
Abstract
The current study aimed at investigating the potential hepatoprotective property and mechanism of meloxicam (MEL) against carbon tetrachloride (CCl4)-induced hepatocellular damage in rats. Subcutaneous administration of CCl4 (2 mL/kg, twice/week for 8 weeks) induced hepatocellular damage substantiated by hematoxylin and eosin staining and significant elevation in serum aspartate transaminase, alanine transaminase, and total bilirubin. In addition, CCL4 treatment led to elevation in liver contents of lipid peroxidation marker (malondialdehyde), prostaglandin E2, active caspase 3, and Terminal deoxynucleotidyl transferase dUTP nick end labeling–positive cells and reduction in the activities of superoxide dismutase, catalase, glutathione-S-transferase, and reduced glutathione in the liver tissue. Prior oral treatment with MEL (5 mg/kg, twice/week) retained the normal liver histology and significantly restored all of these parameters close to normal values. These results demonstrated the hepatoprotective utility of MEL against the CCl4-induced liver injury which might ascribe to its antioxidant, free radical scavenging, antiapoptotic and anti-inflammatory effects.
Introduction
Carbon tetrachloride (CCl4), a well-known hepatotoxin, has been widely used to establish experimental liver heptotoxicity model for studying the mechanisms behind hepatotoxicity, oxidative stress, and the hepatoprotective utility of novel drugs and nutrients. 1,2
CCl4-induced liver damage is mediated via generation of reactive metabolites that play a key role in the pathogenesis of both acute and chronic liver damage.
3,4
Liver microsomal cytochrome P450 metabolizes CCl4 to free radicals that deplete hepatic glutathione and initiate a chain lipid peroxidation (LPO) of the hepatocyte membrane.
5
This ultimately results in the overproduction of reactive oxygen species (ROS) and hepatocyte injuries.
5,6
In normal situation, cellular enzymatic antioxidants including superoxide dismutase (SOD); catalase (CAT); glutathione-S-transferase (GST)
7
; or nonenzymatic antioxidants including reduced glutathione (GSH); vitamins and flavonoids scavenge the toxic ROS such as superoxide anion (O2
Cyclooxygenase (COX)-2 plays a pivotal role in pathogenesis of uncontrolled inflammation, 13,14 and overexpression of COX-2 has been reported in several chronic inflammatory diseases such as rheumatoid arthritis, Crohn disease, and ulcerative colitis. 15 Of particular interest, upregulation of COX-2 has been demonstrated in many inflammatory liver diseases such as human cirrhosis, 16 progressive hepatic fibrosis in chronic hepatitis C infection, 17 and experimental alcoholic-induced liver necroinflammatory injury in rats. 18,19
In contrast to COX-1, COX-2 was not detected in normal liver and it was strongly unregulated in liver cirrhosis in both human
20
and experimental animal models.
21,22
Interestingly, selective COX-2 inhibitors effectively decrease the expressions of COX-2 regulated genes that control the development and progression of fibrosis such as transforming growth factor-beta (TGF-β) and collagen I in human peritoneal mesothelial cells, TGF-β in choline-deficient
Meloxicam (MEL) is an oxicam derivative, belongs to enolic acid group of nonsteroidal anti-inflammatory drugs (NSAIDs), with preferential COX-2 inhibitory activity. 24,25 Meloxicam is a Food and Drug Administration–approved drug for rheumatoid arthritis and osteoarthritis. The drug has shown improved gastric and renal tolerability and higher therapeutic index both experimentally and clinically compared to conventional NSAIDs. 26,27 Moreover, recent study pointed out the potential antioxidant activity for MEL against quinolic acid (QA) induced Huntington (HD) disease. 28
In view of the above mentioned, the aim of this study was to address the potential hepatoprotective utility and mode of action of MEL, a selective COX-2 enzyme inhibitor, against CCL4-induced liver injury in rats.
Material and Methods
Animals
Forty adult male Sprague-Dawley rats weighing 180 to 200 g were obtained from the laboratory animal colony, Ministry of Health and Population (Helwan, Cairo, Egypt). Animals were maintained under standard room temperature (22°C ± 2°C) and relative humidity (60% ± 10%) with a 12-h light/dark cycle at animal center, Faculty of Pharmacy, Al-Azhar University (Cairo, Egypt). All rats were allowed free access to food, water ad libitum throughout the acclimatization and experimental period. The animal experiments were performed in accordance with the protocol approved by Al-Azhar University Ethics Committee.
Chemicals
CCl4 was purchased from Acros organics Co (New Jersey). Caspase 3 antibody was purchased from Thermo Fisher Scientific Inc (Fremont, California). Biotinylated anti-mouse immunoglobulin antibodies were purchased from Zymed (San Francisco, California). Streptavidin-HRP kit was purchased from (Novocastra, UK). Meloxicam was kindly supplied as a yellow pure powder from Amoun pharmaceutical company (Cairo, Egypt).
Experimental Design
After a period of adaptation, we randomized 40 rats equally into 4 groups. Group I received corn oil subcutaneously (SC) twice weekly for 8 consecutive weeks and served as a negative control group (–ve). Group II was challenged with CCl4 in corn oil (50% v/v; 2 mL/kg body weight, SC) twice weekly for 8 consecutive weeks to induce chronic liver damage. 29,30 Group III received MEL orally by gavage at 5 mg/kg body weight 31 twice weekly for 8 consecutive weeks. Group IV administered MEL 1 hour before CCl4 injection using the same mentioned dosing schedule.
We closely monitored the rats daily for any signs of toxicity along the experiment duration and recorded body weights once weekly. At the end of the experimental period, all the animals were euthanized by cervical decapitation and blood was collected from retro orbital plexus and serum was separated by centrifugation at 2500 rpm for 15 minutes. The liver of each animal extracted and weighted to calculate the relative liver weight to 100 g of body weight then divided into 3 parts: first part formalin fixed, paraffin-embedded second part was homogenized; and the third part was preserved frozen at −70°C.
Histopathological examination
Paraffin-embedded specimens were cut into sections of 4 to 6 μ thickness and stained with hematoxylin and eosin (H&E) stain according to the method of Bancroft and Stevens. 32 Two pathologists, blinded to the protocol, performed the histopathological evaluation of the stained tissue sections.
Serum biochemistry
Serum was carefully separated into clean dry Wassermann tubes using a Pasteur pipette and used to determine serum liver function tests; aspartate transaminase (AST), alanine aminotransferase (ALT), and total bilirubin (TLB) using the standard techniques. 33,34
Determination of prostaglandin E2 levels in liver
Prostaglandin E2 (PGE2) in liver homogenates was quantified using the PGE2 enzyme immunoassay system (Amersham Biosciences Corp, New Jersey) per manufacturer's instructions and expressed as pg PGE2/mg protein.
