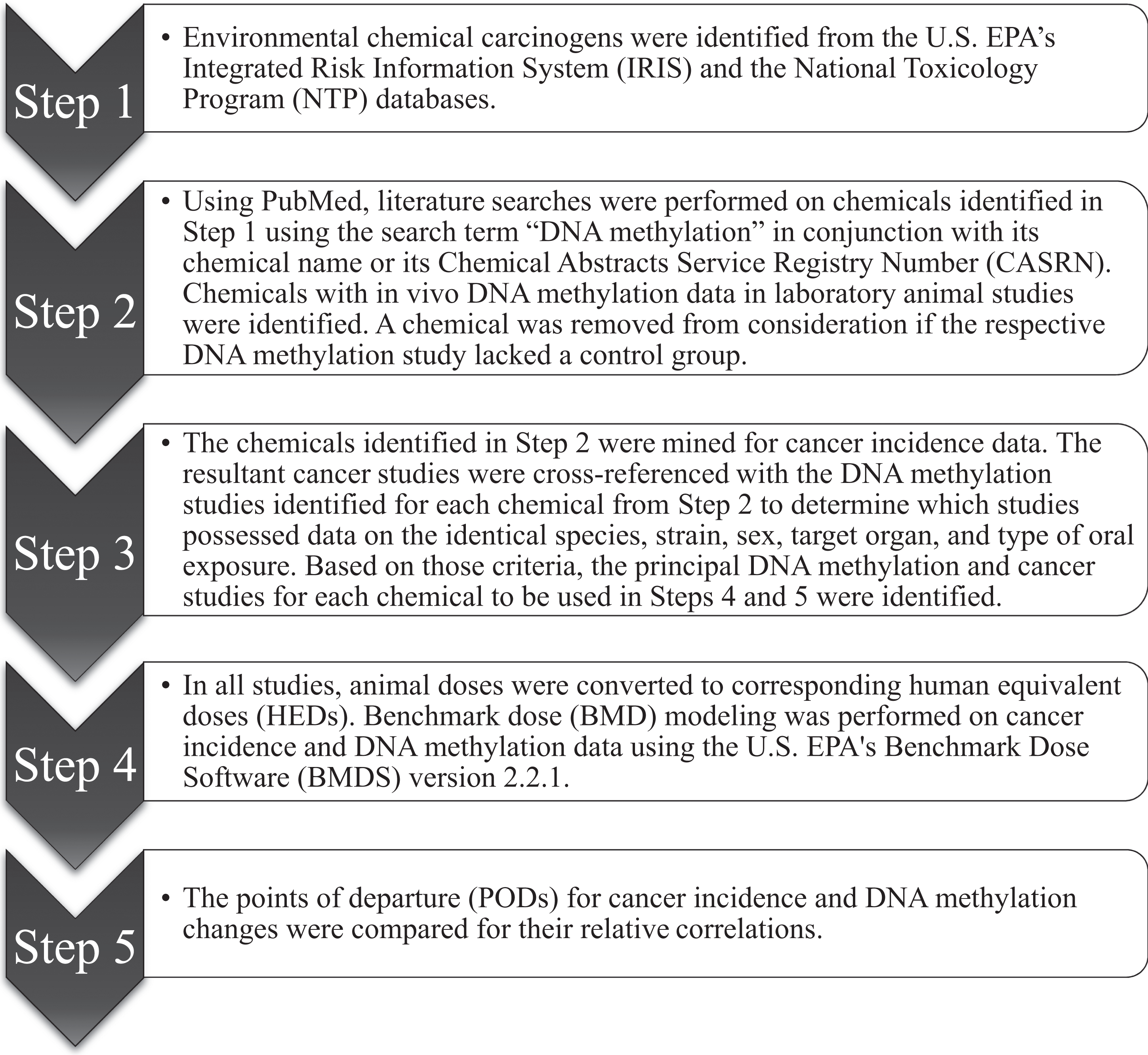
Workflow for selection and analysis of environmental carcinogens associated with DNA methylation changes.
