
Abstract
The timing of carcinogenicity studies in parallel with the clinical development of anti–human immunodeficiency virus (HIV) drugs has been flexible for most cases in the past. This includes postponement of the initiation of the studies and submission of final audited reports to the US Food and Drug Administration (FDA) for a new drug application (NDA) approval. We address this regulatory practice for anti-HIV drugs for which, in the past, there had been no effective treatment. We also examine the correlation of genotoxicity data with carcinogenicity data for the varied subclasses of anti-HIV drugs. We suggest that this regulatory policy regarding the timing of carcinogenicity testing does not compromise the safety standards of FDA’s drug evaluation and the approval process. The policy does facilitate availability of these agents to meet the medical needs of the target population. Our analysis on the profile of carcinogenicity findings of anti-HIV drugs shows trends of class effects. Additionally, both carcinogenicity and genotoxicity data show significant correlations, which provide useful insights into issues involving these 2 important areas of toxicological investigations.
Introduction
In the United States, assessment of the carcinogenic potential of anti- human immunodeficiency virus (HIV) drugs, as well as most other pharmaceuticals, follows both the Food and Drug Administration (FDA) and the International Conference on Harmonization (ICH) guidance. The long-term carcinogenicity bioassays are often conducted in 2 animal species, usually the rat and the mouse, although shorter term alternative studies using transgenic mice (e.g., 6-month mouse transgenic p53+/– assays) or other specific assays as indicated in the guidance may be considered in place of the 2-year bioassays. 1 –4 The objectives of these studies are to identify the tumorigenic potential of new drug products in animals as the basis for relevant human risk assessment. In the past, the timing for initiation and completion of carcinogenicity studies conducted in parallel with clinical evaluation of anti-HIV drugs has been modified from the typical timing seen with other drug products and, in most cases, postponed because of the unmet medical need for the anti-HIV drugs. For example, the study completion and final report submission for some carcinogenicity studies were accepted as a commitment after accelerated approval of the new drug application (NDA). The underlying regulatory view of this decision was that drugs of this category are indicated for life-threatening HIV infection for which there was no effective treatment and there existed an urgency to fulfill an unmet medical need for the target population. Furthermore, most clinical trials on anti-HIV drugs employed surrogate efficacy markers that collected limited human safety data (eg, 6 months), and there may not have been sufficient time to allow for completing the long-term animal carcinogenicity studies. 5
Both industry and regulatory agencies generally recognize that long-term carcinogenicity bioassays are among the most resource-intensive toxicology studies. It is also apparent that a significant need for more effective anti-HIV drugs capable of eradicating HIV infection and curing the disease still exists. Therefore, with the understanding that toxicity concerns will be fully addressed by a sponsor and overall human safety will not be compromised, the current view of delaying the conduct of carcinogenicity studies and final report submissions may continue to benefit anti-HIV drug development.
Due to significant advances in anti-HIV drug development, an increasing number of drug products with varied mechanisms of action has been approved and made available for treatment of HIV infection. For example, the FDA’s Center for Drug Evaluation and Research (CDER) has to date approved (Table 1) 8 nucleoside and nucleotide reverse transcriptase inhibitors (NRTIs), 5 nonnucleoside reverse transcriptase inhibitors (NNRTIs), 10 protease inhibitors (PIs), and 1 each for HIV integrase inhibitor, entry blocker, and fusion inhibitor. 6 It is now generally accepted that HIV infection can be effectively treated using a broad spectrum of anti-HIV drugs such that the infection may no longer be considered acutely life threatening.