Determination of antioxidant enzymes
Supernatants of liver tissues were used for isolation of mitochondria by the method of Johnson and Lardy. 35 The mitochondrial fractions were used for the evaluation of the following parameters: protein was estimated by the method of Lowry et al. 36 Superoxide dismutase was assayed according to the method of Marklund and Marklund. 37 Catalase (CAT) activity was assayed by the method of Sinha. 38 The activity of GST was determined according to Habig et al. 39
Determination of LPO and GSH levels
Hepatic LPO levels were determined by measuring the malondialdehyde (MDA), a thiobarbituric acid reactive substance, level by the method of Ohkawa et al. 40 The hepatic content of GSH was estimated by a colorimetric method using Ellman reagent and glutathione reductase. 41
Evaluation of active caspase 3 content
Active caspase 3 was evaluated immunohistochemically in paraffin-embedded liver tissue sections using active caspase 3 antibody at dilution 1:100, using a sensitive peroxidase-streptavidin method, as described previously. 42,43 Briefly, each block was cut into 4 μm thick sections, which was deparaffinized in xylene and rehydrated in graded alcohols and water. Endogenous peroxidase was blocked, the antigen was unmasked and nontarget proteins were blocked. The slides were incubated, overnight, with the primary antibody at 40°C. After extensive washing, the sections were incubated at room temperature for 10 minutes with biotinylated anti-mouse immunoglobulin antibodies at a 1:20 dilution and subsequently with streptavidinbiotin peroxidase complexes at a 1:25 dilution. The reaction products were visualized by immersing slides in 3, 3′-diaminobenzidine tetrahydrochloride and finally counterstained with hematoxylin. We used tonsil as the positive controls for caspase-3 and performed negative control by replacing the primary antibody with dilution buffer. The percentage of immunoreactive cells in this assay was calculated by counting both positive stained nuclei (brown color) against the total number of cells in 3 random high-power fields for each tissue section. Three tissue sections, with at least 2000 liver cells, per animal were used and 6 animals were randomly included from each group.
Determination of apoptosis
Terminal deoxynucleotidyltransferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay was performed as a quantitative index of apoptosis. Apoptosis was evaluated with the ApopTag Kit (Intergen, Purchase, New York) following the manufacturer’s instructions. The percentages of apoptotic cell expression level was established by counting the number of ApopTag-labeled nuclei (positively stained cells with brown colored) in 3 ×400 microscopic fields for each tissue, and these numbers were compared to the total number of cells. At least 2000 cells were counted per specimen, and the numbers shown represent the average of 6 animals per group.
Statistics
All data are presented as mean ± standard error of the mean (SEM). 44 Group differences were analyzed using 1-way analysis of variance (ANOVA) followed by Tukey test as a post-ANOVA multiple comparison test on raw data (Sigma Stat version 3; SPSS Inc, Chicago, Illinois). Scores for the active caspase 3 protein and TUNEL-positive cells were subjected to Kruskal-Wallis test, followed by the Dunn test (Sigma Stat version 3; SPSS Inc). A P value of <.05 was considered statistically significant.
Results
Effect of MEL on Relative Liver Weight to Total Body Weight
Figure 1 illustrates the relative liver weight to 100 g of body weight in the different experimental groups of rats that were sacrificed at the end of the study. Apparently, treatment with CCl4 significantly (P < .05) increased the relative liver weight to 100 g of body weight compared to the control group. Administration of MEL alone did not change the relative liver weight to 100 g of body weight compared to control group. While prior administration of MEL to CCl4 resulted in a significant decrease in liver weight to body weight compared to CCl4-treated group.
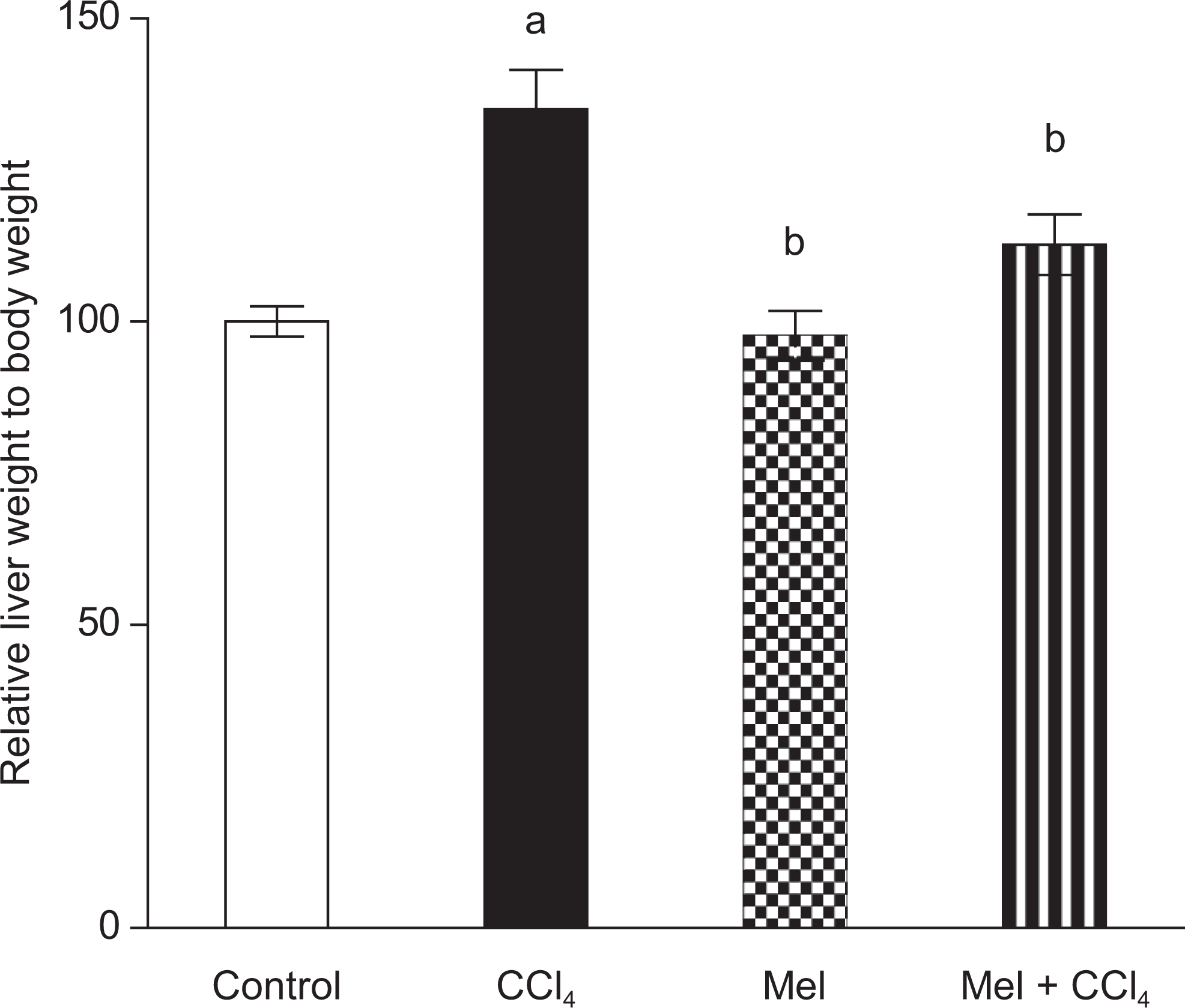
Effect of carbon tetrachloride (CCl4), meloxicam (Mel), or a combination of meloxicam and CCl4 on relative liver weight to total body weight in rats. Animal treatment was continued for 8 weeks. Data were calculated as relative weight of liver to 100 g animal body weight at the end of experiment and plotted as percentage of control value. Data are presented as mean ± standard error of 10 animals/group; a or b indicates significant difference from control or CCl4 respectively at P ≤ .05 using Tukey test as post-ANOVA test. ANOVA indicates analysis of variance.
In addition, CCl4 induced a remarkable decrease in the total body weight gain which is restored upon prior administration of MEL (Supplemental Figure 1).