For all studies, animal doses were duration adjusted and converted to corresponding human equivalent doses (HEDs). The HEDs were calculated by using a BW3/4 cross-species scaling factor approach to extrapolate toxicologically equivalent doses of an orally administered dose from laboratory animals to humans. Cross-species scaling factors were calculated 31 –33 as BWa 1/4 ÷ BWh 1/4, where BWa is the average body weight of an animal and BWh is the average body weight of a human. Because laboratory animal body weights were not reported in the DNA methylation studies, average default body weights were used to calculate HEDs from these studies. 34 The default body weights of the laboratory animals were specific for the study duration. Time-weighted average body weights were calculated from the cancer studies to further calculate HEDs (see Supplemental Table S3). The resulting cross-species scaling factor was multiplied by the laboratory animal dose to yield a HED as follows:
HED = laboratory animal dose (mg/kg/d) × cross-species scaling factor
For di(2-ethylhexyl) phthalate (DEHP), the corresponding HEDs were based on the parental body weight for the DNA methylation studies. The BMD modeling of cancer incidence and DNA methylation data was performed using US EPA’s BMD software (BMDS) version 2.2.1 (step 4). Cancer incidence data (Supplemental Table S1) were modeled using a 10% benchmark response (BMR) to derive a POD with a multistage cancer model. 31 DNA methylation data were BMD modeled as continuous data. Because no empirical data or specific guidance exist to determine the appropriate BMR for DNA methylation data, BMRs of both 10% and 1 standard deviation were calculated. If the DNA methylation data could not be BMD modeled, a no-observed-effect level (NOEL) was identified and used as the POD for DNA methylation. In the absence of a NOEL, a factor of 10 was applied to a lowest-observed-effect level (LOEL) to approximate a NOEL. 35 –37 The LOELs for DNA methylation were characterized based on statistically significant changes from control values. In the absence of statistical analysis, any demonstrative change in Southern blot band intensity was used to determine a LOEL. For each chemical, the resulting POD for cancer incidence was compared with the POD for DNA methylation changes to determine their relative correlation (step 5). All the cancer studies investigated in this manuscript used a statistical significance level (P value) ≤ 0.05.
Results
Data Mining of Chemicals and Animal Studies for Cancer Incidence and DNA Methylation Changes
Based on the criteria discussed earlier in the Methods section, 91 chemicals from the IRIS database and 84 chemicals from the NTP database were identified in step 1 (Figure 1). From these 175 chemicals, 16 chemicals met the selection criteria listed in step 2 (ie, in vivo DNA methylation data from laboratory animal studies). Seven chemicals were further excluded because they did not have the appropriate cancer incidence data in the identical species, strain, sex, target organ, and exposure type used in the DNA methylation studies, resulting in a final total of 9 chemicals as follows: arsenic, bromodichloromethane, dibromochloromethane, chloroform, hydrazine, trichloroethylene, benzidine, trichloroacetic acid, and DEHP.
Although laboratory animal data for cancer incidence and DNA methylation were found in the same species (mice), strain (C3H/HeN), sex (male), target organ (liver), and exposure route (drinking water) for arsenic, there were inconsistent findings with respect to cancer incidence and cancer type following the same experimental protocol in arsenic-treated C3H/HeN mice as reported by Waalkes et al 38 and Nohara et al. 39 Specifically, Waalkes and colleagues reported a statistically significant increase in the incidence of hepatocellular carcinoma (14/23), whereas no such increase (only 1/12) was observed by Nohara and colleagues at the same dose. Additionally, the background rate of cancer incidence in the control group from both studies was elevated (42%) compared to the other 8 chemicals in our analysis. With respect to the epigenetics data, hypomethylation of the promoter region of the estrogen receptor alpha gene reported by Waalkes et al 40 could not be reproduced by Suzuki et al 41 at the same dose. Similarly, Cui and colleagues measured both cancer incidence and DNA methylation in the same species (mice), strain (A/J), sex (male), target organ (lung), and exposure route (drinking water) following the administration of arsenic. 42 Although statistically significant DNA methylation changes (ie, hypermethylation of the promoter region of the tumor suppressor Ras association domain family member 1 isoform A [Rassf1a] gene) were observed at the highest dose, no significant increases in cancer incidence were observed in this study. The background rate of cancer incidence in the control group was also elevated (47%). Thus, due to the discordance in experimental findings on cancer incidence, cancer type, and DNA methylation between different studies following oral arsenic administration, as well as a high rate of background cancer incidence in the respective control groups, arsenic was not included in our analysis.