Timing of Conduction of Carcinogenicity Studies of Anti-HIV Drugs in Reference to NDA Approvals and Findings of Carcinogenicity and Genotoxicity
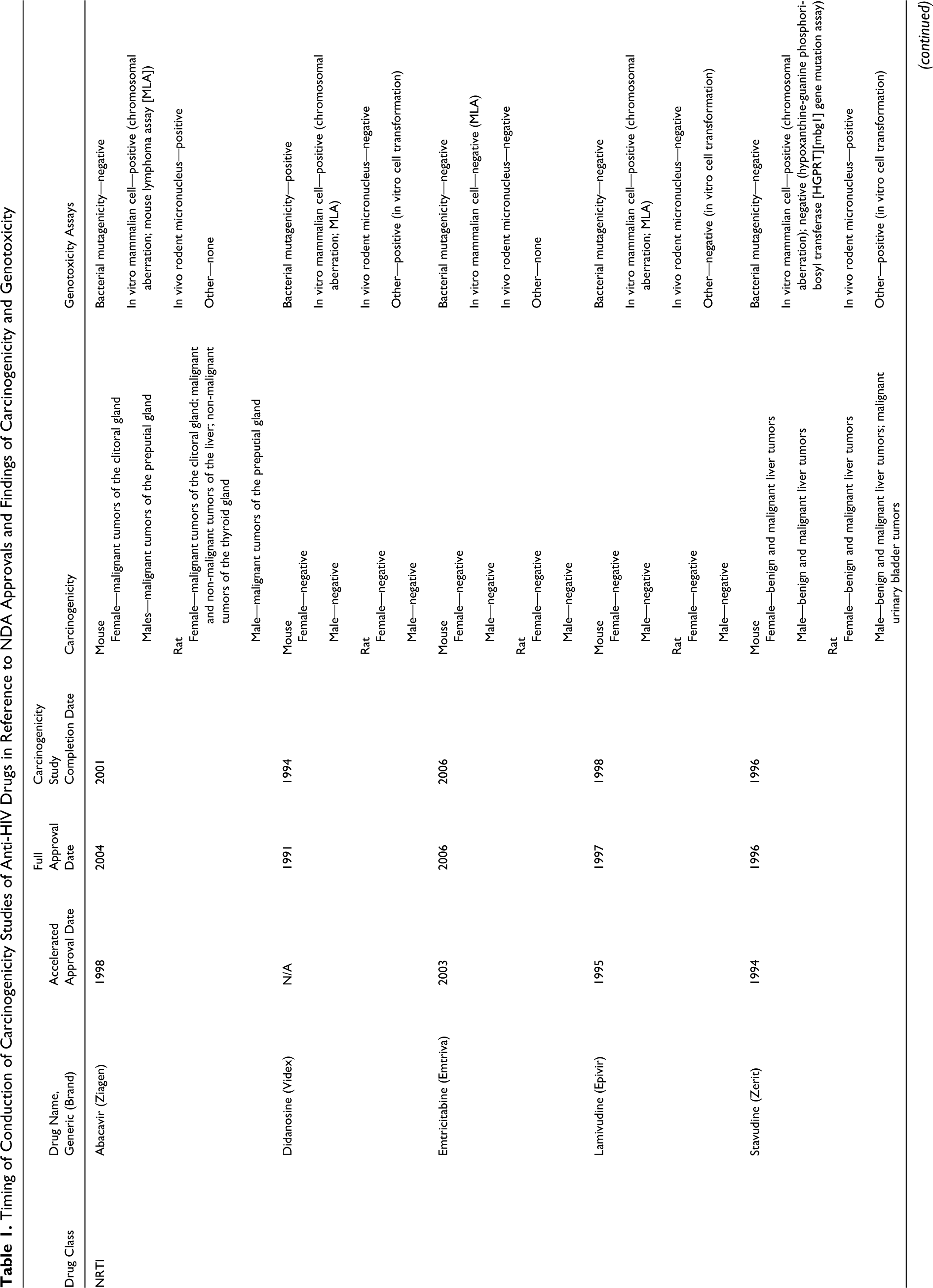
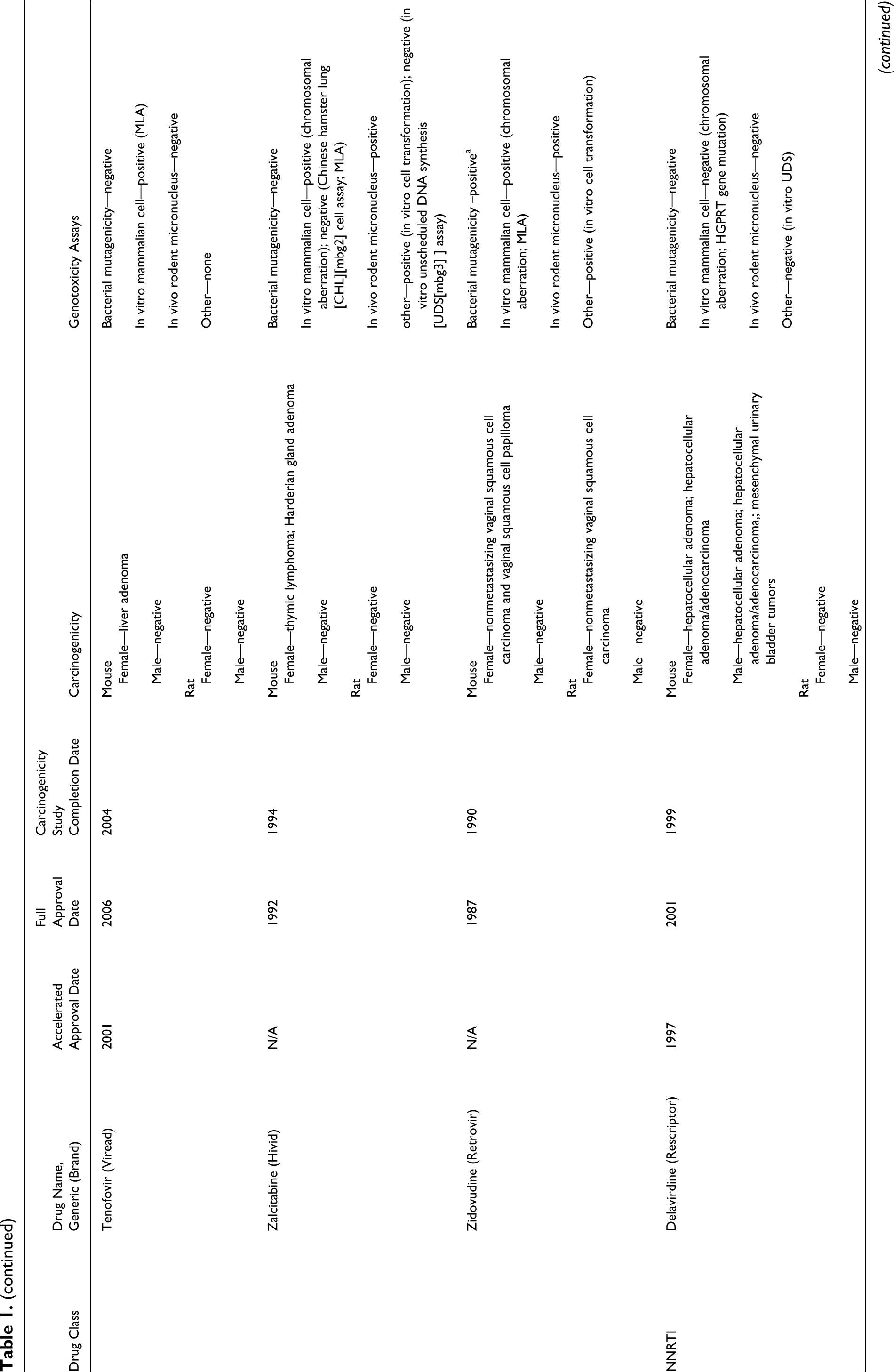
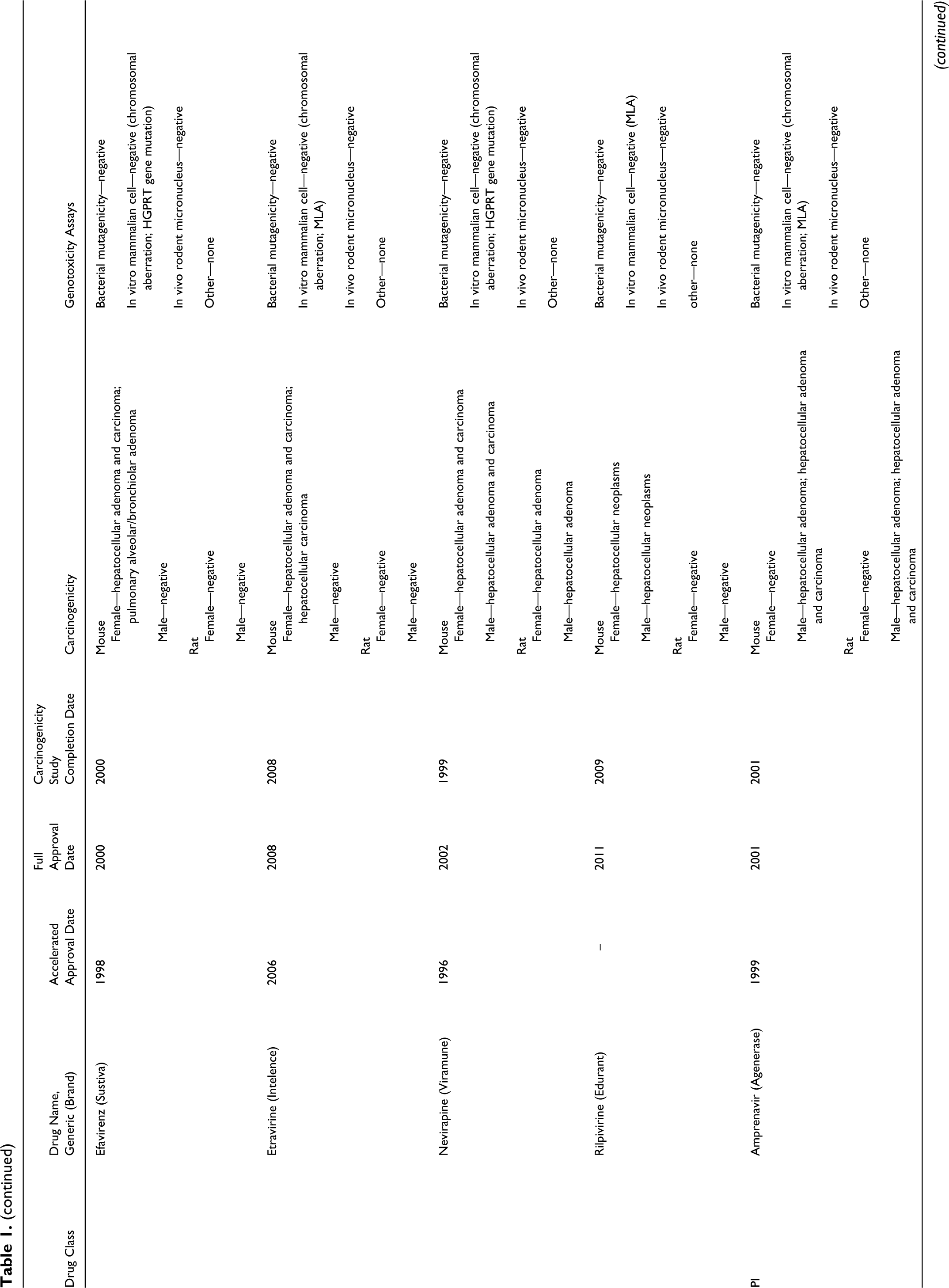
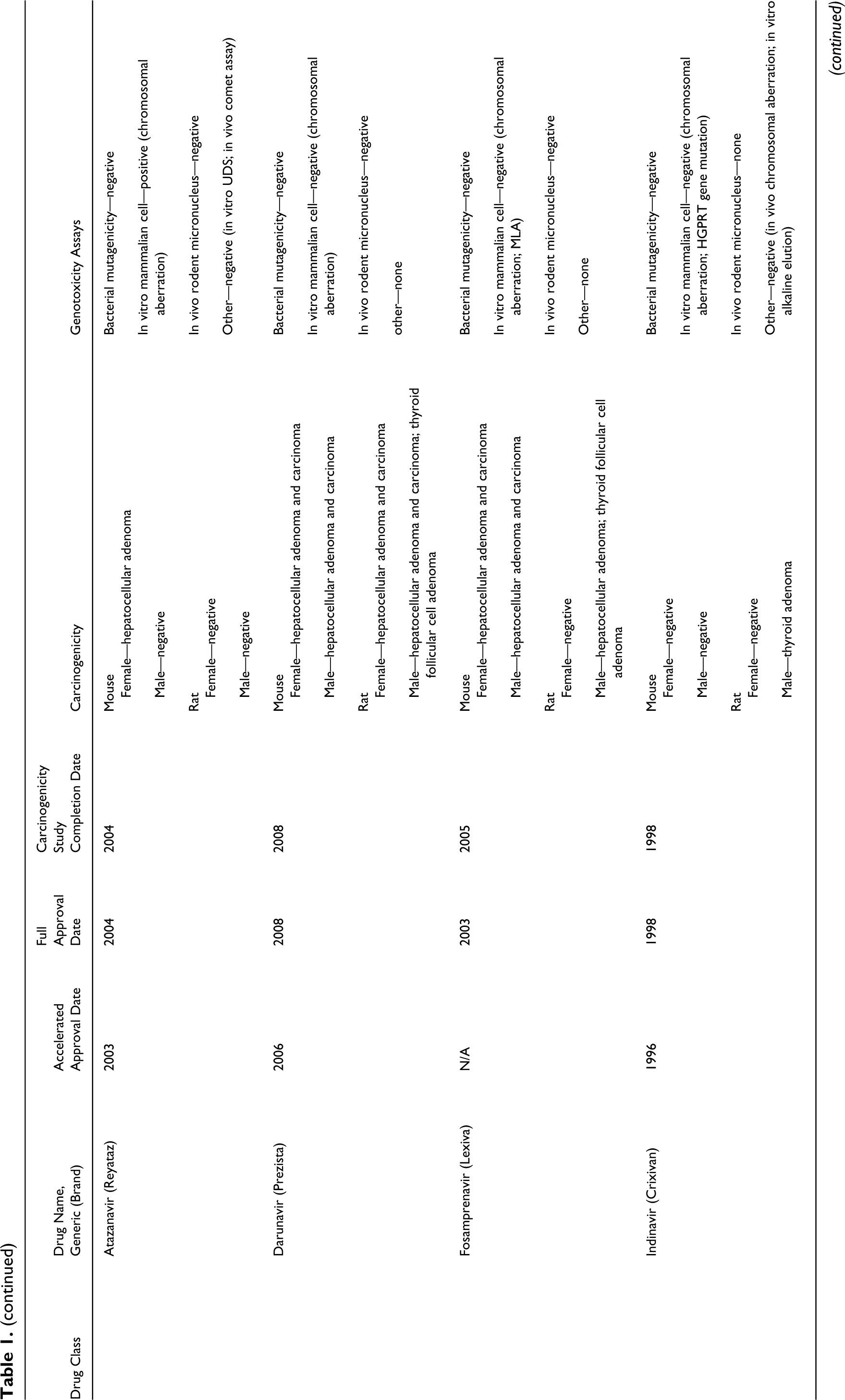
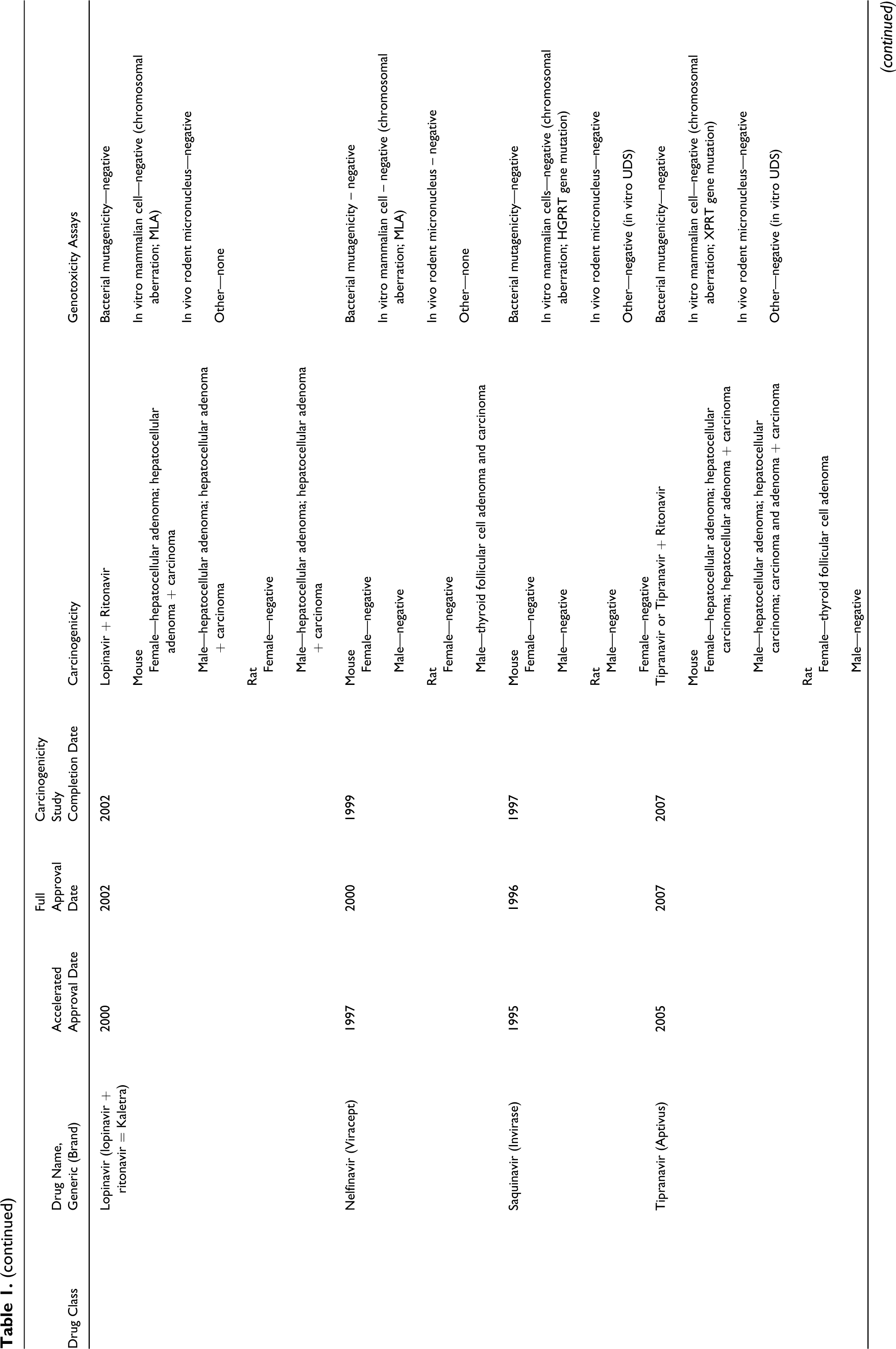
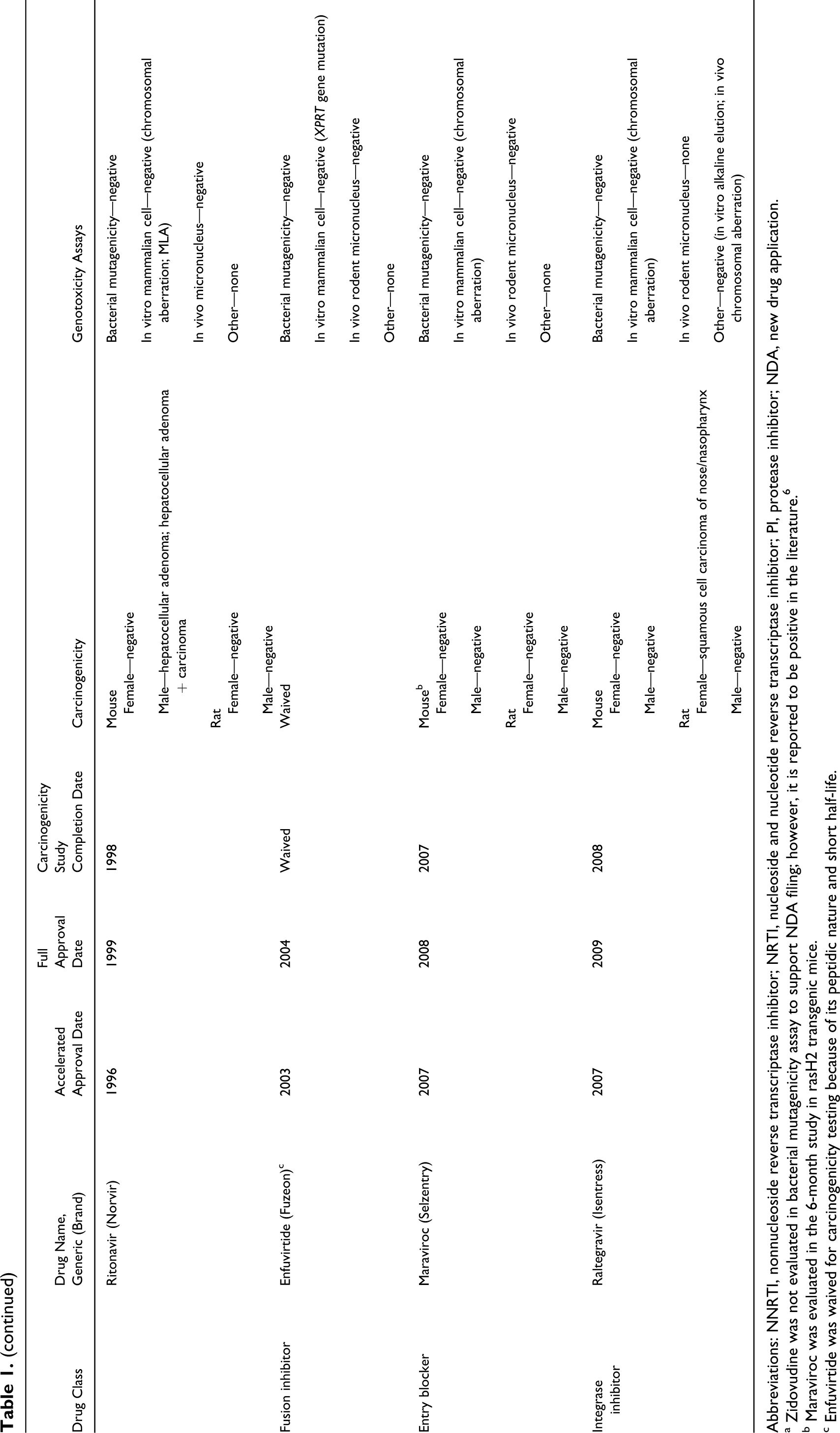
Abbreviations: NNRTI, nonnucleoside reverse transcriptase inhibitor; NRTI, nucleoside and nucleotide reverse transcriptase inhibitor; PI, protease inhibitor; NDA, new drug application.
a Zidovudine was not evaluated in bacterial mutagenicity assay to support NDA filing; however, it is reported to be positive in the literature. 6