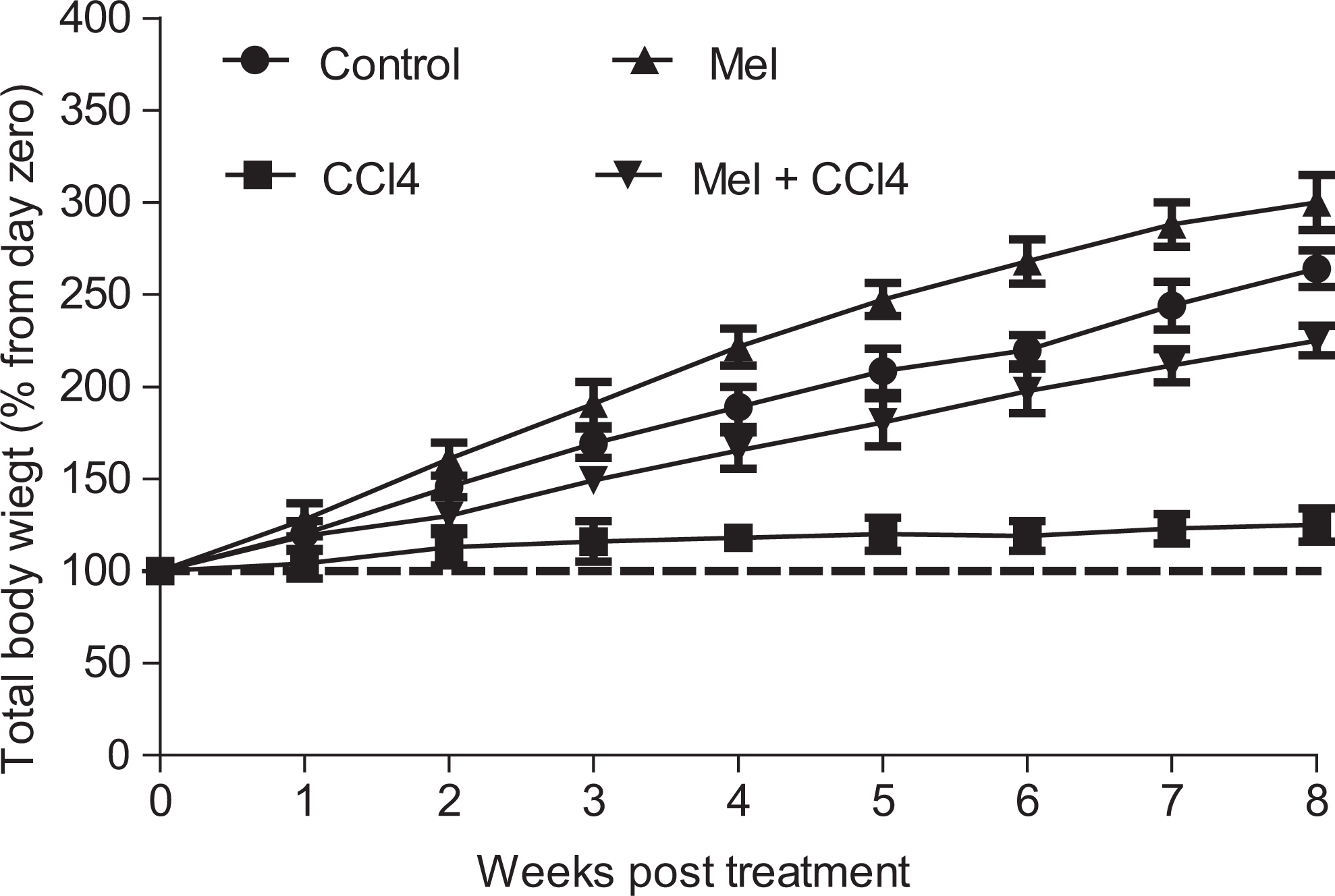
Effect of carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4 on rat total body weight. Meloxicam abrogated CCl4-induced inhibition of rat body weight gain. Body weight were recorded once weekly for 8 weeks. Data are plotted as percentage from corresponding day zero (initial day of treatment) value and are presented as mean standard error of 10 animals/group
Effect of MEL on Serum Biochemical Parameters
The serum levels of AST, ALT, and TLB are presented in Figure 2A, B, and C. In the CCl4-treated group, the serum levels of AST, ALT, and TLB significantly, P < .05, increased to 226.7 ± 8.8, 94.5 ± 3.3, and 6.8 ± 0.42, respectively, compared to negative control group values of 92.4 ± 4.2, 21.6 ± 2, and 0.8 ± 0.03, respectively. Pretreatment of CCl4-treated rats with MEL significantly, P < .05, decreased the CCl4-induced elevation of these marker levels to 109 ± 3, 26.6 ± 2, and 3.2± 0.3, respectively. Interestingly, MEL by itself, had no significant increase in the serum levels of AST, ALT, and TLB, 94 ± 3, 24.6 ± 2, and 1.2 ± 0.27, respectively, compared to control group.
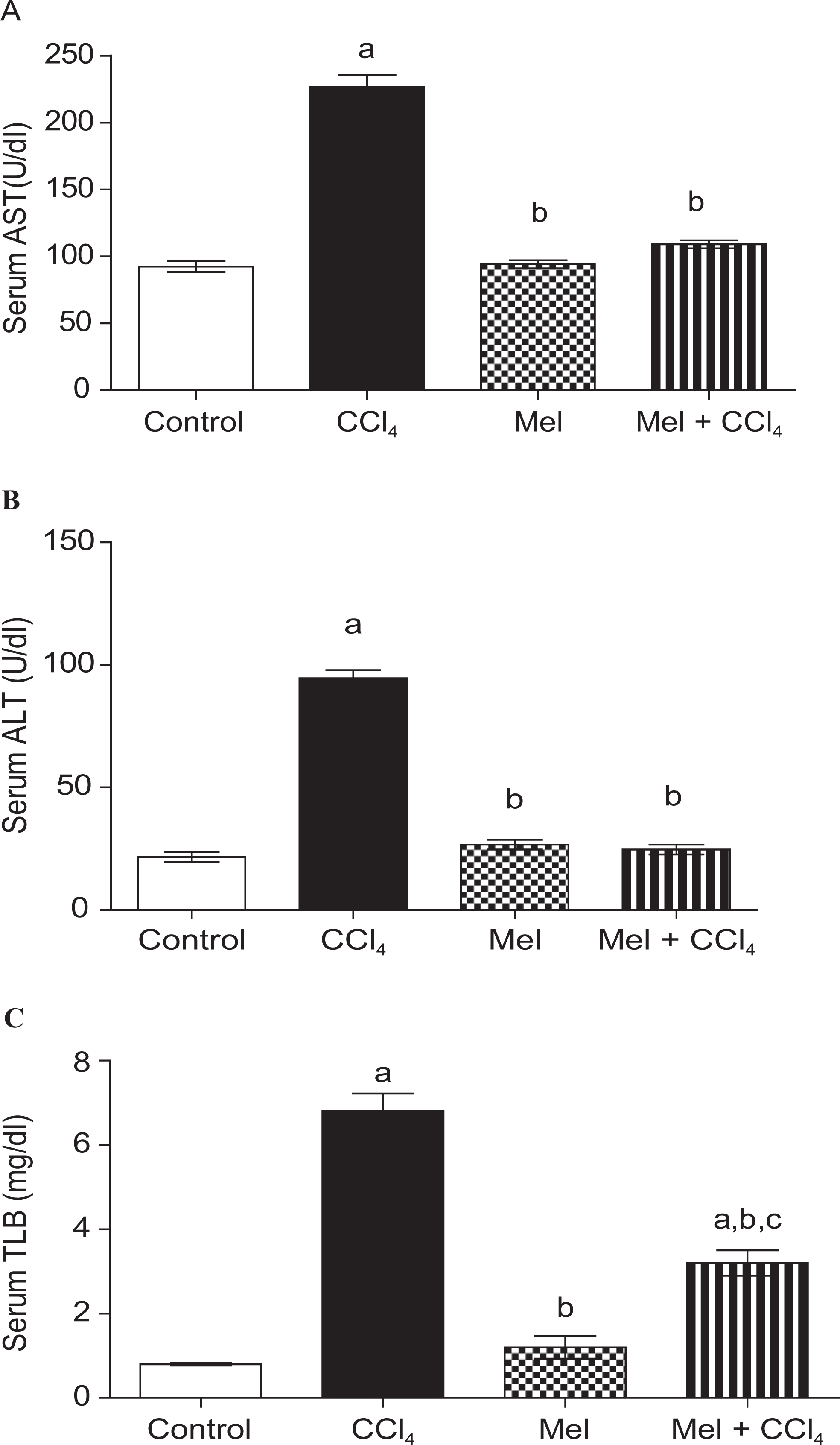
Effects of carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4 on liver function test in rats after 8 weeks of treatment. Meloxicam abrogated CCl4-induced liver enzymes close to normal as manifested in figures; (A) serum aspartylaminotransferase (AST), (B) serum alanine aminotransferase (ALT), and (C) serum total bilirubin (TLB). Data were presented as mean ± standard error of 10 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P ≤ .05 using Tukey test as post-ANOVA test. ANOVA indicates analysis of variance.