The principal cancer and DNA methylation studies selected for data mining were conducted in mice, rats, and hamsters (Table 1). Trichloroethylene was the only chemical that has cancer and DNA methylation studies in both sexes of mice that also met all other step 3 criteria. Durations of the cancer studies ranged from 526 days to over 24 months, and durations of the DNA methylation studies ranged from 5 days to 21 months. There were 3 chemicals (bromodichloromethane, trichloroethylene, and trichloroacetic acid) with multiple time points at which DNA methylation was measured. For these chemicals, DNA methylation data from the longest treatment duration were used (ie, 28 days for bromodichloromethane in male B6C3F1 mice; 33 days for trichloroethylene in female B6C3F1 mice; 5 days for trichloroacetic acid in female B6C3F1 mice). It should be noted that there were no differences in the magnitude of DNA methylation across treatment durations, as well as no variations across different methylation end points within a given study. The most sensitive DNA methylation change (ie, the one that occurred at the lowest dose in the principal DNA methylation study) was identified as the POD for each chemical. Additional details on the DNA methylation studies evaluated for each chemical are presented in Supplemental Table S2. The types of oral exposures included both gavage and drinking water (Table 1). The liver was determined to be the primary target organ for both cancer incidence and DNA methylation changes for all chemicals analyzed except for bromodichoromethane (kidney) and DEHP (testes; Table 2).
Principal DNA Methylation and Carcinogenicity Studies Resulting From a Data Mining Experimental Approach.
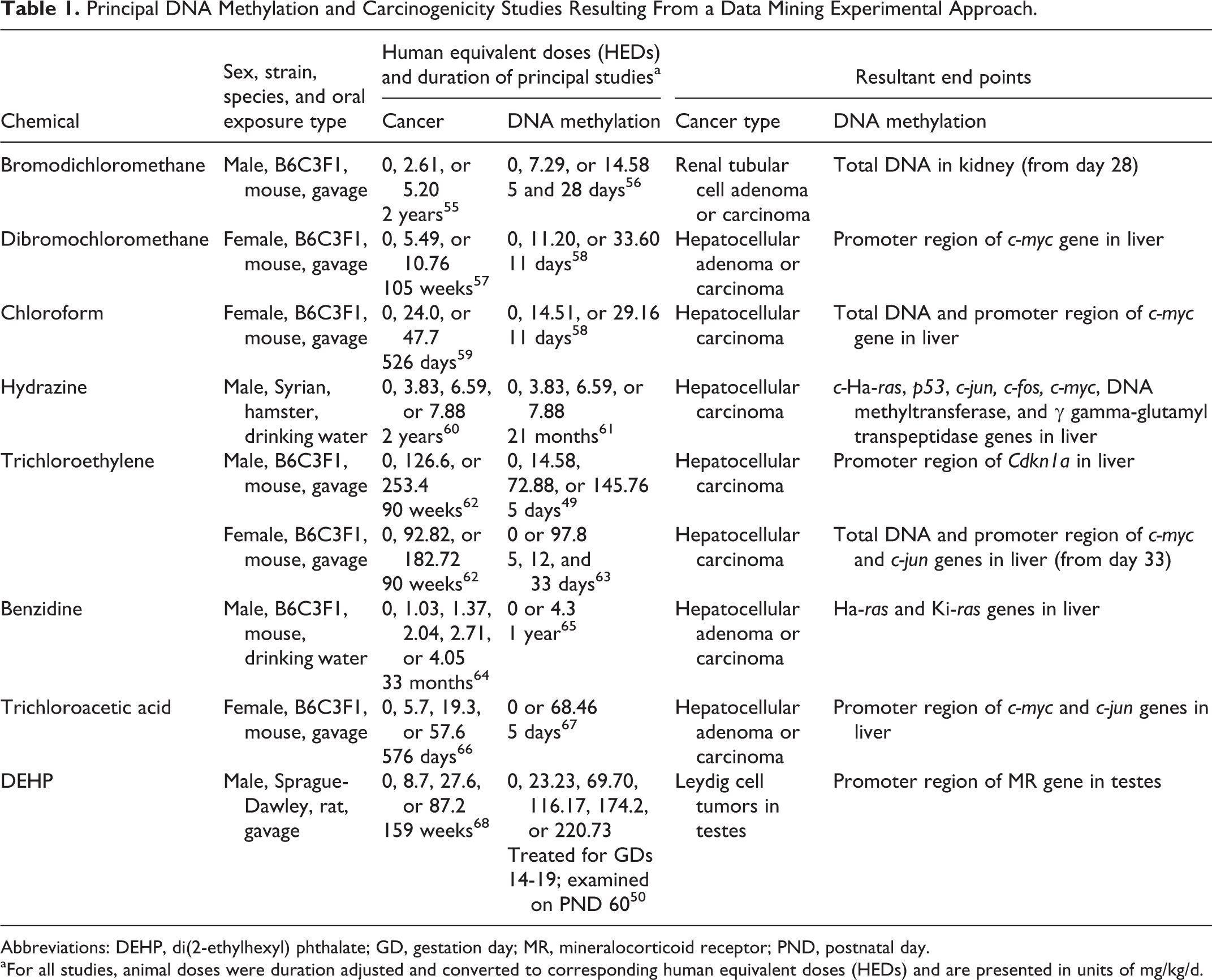
Abbreviations: DEHP, di(2-ethylhexyl) phthalate; GD, gestation day; MR, mineralocorticoid receptor; PND, postnatal day.