b Maraviroc was evaluated in the 6-month study in rasH2 transgenic mice.
c Enfuvirtide was waived for carcinogenicity testing because of its peptidic nature and short half-life.
Some categories of antiretrovirals such as NRTIs, NNRTIs, and PIs have more drugs approved and may provide more options for anti-HIV therapy than other newer subclasses (eg, entry/fusion blockers). As drug development strategies and priorities evolve, it is timely to revisit the history of drug approvals for this therapeutic category and to review how industry has applied the unique policy of deferring animal carcinogenicity studies to determine whether the past practice of deferring carcinogenicity studies as a postapproval commitment continues to be necessary. Because genotoxicity testing and animal carcinogenicity studies are thought to be correlated with direct-acting genotoxic agents, both data types were examined.
A Survey of Anti-HIV Drug Approvals and the Timing of Completion of Carcinogenicity Studies
The results of our survey of anti-HIV drugs are listed in Table 1. Key findings on both carcinogenicity and genotoxicity are also included. This information was obtained from FDA’s approved drug package inserts and reviews. 6 –8 The studies included in Table 1 were conducted according to FDA and ICH recommendations . 1 –4,9 –11
In the beginning era of anti-HIV drug development (in the early 1990s), zidovudine, didanosine, and zalcitabine gained FDA marketing approval through expedited review processes, although a formal accelerated approval mechanism was not in place at that time. Subsequently, most of the anti-HIV agents, with the exception of fosamprenavir and rilpivirine, were reviewed under the accelerated approval processes. For those drugs subjected to an accelerated review, the animal carcinogenicity studies were generally completed post-accelerated approval and prior to full NDA approval.
To date, the approved drugs in the NRTI, NNRTI, and PI classes provide different safety profiles and predictable efficacy. Future development of new anti-HIV drugs are expected to have ample margins of safety (eg, limited target organ of toxicity, minimal reproductive toxicity, and genotoxic findings), or to have superior and/or more favorable human pharmacokinetics, formulations, and efficacy profiles (eg, once a day dosing, low potential to induce resistance). All of these factors should support an accelerated review status and a continued policy for a delay of carcinogenicity studies for potential candidates in these drug classes.