Histopathological Examination
The histopathological observations also support the results obtained from serum biochemical assays. The liver lobules of the control animals showed a classical lobular structure with hepatocyte plates directed from the portal triads toward the central vein, where they freely anastomose and the liver sinusoids normally irregularly dilated (Figure 3I). In contrast, liver sections from the rats treated with CCl4 alone showed fatty change, massive ballooning degeneration, and apoptosis with a predominant centrilobular distribution. Focal centrilobular hepatocyte collapse was associated with the collapse of the intervening stroma. Few inflammatory cells were observed (Figure 3II) and the inflammatory reaction divided the hepatic parenchyma into lobules (Figure 3II). Conversely, there was no histopathological alteration detected in the MEL sections (Figure 3III). Sections showed preserved lobular architecture, no remarkable inflammation, and absent hepatocyte degenerative changes (Figure 3III). Administration of MEL ahead of CCl4 challenge revealed better preservation of the normal liver architecture and milder ballooning hepatocyte changes (Figure 3IV).
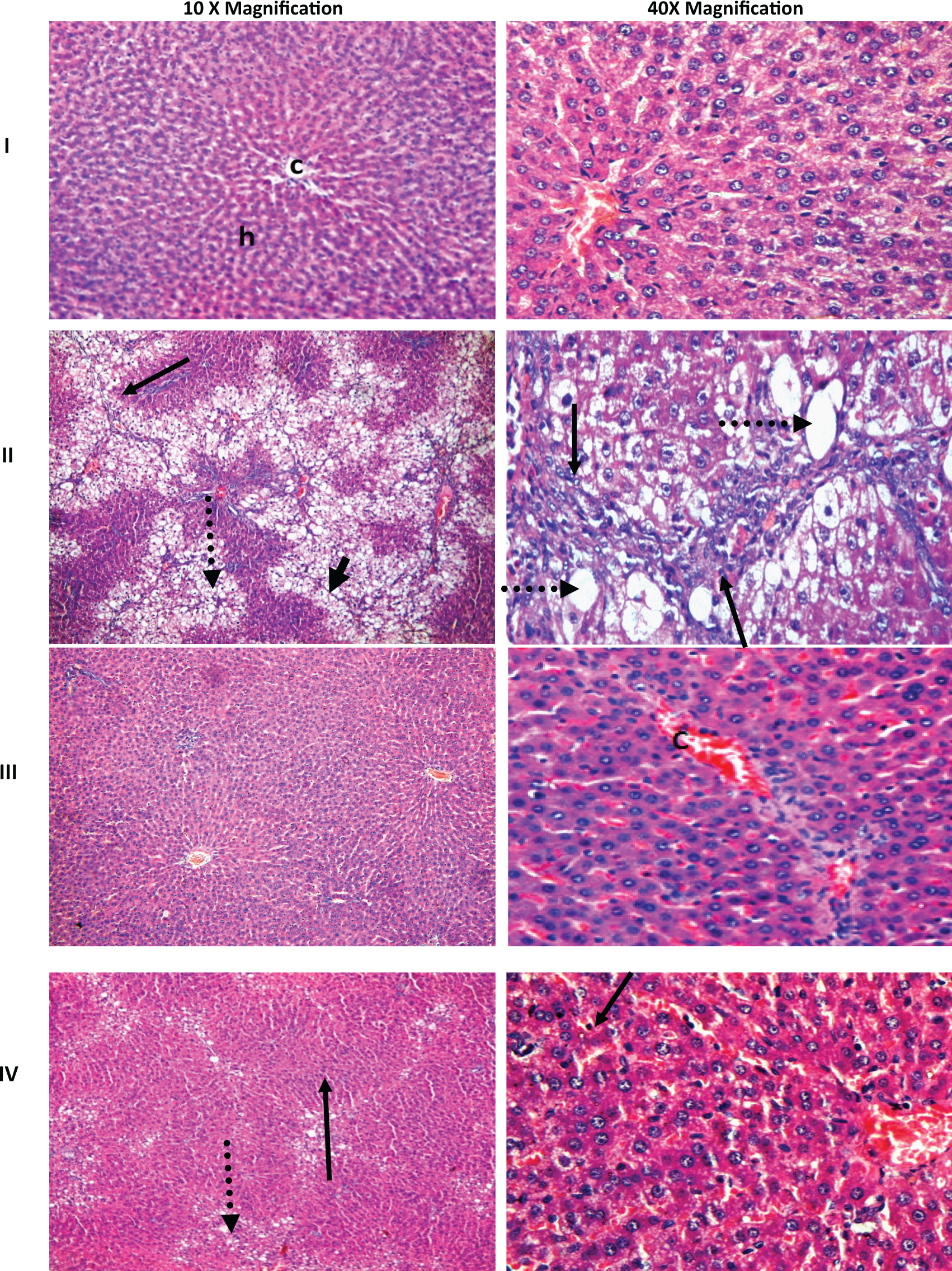
Representative H&E micrographs from liver tissues collected from rats treated with; (I) corn oil (control), (II) carbon tetrachloride (CCl4), (III) meloxicam, and (IV) combination of meloxicam and CCl4. (I) Control section showing normal histological structure of the central vein (c) and surrounding hepatocytes (h), (II) CCl4 treatment for 8 weeks induced histological changes in rat liver manifested by massive ballooning (vacuolar) degeneration, necrosis, and apoptosis in most of the hepatic parenchyma with predominant centrilobular distribution (dotted arrow). Focal centrilobular hepatocyte collapse was associated with the collapse of the intervening stroma (short thick arrow). Inflammatory cells were observed (long arrow). (III) Conversely, meloxicam treatment did not show any significant histopathological alteration (h), while (IV) prior treatment with meloxicam before CCl4 prevented extensive hepatocellular degeneration, although there was slight ballooning degeneration in centrilobular hepatocytes compared to CCl4 alone. Abbreviations and symbols: hepatocytes (h); massive ballooning degeneration (dotted arrow); focal centrilobular hepatocyte collapse (short thick arrow); inflammatory cells infiltrations (long arrow).
Evaluation of PGE2 Level in Liver
The effects of MEL and/or CCl4 on PGE2 level are compiled in Figure 4. Administration of CCl4 resulted in a significant increase in PGE2 level to 590 pg/mg protein ± 7.5 (218.3% of control value) compared to control animals that exhibited 270 ± 6. Treatment with MEL had no significant decrease in PGE2 level to 267 ± 3.7 (98.9% of control value) compared with the control group. Treatment with Mel, ahead of CCl4, prevented a significant rise in PGE2 level. The combination group exhibited PGE2 level of 306 ±3.9 which is nearly close to the control value (113% of control value).