aFor all studies, animal doses were duration adjusted and converted to corresponding human equivalent doses (HEDs) and are presented in units of mg/kg/d.
Points of Departure for Cancer Incidence (BMD10HEDs) and DNA Methylation Changes (NOELHEDs).
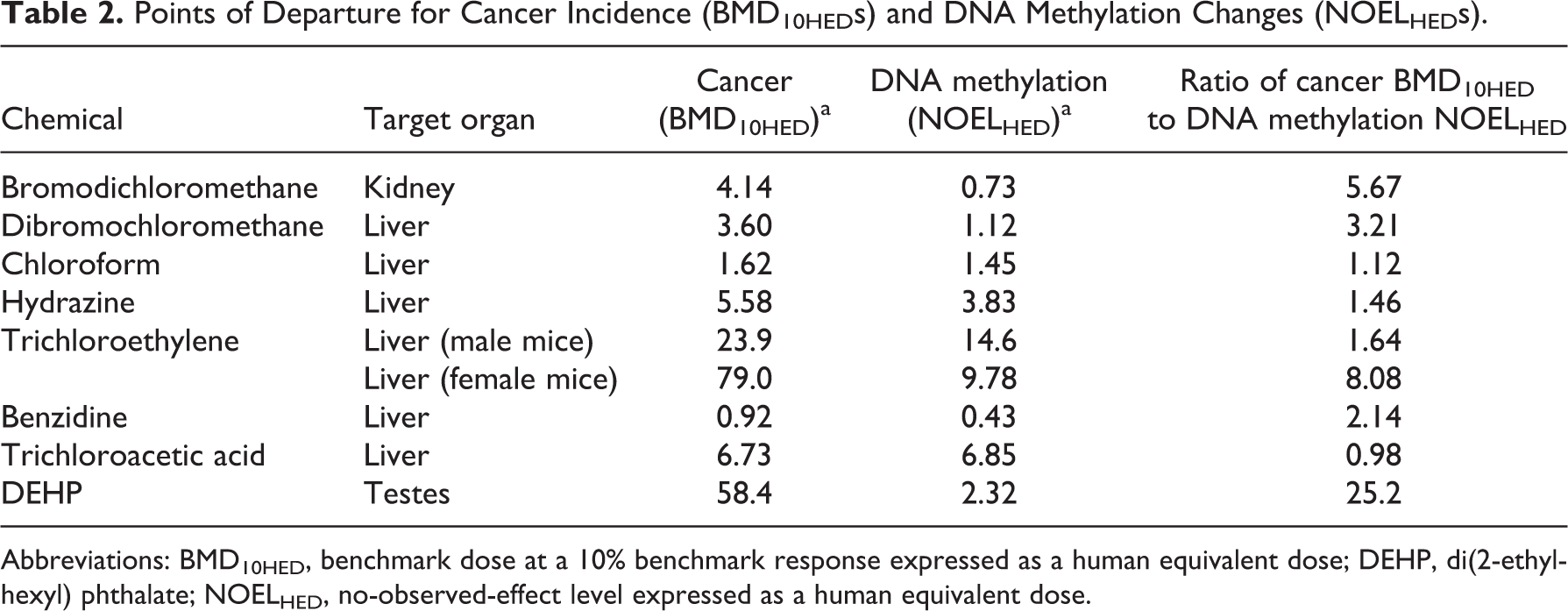
Abbreviations: BMD10HED, benchmark dose at a 10% benchmark response expressed as a human equivalent dose; DEHP, di(2-ethylhexyl) phthalate; NOELHED, no-observed-effect level expressed as a human equivalent dose.