We also need to consider emerging classes of anti-HIV drugs, such as integrase inhibitors and entry/fusion blockers. These drugs exhibit novel mechanism of action and have the potential to further provide the unmet medical needs in this therapeutic area. It is important to continue current policy of delaying the conduct of carcinogenicity studies for these new classes of anti-HIV drugs, as historically carried out, to facilitate and benefit rapid strategic drug development.
Predictability of Genotoxicity on Tumorigenicity of Anti-HIV Drugs
The predictive nature of individual genotoxicity assays may be considered when correlating genotoxicity with carcinogenicity data for anti-HIV drugs. The sensitivity of an overall genotoxic evaluation is increased by evaluating multiple end points as recommended by FDA and ICH, 9 –11 whereas specificity may be concurrently reduced (ie, increase in false positives). 12,13 However, many rodent carcinogens exert their effects through nongenotoxic mechanisms. A review of the FDA’s approved prescription drug listing showed that almost two thirds of agents shown to be rodent carcinogens tested negative in all genotoxicity assays conducted. 8
Liver or thyroid tumors via an epigenetic mechanism, for example, hepatic enzyme induction, have been shown for some xenobiotics including phenobarbital. 14 –16 A potent hepatic CYP450 inducer may cause liver hypertrophy and hyperplasia, as is often observed in rodent repeat-dose toxicity studies. The role enzyme induction plays in tumor formation has been the subject of speculation and research for many years and is especially critical when drug administration is extended throughout the lifespan. 16 –18 A similar possibility exists for hepatic thyroid hormone metabolizing enzyme inducers (eg, uridine 5'-diphospho-glucuronosyltransferase) that could disturb the neuroendocrine feedback axis and produce thyroid follicular cell neoplasia in rodents. 15,19
Agents with these or other modes of action may result in tumor formation in rodents under lifetime drug administration. However, tumors resulting from either mode of toxic mechanisms in rodents may not be considered relevant during human risk assessment. 16 –20 While some anti-HIV drugs have been shown to be hepatic enzyme inducers, definitive conclusions regarding the precise mode of action and human relevance are not routinely available during clinical development of anti-HIV drugs.
Genotoxicity and Carcinogenicity Data on Anti-HIV Drugs
Nucleoside Reverse Transcriptase Inhibitor
For the NRTIs, the data in Table 1 show that 7 of 8 agents tested positive in 1 or more genotoxicity assays, with the exception of emtricitabine. Five of 8 were shown to be rodent carcinogens (abacavir, stavudine, tenofovir, zalcitabine, and zidovudine). All of these drugs tested positive in genotoxicity assays, either in the in vitro human lymphocyte or mouse lymphoma assays (note: zidovudine was positive in the Ames [bacterial reverse mutagenicity], in vivo micronucleus, and in vitro hamster ovary cells assays, as reported in the published literature). 7 Additionally, abacavir, stavudine, zalcitabine, and zidovudine also induced micronuclei in vivo. Didanosine was the only NRTI drug which tested positive in the Ames assay. These study data are consistent with the view that nucleoside analogues such as NRTIs, acting as chain terminators or through alteration of the nucleotide pool, are likely to be genotoxic. 21
Abacavir, stavudine, tenofovir, zalcitabine, and zidovudine were identified as rodent carcinogens. In certain cases, the tumors seemed to be mediated through hepatic enzyme induction mechanisms. Furthermore, all but tenofovir produced tumors that involved tissues at the nonhepatic and nonthyroid sites. While positive for genotoxicity, both didanosine and lamivudine tested negative in the carcinogenicity studies. This limited data would imply that genotoxicity data are not entirely predictive of tumorigenicity for these 2 cases and this subclass of drugs. Emtricitabine is the only NRTI that tested negative for both genotoxicity and carcinogenicity. All NRTIs that produced tumors in tissues other than liver or thyroid tested positive in the in vivo micronucleus assay.
Zidovudine, the first anti-HIV drug made available to the public during the early history of the AIDS epidemic period, received much attention during its development. In the study with this drug, unique malignant vaginal tumors were reported in both adult rats and mice. Because of concerns for tumorigenicity across multiple generations, 2 follow-up transplacental carcinogenicity studies were performed in pregnant mice. The results 6,7 again showed an increased tumor rate in the female reproductive tracts of the offspring. Because zidovudine is genotoxic (Table 1), the genotoxicity was predictive of carcinogenicity for this compound.