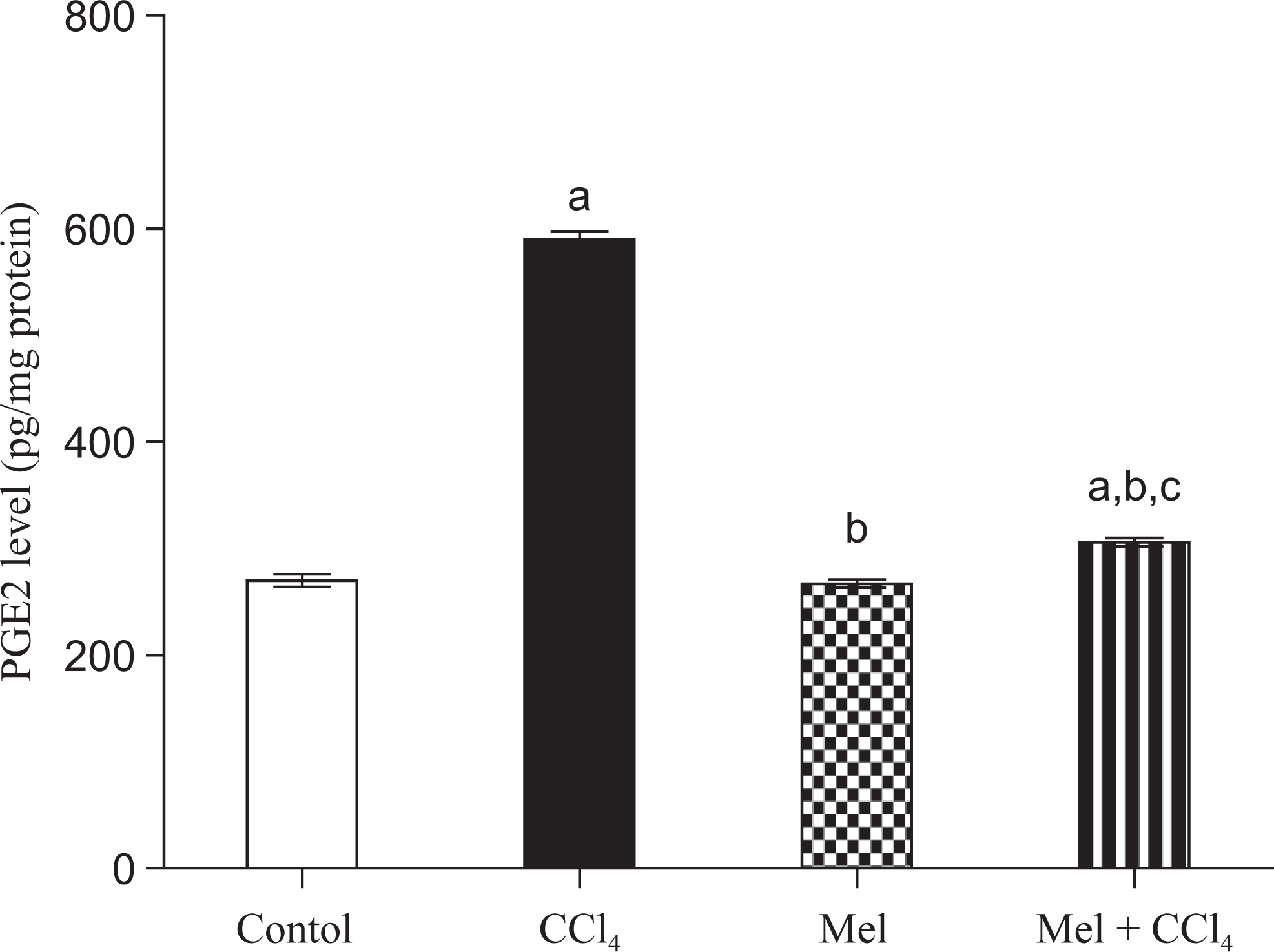
Evaluation of prostaglandin E2 (PGE2) level in rat liver after treatment with carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4. Data are presented as mean ± standard error of 10 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P ≤ .05 using Tukey test as post-ANOVA test. ANOVA indicates analysis of variance.
Effect on LPO and GSH Level
CCl4 caused a substantial increase in liver MDA content to 1033% ± 45.3%, with concomitant depletion in GSH content 47.2% ±1.26% compared to the negative control group (Figure 5). Administration of MEL alone showed a nonsignificant decrease in liver MDA content 128.2 ± 8.8 (Figure 5A), while exhibited a significant decrease in GSH content 86.5 ± 3.45 compared to the −ve control group (Figure 5B). Consistent with the serum level of AST, ALT, and TLB, combined administration of MEL and CCl4 resulted in a significant reduction in the liver MDA content 192 ± 16.05 with marked increase in liver GSH content 69.9 ± 3.21 compared to CCl4-treated group (Figure 5).
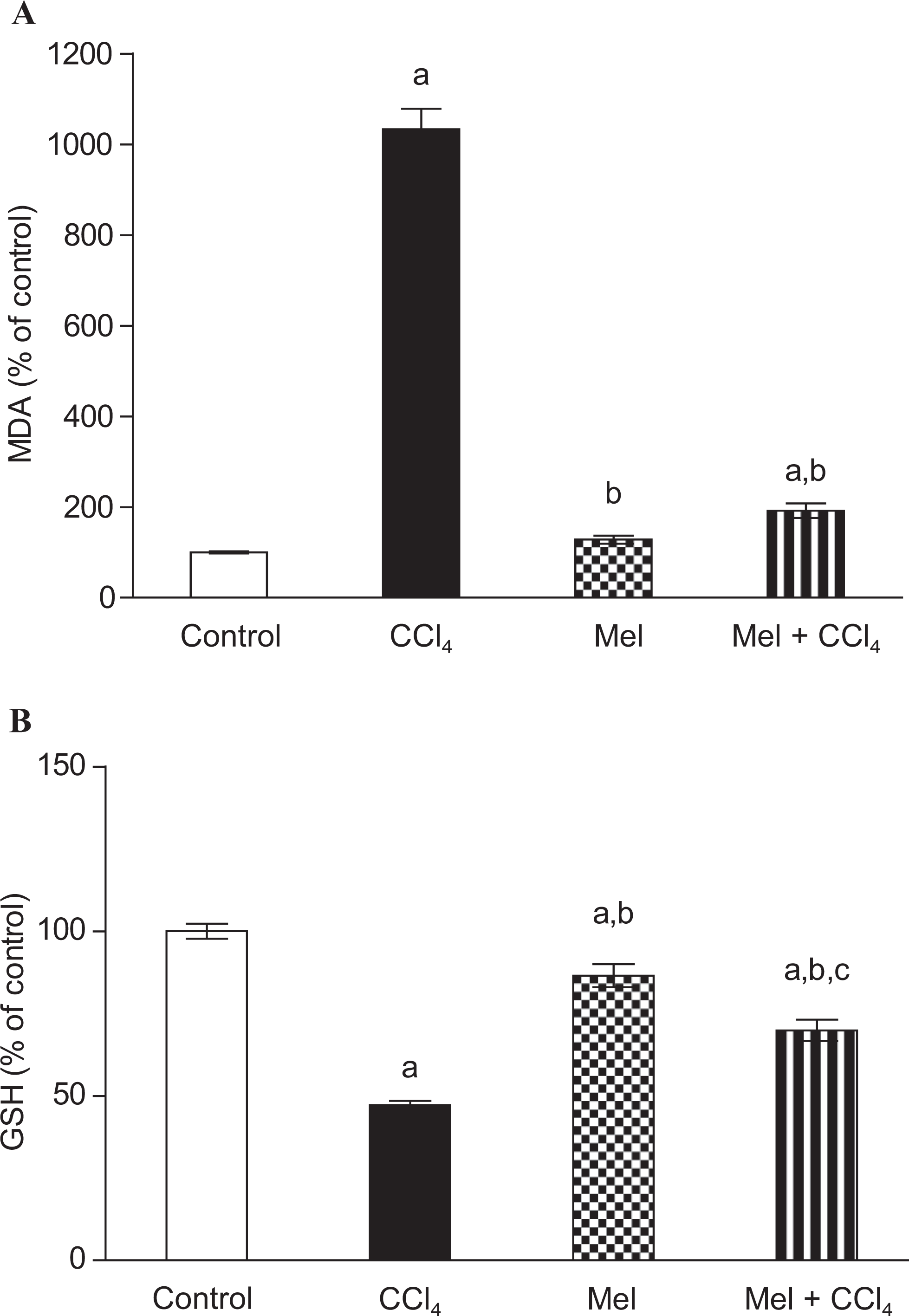
Effect of carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4 on the liver content of malondialdehyde (MDA) and reduced glutathione (GSH) level in rats. Meloxicam modulated CCl4-induced changes in (A) malondialdehyde (MDA) content; and (B) reduced glutathione (GSH) content. MDA was taken as indicator for lipid peroxidation. Animal treatment protocol implies twice weekly injection for 8 weeks. Data are plotted as percentage of corresponding control value. Data are presented as mean ± standard error of 10 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P ≤ .05 using Tukey test as post-ANOVA test. ANOVA indicates analysis of variance.