Benchmark Dose Analysis of Cancer Incidence and DNA Methylation Changes
All cancer incidence data were successfully BMD modeled, and the PODs (ie, BMD10s) for the most sensitive cancer effects in the same target organ as the observed DNA methylation changes were identified (Table 2). However, DNA methylation data were not amenable to BMD modeling for 5 of the 8 chemicals analyzed because these data either had no associated variance or could not be quantified (eg, presented only as a Southern blot). For the remaining 3 chemicals, BMD models did not adequately fit the DNA methylation data. Thus, an initial attempt was made to identify NOELs to serve as PODs for DNA methylation changes. However, NOELs for DNA methylation changes could not be identified for 6 of the 8 chemicals, thus a factor of 10 was applied to the LOEL to approximate a NOEL in these cases as described earlier.
Comparison of PODs for Cancer Incidence and DNA Methylation Changes
Because BMDs associated with BMRs of 1% to 10% have been shown to approximate NOELs 43 , BMD10s for cancer incidence and NOELs for DNA methylation changes were compared to examine their correlation. The ratio of cancer incidence POD values to DNA methylation POD values was within a factor of 10 for all chemicals except DEHP (Figure 2). The ratio of the cancer BMD to the DNA methylation NOEL for female mice is 5-fold higher than for male mice following exposure to trichloroethylene, but still within a factor of 10. This result suggests that the PODs for cancer incidence and DNA methylation changes are relativity similar in sensitivity across different chemical classes. However, for DEHP, a known reproductive toxicant, the POD for DNA methylation changes is 25-fold more sensitive than the POD for cancer incidence. It should be noted that DNA methylation status was measured in the testes of male offspring (at postnatal day [PND] 60) from pregnant rats administered DEHP on gestation days (GDs) 14 to 19, whereas cancer incidence in the testes was measured in adult rats administered DEHP for 159 weeks.
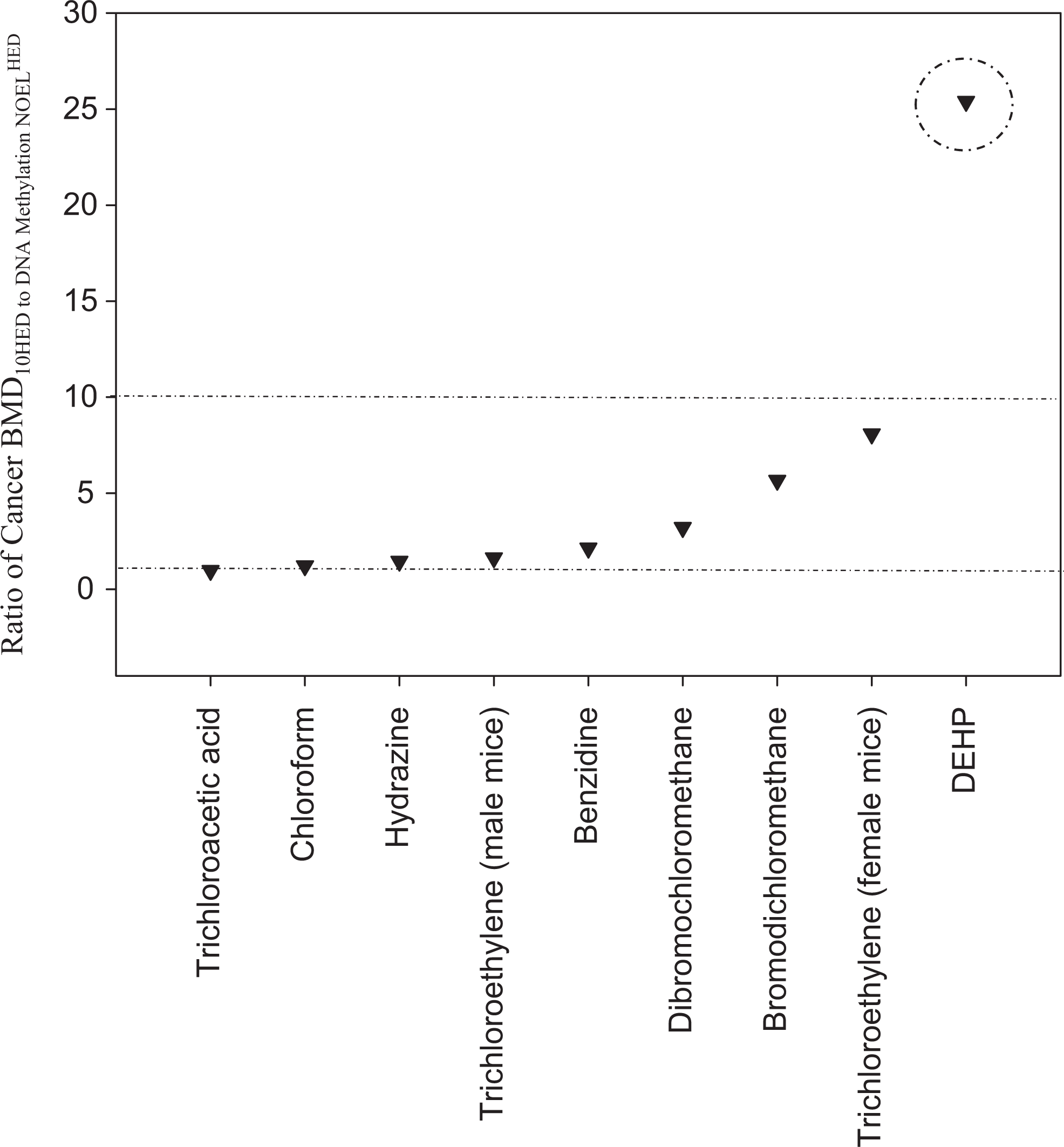
Comparison of points of departure for cancer incidence and DNA methylation.
Discussion
The current HHRA paradigm cannot keep up with the pace at which new chemicals are introduced into commerce. 2 Hence, there is a need to increase the efficiency of the current HHRA process by incorporating molecular toxicology testing data in the evaluation of potential health effects induced by environmental chemical exposures. Although molecular toxicology studies, specifically those examining DNA methylation changes, have contributed significantly to our current understanding of the cellular processes involved in chemical-induced toxicity, the use of this information in the current HHRA paradigm is extremely limited. In this study, the validation of the dose-response concordance between cancer incidence and DNA methylation changes was evaluated by comparing their respective PODs. This type of epigenetic information could possibly be used in health assessments of chemicals that have little or no toxicity and/or risk assessment information. These kinds of health assessments would highlight the significance of using molecular toxicity testing to increase the efficiency of the current HHRA paradigm. 4,5