Nonnucleoside Reverse Transcriptase Inhibitor
Unlike NRTIs, all NNRTIs tested negative in genotoxicity assays, regardless of their DNA synthesis chain termination properties. The results may be related to the characteristics of NNRTI’s noncompetitive (vs NRTIs’ competitive) inhibition toward nucleoside phosphorylation. In contrast to the negative genotoxicity data, all NNRTIs were shown to be rodent carcinogens (efavirenz, etravirine, delavirdine, nevirapine, and rilpivirine), resulting in hepatocellular adenoma/carcinoma. These liver tumors may be related to the dose-limiting hepatotoxicity seen in chronic toxicity studies on NNRTIs. Hepatic enzyme induction mechanism as mentioned in the previous section may also play a role in tumor formation as all 5 agents in this class are liver enzyme inducers. Tumors were also noted in urinary bladder and lung for delavirdine and efavirenz, respectively. Overall, NNRTIs may be viewed here as nongenotoxic rodent carcinogens, as based on sponsor’s 2-year bioassays.
Protease Inhibitor
In regard to PIs, the data in Table 1 show that only 1 out of 10 agents tested positive in genotoxicity assays, whereas 9 of 10 were shown to be carcinogenic in animals (amprenavir, atazanavir, daurunavir, fosamprenavir, lopinavir, indinavir, nelfinavir, ritonavir, and tipranavir). Among all the PIs, atazanavir was the only agent that was genotoxic (ie, tested positive by chromosomal aberration assay) and that saquinavir was the only PI that was not a rodent carcinogen. For anti-HIV drugs from other classes, most tumors were detected in liver or thyroid gland, implying nongenotoxic carcinogenicity by mechanisms with questionable human relevance.
Integrase Inhibitor, Entry Blocker, and Fusion Inhibitor
Finally, newer drug classes such as HIV integrase inhibitor, entry blocker, and fusion inhibitor (ie, raltegravir, maraviroc, and enfuvirtide, respectively) all tested negative in genotoxicity assays. Raltegravir produced squamous cell carcinoma in the nose/pharynx of female rats (possibly due to local deposition and/or aspiration of drug during dosing), 6 whereas enfuvirtide was waived for carcinogenicity testing because it is a peptide (36-amino acid) with a short half-life. Currently, there are not enough cases to determine a class effect that could be examined for a trend for these 3 novel classes of anti-HIV drugs.
Summary
Of the 26 anti-HIV drugs approved for marketing, 8 tested positive in at least 1 genetic toxicology assay compared to 20 of 25 (excluding the waiver of enfuvirtide) that were shown to be rodent carcinogens. Thirteen of 20 drugs produced hepatocellular and/or thyroid tumors only. Although not demonstrated experimentally, these tumor profiles appear to be consistent with carcinogens mediated through nongenotoxic mechanisms. The genotoxicity data support this reasoning as only 2 out of these 13 agents (ie, tenofovir and atazanavir) demonstrated genotoxic potential. In both cases, the drugs were shown positive in only 1 of genotoxic assays conducted (ie, in vitro mammalian cell assays). It is also interesting to note that among the positive genotoxicity cases discussed above, only didanosine was positive in the Ames test, an assay with high specificity relative to its comparably low sensitivity. 22 In viewing the fact that didanosine was not carcinogenic, the Ames test did not accurately predict the bioassay outcome.
Approximately 70% of the rodent carcinogens described in this article were negative in the genetic toxicology assays. The low rate of detection by genotoxicity testing is consistent with previously published reports 8 and implicates the predominance of nongenotoxic carcinogens identified in standard 2-year bioassays with these drugs. Alternative speculation that can partly provide an explanation on this phenomenon would be consideration of the early termination of drug development by some sponsors for agents that are clearly genotoxic (eg, Ames positive). In short, although there are many factors affecting a correlation between genotoxicity and carcinogenicity study results, 23 our analysis of the data for this therapeutic group of structural similarities with shared mechanisms of action has provided useful insights into issues involving in these 2 important areas of toxicological investigations.
Conclusion
Most antiretrovirals for HIV infection are made available to the public through expedited development processes that include delay of initiating carcinogenicity studies and submission of the final carcinogenicity reports after accelerated approval is granted. We feel that this practice does not compromise safety standards of FDA CDER’s drug evaluation and the approval process but does facilitate the availability of these agents to meet urgent medical requirements. Animal toxicity study results obtained after full NDA approval showed that (1) there were trends of class effects, regarding NRTIs, NNRTIs, and PIs, and (2) certain agents identified as rodent carcinogens tested negative in all genotoxicity assays and/or exhibited tumor profiles consistent with nongenotoxic modes of action. The percentage of potentially nongenotoxic carcinogens from anti-HIV drug category appears to be in line with literature reports for drugs in general. The integrated analysis of genotoxicity and carcinogenicity studies provides useful insights in correlating findings in this unique category of therapeutic agents.
Footnotes
Acknowledgment
This article is not an official FDA guidance or policy statement. No official endorsement by the FDA is intended. The authors wish to thank Drs William Taylor, Debra Birnkrant, Steven Gitterman, and Abby Jacobs for their valuable inputs and summer intern Mr Benjamin Col for his document research.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.