Effect of MEL on Antioxidant Enzymes
Figure 6 shows the activities of enzymatic antioxidants (SOD, CAT, and GST) in the liver tissue of rats. A significant fall, P < .05, in the activities of the above-mentioned enzymatic antioxidants was observed after CCl4 administration (30.5% ± 8.1%, 26.6% ± 3.5% and 35.9% ± 2.2% of control values, respectively). Administration of MEL 1 hour ahead of CCl4 injection significantly prevented a significant reduction in these enzyme activities. MEL-alone treated group showed a significant increase in liver SOD and GST activities; 118 ± 5.32 and 362 ± 14.16, respectively, nevertheless MEL exhibited a nonsignificant decrease in liver catalase activity, 94.25 ± 10.06, when compared with negative control group.
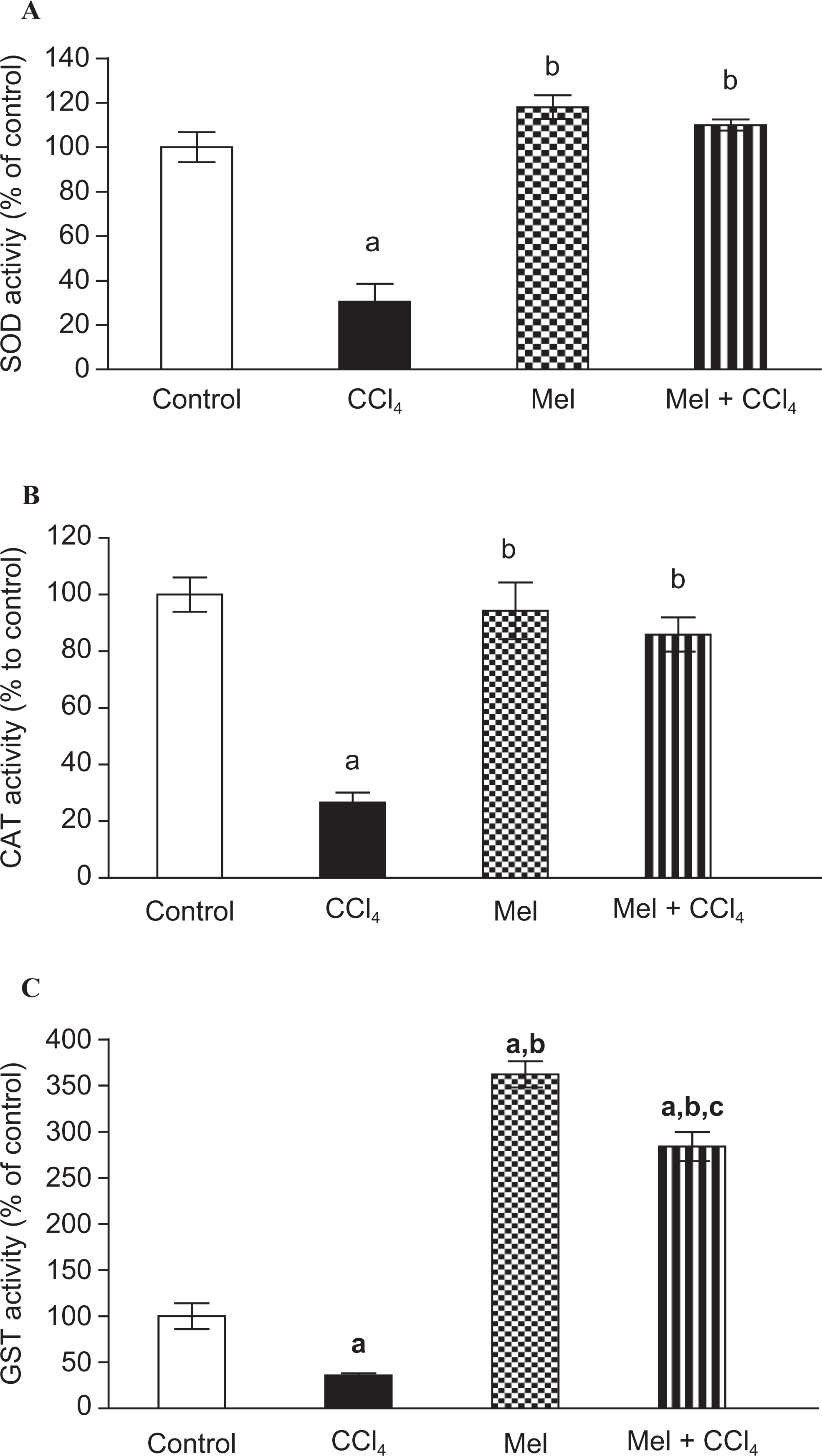
Evaluation of enzymatic antioxidants defense system in rat liver after treatment with carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4. Meloxicam reversed CCl4-induced inhibition of antioxidant enzymes; (A) superoxide dismutase (SOD), (B) catalase (CAT), and (C) glutathione-S-transferase (GST). Enzyme activities are plotted as percentage of corresponding control value. Data are presented as mean ± standard error of 10 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P ≤ .05 using Tukey test as post-ANOVA test.
Determination of Hepatic Active Caspase 3 Content and Induction of Apoptosis
We further evaluated active caspase 3 content in liver sections using immunohistochemical technique as a potential mechanistic tool. The CCl4-treated group exhibited a significant (P < .05) increase in active caspase 3 content to 354.3% ± 8.35% (Figure 7). Meloxicam alone induced a nonsignificant decrease in caspase 3 content; 126% ± 7.87%, conversely combination group showed a significant (P < .05) suppression in caspase 3 content, 190.28% ± 5.11%, compared to CCl4-treated group (Figure 7).