In a similar analysis to that described herein, Alyea et al 24 compared the dose-response data for apical end points with epigenetic end points for 1,3-butadiene, arsenic, and diethylstilbesterol. DNA methylation changes were observed at a lower (1,3-butadiene), higher (arsenic), and similar (diethylstilbesterol) dose level compared with that for cancer incidence. The study authors suggested that their analysis does not warrant change in the current practice of deriving toxicity values from apical end points because epigenetic variations mostly occur within the dose-response curve for apical effects. In the present study, we performed extensive data mining of selected environmental chemical carcinogens from the US EPA and NTP databases. Because epigenetic changes do not correlate well across different strains as mentioned above, it is important to compare epigenetic changes in the same strain of animals. 29 Analysis of the 3 chemicals by Alyea et al 24 was not made across the same strains of animals. Conversely, we not only examined the same species and strains, but also examined the same sex, oral exposure route/type, and target organ. Additionally, the NOEL/LOEL approach was used to identify PODs for cancer incidence by Alyea et al. 24 Because regulatory agencies, including the US EPA, generally do not use the NOEL/LOEL approach for identifying PODs to derive cancer slope factors when 1 or more BMD models fit the cancer incidence data, we used the multistage cancer BMD model to derive PODs for cancer incidence. 44
In our analysis, hypomethylation was the predominant epigenetic change observed. Specifically, hypomethylation of total DNA as well as the c-myc, c-jun, Ha-ras, and Ki-ras proto-oncogenes were identified (Table 3). Genome-wide DNA hypomethylation promotes chromosomal instability and carcinogenesis. 16 C-myc, c-jun, Ha-ras, and Ki-ras proto-oncogenes have been shown to be associated with increased protein expression, enhanced cell proliferation, and carcinogenesis (eg, in liver, lung, and colon) by regulating cellular signal transduction, cell growth, differentiation, adhesion, survival, and apoptosis. 45 –47 Likewise, hypermethylation of c-Ha-ras and DNA methyltransferase as well as hypomethylation of the p53 and Cdkn1a tumor suppressor genes, all of which have been positively correlated with carcinogenesis 48 –50 , was also observed. In addition to proto-oncogenes and tumor suppressor genes, hormonal imbalances that can induce cancer were also observed. Loss of methylation in the mineralocorticoid receptor gene leads to reduced testosterone synthesis (as observed following DEHP exposure), 51 which interferes with the negative feedback control, thereby inducing the overproduction of follicle-stimulating and luteinizing hormones and increasing the proliferation of Leydig cells. 52,53
Specific DNA Methylation Changes Following Environmental Chemical Exposure.
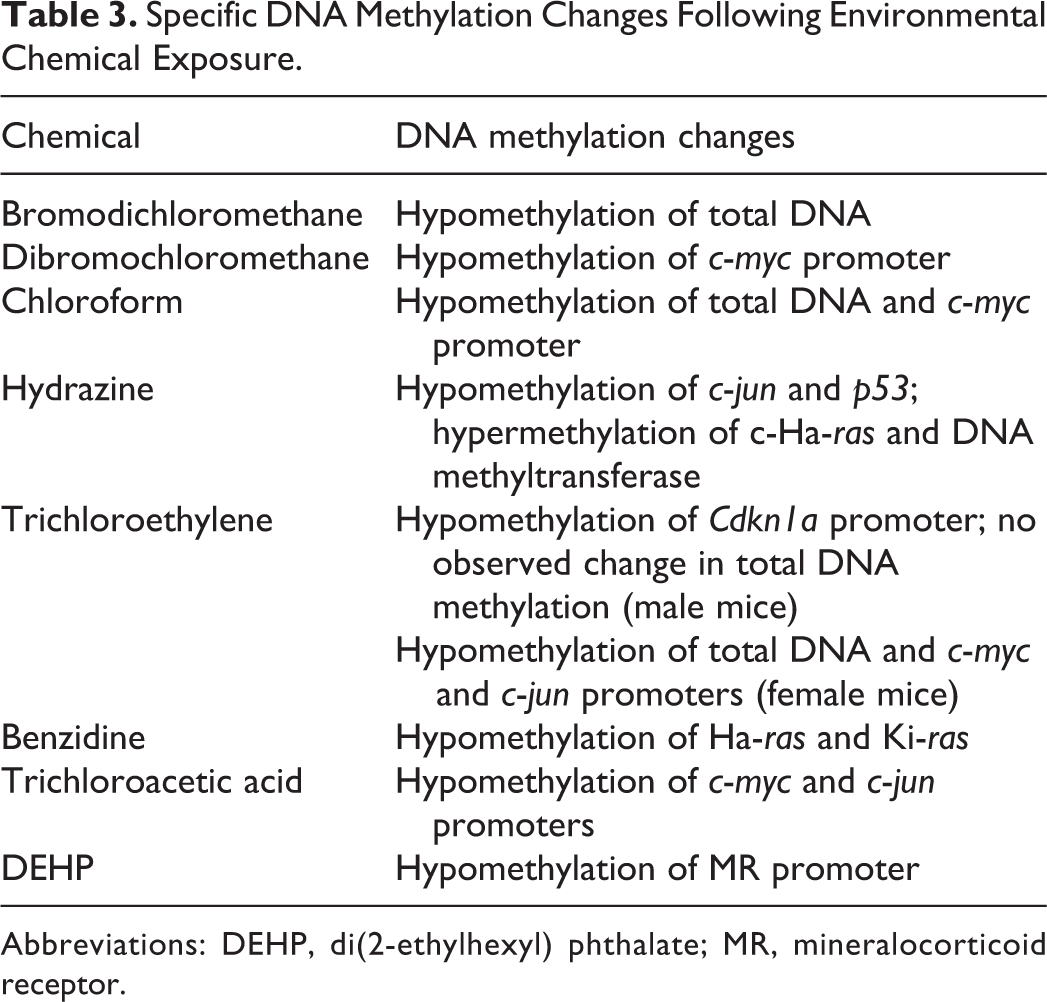
Abbreviations: DEHP, di(2-ethylhexyl) phthalate; MR, mineralocorticoid receptor.