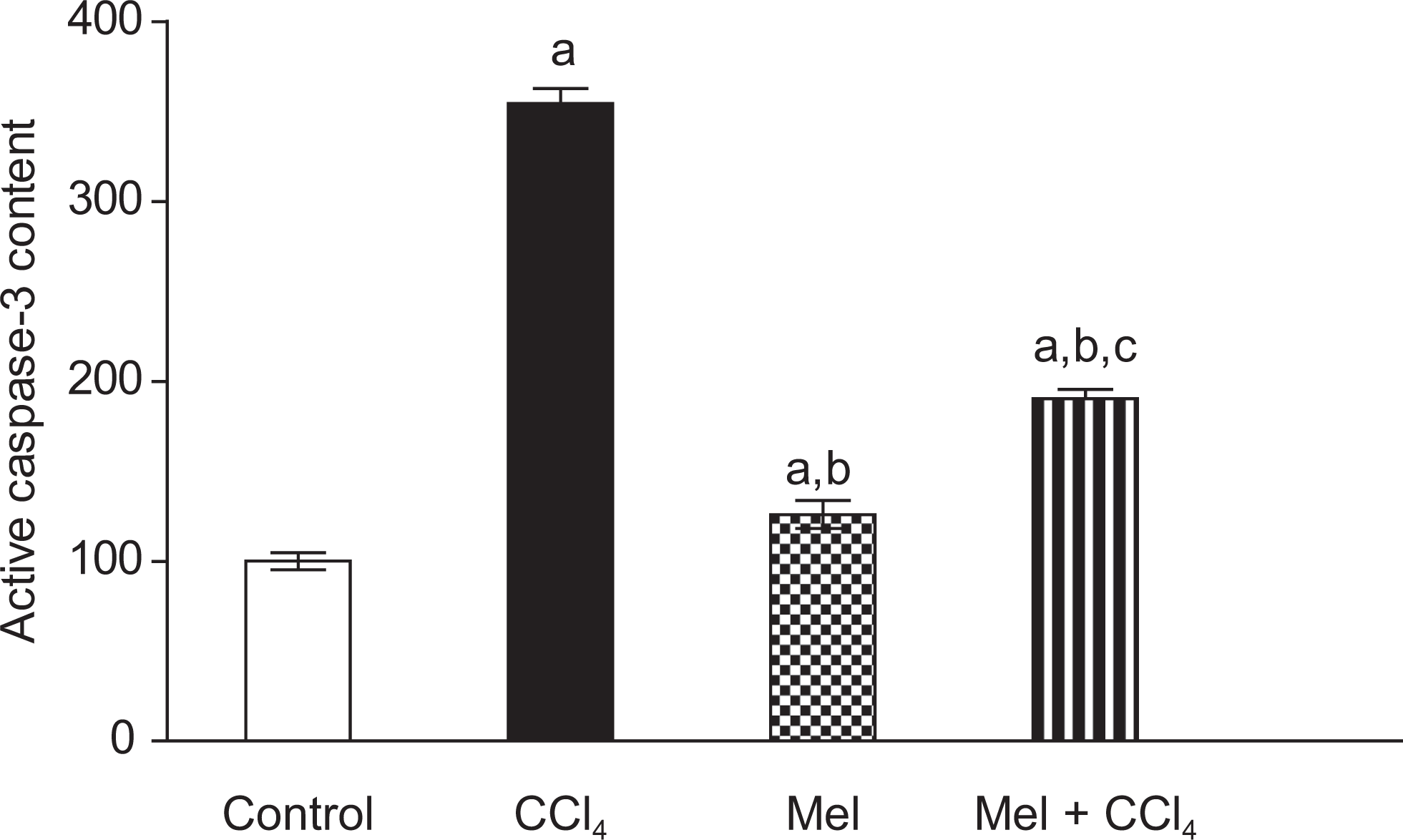
Effect of carbon tetrachloride (CCl4), meloxicam (Mel), or both meloxicam and CCl4 on active caspase 3 content in rat liver. Active caspase 3 content was evaluated by immunohistochemical technique and semiquantified by 2 histopathologists. Data are calculated as percentage expression within the liver sections and plotted as percentage from negative control group. Data are presented as mean ± standard error of 6 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P < .05 using Kruskal-Wallis test, followed by the Dunn test.
To confirm the possibility of liver cell death through apoptosis and possible protection using MEL, we evaluated apoptosis induction using the TUNEL assay. The CCl4-treated liver sections exhibited strong positive tunnel reaction; 30% ± 2% (Figure 8). This was significantly higher (P < .05) compared to vehicle-treated group (7.5% ± 2%). Interestingly, liver sections collected from animals treated with MEL prior to CCl4 showed significant decrease of TUNNEL-positive cells of 12.3% ± 2.4% compared to CCl4 group, while MEL alone did not induce any significant change compared to the control value (Figure 8).
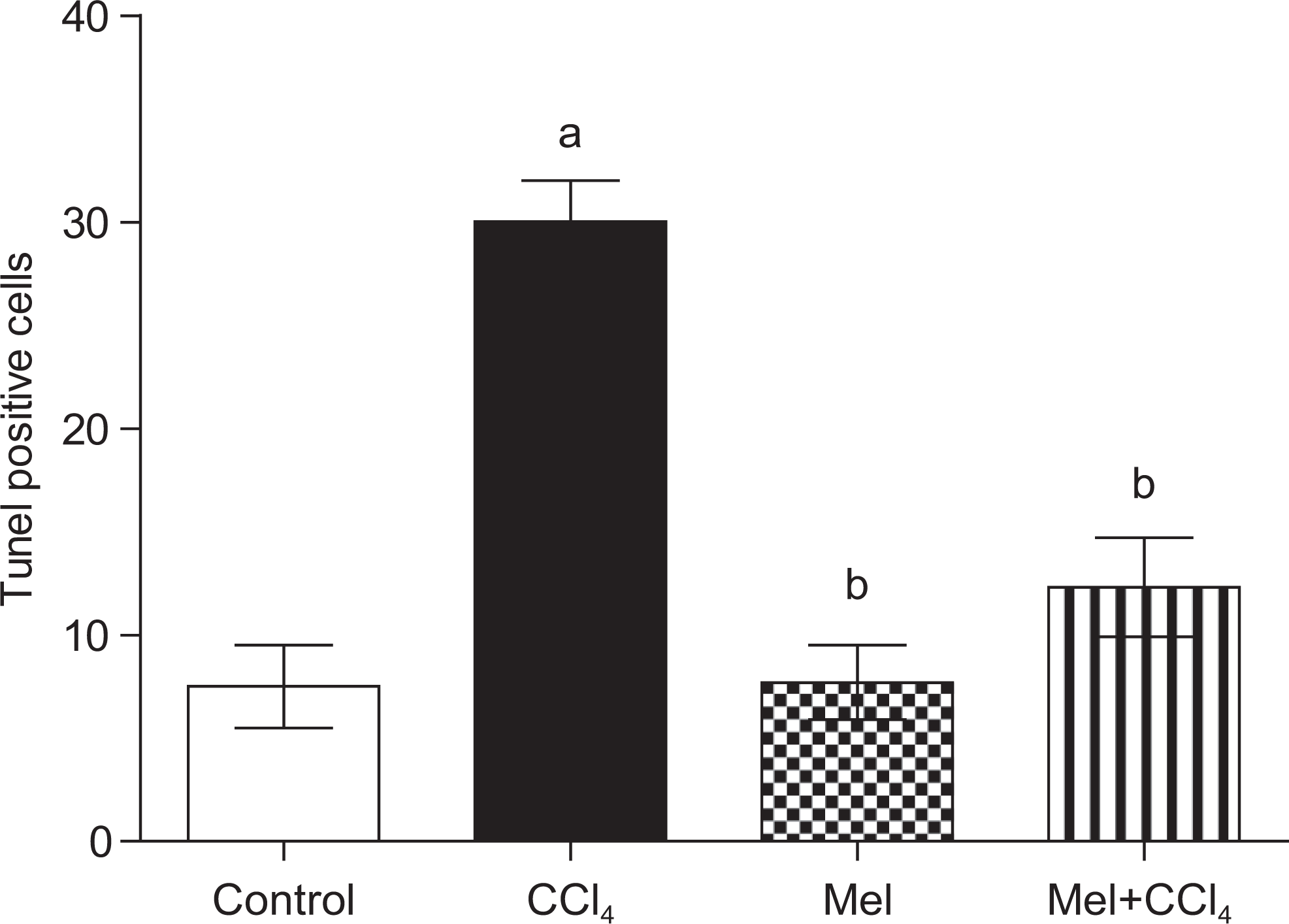
Carbon tetrachloride (CCl4) induces apoptosis in rat liver cells, while prior treatment with meloxicam (Mel) abrogated this effect. Data are calculated as percentage expression within the liver sections and are presented as mean ± standard error of 6 animals/group; a, b, or c indicates significant difference from control, CCl4, or meloxicam, respectively, at P < .05 using Kruskal-Wallis test, followed by the Dunn test.
Discussion
CCl4 is a classical hepatotoxicant that causes rapid liver damage progressing from steatosis to centrilobular necrosis. 5 The mechanism of liver injury induced by CCl4 is thought to involve free radical generation such as trichloromethyl radical (CCl3 •) and proxy trichloromethyl radical (OOCCl3 •) and thus LPO and induction of inflammatory response. 5,45 –47 Therefore, it is conceivable that antioxidative activity and/or the inhibition of free-radical generation or anti-inflammatory action are important in terms of protecting the liver from CCl4-induced damage. 48
In line with this notion, the current study was aimed at protecting liver against injury induced by CCl4 utilizing MEL, a selective COX-2 inhibitor, with both anti-inflammatory and antioxidant activity. Our study demonstrated that, exposure to CCl4-induced liver cell injury manifested by (1) increased relative liver weight to 100 g body weight and (2) increased serum levels of liver enzymes and total serum bilirubin (Figures 1 and 2). The occurrence of hepatic injury is further confirmed via histopathological evaluation (Figure 3). Moreover, liver cell death was also confirmed by increased percentage of TUNEL-positive cells in CCl4-treated groups compared to the control group (Figure 8). CCl4-induced liver toxicity is mediated by (1) GSH depletion (Figure 5B); (2) decreased activities of antioxidant enzymes: SOD, CAT, and GST (Figure 6); (3) increased liver level of MDA, an LPO marker (Figure 5 A), which confirm the induction of oxidative stress 49 ; (4) increased level of PGE2 (Figure 4) indicating induction of COX-2 enzyme and led to a strong inflammatory response reported in the histopathological finding (Figure 3); (5) induction of apoptosis in liver tissue (Figure 8) which is explained by activation of caspase 3 (Figure 7); or (6) some or all of the above. An overproduction of COX-2 has been demonstrated in chronic liver disease of both human and experimental animals. 19,50 –52 Moreover, COX-2 activity generates ROS which are related to oxidative stress. 53 In addition, previous studies reported a strong association between administration of CCl4 and both increased expression of COX-2 and infiltration of inflammatory cells into the liver. 54,55 An effect, that results in the release of various free radicals causing cellular damage through oxidative stress and LPO. 21,54,56 –58
The cascade of oxidative stress and inflammation from CCl4 intoxication could disrupt the structural integrity of the hepatic cell membrane and lead to leakage of AST and ALT hepatic cellular enzymes into the blood from dead hepatocytes and possible hepatic dysfunctions manifested by increased level of serum TLB (Figure 2), thus producing signs of hepatotoxicity. 59,60 This inflammatory response could be responsible for the increased ratio of liver weight to total body weight in CCl4-treated group. 56,58
The relationship between ROS and apoptosis has been under investigation for a number of years. The ROS play important roles in apoptosis initiated in mitochondria. 61 –63 CCl4 intoxication has been suggested to cause severe apoptosis. 5,61,63 Consequences from the toxin-induced excessive oxidative stress, depletion of antioxidant enzymes, and induction of membrane LPO may prompt the extrinsic or intrinsic apoptotic pathways. 64,65 These pathways eventually lead to activation of caspases pathway for apoptosis that ends up with caspase 3 activation, the real executioner of apoptosis, once triggered; the active caspases initiates cell apoptosis. 66 –68 Coping with this scenario, our results collectively indicate that CCl4 causes apoptosis in the liver by activating caspase 3 (Figures 7 and 8) and that the apoptosis and necrosis proceed simultaneously in the liver as shown by the high AST and ALT serum levels (Figure 2) and confirmed in H& E histological analysis (Figure 3) and TUNEL assay (Figure 8).
Prior MEL administration significantly mitigated CCL4-induced hepatotoxicity. This was clearly manifested by (1) decreased serum levels of ALT, AST, and TLB 69 (Figure 2); (2) improvement in liver weight to body weight (Figure 1); (3) restoration of liver histology close to normal (Figure 3). These hepatoprotective effect are mediated via (1) inhibition of PGE2 production (Figure 4); (2) induction of antioxidant defense system including GSH, SOD, CAT, and GST (Figures 5B and 6); (3) reduction of LPO or antiradical effect indicated by lower MDA level 70 (Figure 5A); (4) reduction of active caspase 3 expression (Figure 7); or (5) some or all of the above.
The antioxidant activity for MEL in this study supports the previous finding by Gurocak et al 71 in acute pyelonephritis model and results of Cimen and his coworkers 72 utilizing MEL as a protector against osteoarthritis-induced free radical generations in human erythrocytes. Arafa et al declared the partial role of antioxidant activity for MEL against bleomycin-induced lung fibrosis in mice. 73
Interestingly, the amelioration in the biochemical enzymatic and oxidative stress markers in the combination group compared with CCl4-treated group was accompanied by an alleviation of the histopathological changes. This effect might be in part ascribed to the COX-2 inhibitory effect of MEL demonstrated in our study via reduction in PGE2 level (Figure 4). 74
An important finding of the current study was that MEL substantially abrogated CCl4-induced caspase 3 activation and reduced apoptotic changes in the liver sections (Figures 7 and 8). We suggest that the inhibition of caspase 3 by MEL could be effective in preventing CCl4-induced hepatocyt apoptosis. 28 In addition to its antioxidant and free radical scavenging activities, the well-established anti-inflammatory potential 75 might be implicated in abrogation of caspases cascade activation. 76,77
Interestingly, the global finding of our study are in accordance with the previous reports by Chávez et al 50 and Planaguma et al 15 who reported the success of selective COX-2 inhibitors, celecoxib, and a SC-236, respectively, to protect against liver injury induced by CCl4.
Conversely, our results are in contrast to the study of Bhave et al. 78 Whereas they have shown that COX-2 inhibition exacerbates CCl4 liver injury. 78 This difference might be ascribed to difference in (1) the type of animals used; (2) protecting agents; (3) dose regimen; (4) liver injury models utilized; or (5) all of the above.
Although the study presented in this article succeeded to fulfill its aim and prove the hepatoprotecive concept and explored some of the possible mechanisms behind the hepatoprotection by MEL, Further studies are still required to establish the exact mechanism behind amoleriation of CCl4-induced liver insult. Some of such mechanisms include evaluation of metabolizing enzymes in both phase I (responsible for generation of toxic metabolite from CCL4) and phase II (responsible for detoxification of CCl4 metabolites). In addition, COX-2 downstream genes controlling apoptosis and fibrosis in liver should be also evaluated.
Conclusion
The results of the current study bring to light the hepatoprotective potential of MEL, a selective COX-2 inhibitor. The protective role of MEL against CCl4-induced hepatocellular damage could possibly be ascribed to its antioxidant potential, free radical scavenging properties, anti-inflammatory effect, and abrogation of apoptosis via suppression of active caspase 3. These results may also open new avenues toward further studies for the possibility of using COX-2 inhibition approach as an adjuvant therapy for liver inflammation and fibrosis.
Footnotes
Acknowledgment
We are deeply thankful to Dr Adel Bakeer Kholoussy, Professor of Pathology, Faculty of Medicine, Cairo University, Egypt, and his highly respected laboratory team for their invaluable contribution in histopathological examination part of this work.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.