Increasing evidence indicates that the epigenome is particularly vulnerable for lifelong alterations during developmental periods (eg, gestation). The current analysis revealed that for DEHP, the POD for DNA methylation changes is 25-fold more sensitive than the POD for cancer incidence. This ratio was substantially higher compared to that of the other chemicals analyzed that had ratios ranging from approximately 1 to 8. A possible explanation for this observed difference in sensitivity could be due to the administration of the chemical during different life stages (ie, DEHP was administered to pregnant animals in its principal DNA methylation study, compared to all other chemicals being administered to adult nonpregnant animals). This explanation is supported by the observation that the epigenome is most sensitive to environmentally induced changes due to an increase in DNA synthesis and formation of DNA methylation patterns during early development. 54 In the principal DNA methylation study for DEHP, male rats were treated in utero from GDs 14 to 19. 51 Thus, the life stage during which treatment occurs should be taken into account when considering the potential application of epigenetic end points as a screening tool to predict the potential toxicity/carcinogenicity of a chemical.
In summary, our results demonstrate that DNA methylation alterations of total DNA as well as that of individual genes are biologically plausible key events in putative modes of carcinogenic action for the analyzed chemicals. The high degree of correlation between PODs for cancer incidence with those for DNA methylation suggests that measurement of DNA methylation end points may be used as a screening tool to assist in predicting potential toxicity/carcinogenicity and in prioritizing large numbers of chemicals that have little or no toxicity data and need more comprehensive toxicological evaluation. Additionally, the DNA methylation studies combined with other high-throughput in vitro screening assays (eg, genotoxicity) may help to identify potential modes of action of chemicals and facilitate time and cost-efficient chemical risk assessments.
Current Research Needs and Limitations
Interest in environmental epigenetic research has increased significantly in recent years, as exemplified by an exponential increase in PubMed-indexed publications. 55 Research funding agencies such as the National Institute of Environmental Health Sciences and the National Cancer Institute, among others, have also shown tremendous support for environmental epigenetic research. Thus, this rapidly growing field is likely to produce more environmental epigenetic data in the future. Although the present analysis shows that the incorporation of epigenetic data into the current HHRA paradigm looks promising, many data gaps need to be filled before conclusions can be drawn about this possible integration. For example, all of the molecular toxicology studies mentioned herein are focused on the methylation status of only a few genes. Only 8 of 170 chemicals from the IRIS and NTP databases could be included in this analysis because appropriate studies that looked for both cancer incidence and DNA methylation changes in the same species, strain, sex, target organ, and type of oral exposure were not available for the other chemicals. Therefore, further studies are warranted that include (1) evaluation of additional genes and other epigenetic end points (ie, histones and noncoding RNAs) following chemical exposure, (2) analyses of epigenetic alterations on a larger number of environmental toxicants, (3) analyses using in vitro DNA methylation data from animal and/or human cell lines in lieu of DNA methylation data from laboratory animals, and (4) evaluation of the causal link between epigenetic modifications and health effects in chemical-induced toxicities. Despite these limitations, the current work provides an analysis of the POD concordance between cancer incidence and DNA methylation changes for environmental chemical carcinogens.
Conclusion
Taken together, this work shows that POD values for cancer incidence and DNA methylation changes are highly correlated for environmental chemical carcinogens. Another notable finding is that DNA methylation changes are highly sensitive following exposure to a reproductive toxicant. This study also demonstrates that the correlation between cancer incidence and DNA methylation changes is valid across different species (rats, mice, and hamsters), sexes, organs (liver, kidney, and testes), and classes of chemicals. Furthermore, the exposure durations of all the DNA methylation studies were shorter than a typical 2-year carcinogenicity study, suggesting that DNA methylation studies may be more time- and cost-efficient. This work identifies the potential utility of epigenetic testing data, particularly DNA methylation data, in the HHRA process for environmental chemical carcinogens that currently lack health assessment information. Furthermore, by adopting the data mining approach used in this manuscript, one could elucidate possible correlations of other epigenetic end points (ie, histone modification and noncoding RNA regulation) with cancer and noncancer end points.
Footnotes
Acknowledgments
The views expressed in this article are those of the authors and do not necessarily represent the views and policies of the US Environmental Protection Agency.
Authors’ Note
Kuppusamy, S. contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically revised the manuscript; Kaiser, P. contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript; Wesselkamper, S. contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy.
Notes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.