
Abstract
There are several recent examples where clinically significant, safety-related, drug effects on hemodynamics or cardiac function were not apparent until large clinical trials were completed or the drugs entered the consumer market. Such late-stage safety issues can have significant impact on patient health and consumer confidence, as well as ramifications for the regulatory, pharmaceutical, and financial communities. This manuscript provides recommendations that evolved from a 2009 HESI workshop on the need for improved translation of nonclinical cardiovascular effects to the clinical arena. The authors conclude that expanded and improved efforts to perform sensitive yet specific evaluations of functional cardiovascular parameters in nonclinical studies will allow pharmaceutical companies to identify suspect drugs early in the discovery and development process while allowing promising drugs to proceed into clinical development.
Introduction
Background
Preclinical evaluation of the cardiovascular effects of human drug candidates has evolved significantly over the last 20 years. Before the 1990s, pharmaceutical companies typically screened drug candidates in anesthetized dogs, often using the same laboratory group engaged in cardiovascular efficacy research. In 1991, the first regulatory guidance on safety pharmacology was developed by the Japanese Ministry of Health and Welfare, 1 recommending that test substances in drug studies be assessed for an overall profile of pharmacological effects. 2
In the late 1990s and early 2000s, guidelines were expanded to cover issues around QT interval prolongation. Recommendations included the in vivo assessment of blood pressure, heart rate and electrocardiogram (ECG), and the in vitro hERG voltage clamp assay. Adding electrophysiological endpoints to nonclinical safety testing has increased the quantity and quality of cardiovascular information available and reduced the risk of drug-induced QT prolongation. However, with comparatively less regulatory focus on the measurement of hemodynamic parameters, their potential impact on the drug development process has not been fully recognized. Furthermore, the assessment of potential cardiovascular effects after multiple days of drug dosing has not been the focus of the guidelines and has therefore not received much attention. This is despite the obvious potential for delivering valuable information on drug-induced effects that might emerge with prolonged treatment. The potential benefit of regularly testing for cardiovascular drug-induced effects following multiple day treatment is therefore unknown.
There are several recent examples where clinically significant drug effects on hemodynamics or cardiac function were not apparent until large clinical trials were completed or the drugs entered the consumer market. 3,4 Such late-stage observations can have significant impact on patient health and consumer confidence, as well as ramifications for the regulatory, pharmaceutical, and financial communities. An improved understanding of how preclinical cardiovascular effects translate to the clinical arena is clearly needed. Would an enhanced understanding of preclinical cardiovascular findings lead to superior decision making in drug development? Would certain nonclinical study designs better promote more timely and appropriate go/no go decisions in drug development?
These types of questions spawned a “think-tank” under the auspices of the nonprofit International Life Sciences Institute—Health and Environmental Sciences Institute (ILSI-HESI). A consortium of industrial, academic, and government scientists met in Washington, DC (USA) on June 2-4, 2009, to develop integrated approaches for the evaluation of cardiovascular structure and function in drug development. A list of organizations represented at this meeting is included in Table 1 . Several HESI working groups, including ours, were formed at this meeting. We (the authors) accepted the task of representing our working group in the exploration of functional cardiovascular safety assessment in drug development. This manuscript is the product of our working group’s subsequent and ongoing discussion on this important topic.
Organizations Represented at the June 2009 HESI Meeting “Current Practice in Structural and Functional Assessment of Cardiovascular Toxicity: Issues and Opportunities”

Overview
Specific topics addressed herein include: peripheral cardiovascular hemodynamics (particularly arterial blood pressure), cardiac contractility (inotropy), functional cardiovascular endpoints (ECG, blood pressure, and contractility) in nonrodent toxicology studies, and the role of assay sensitivity in both nonclinical acute safety pharmacology and repeat-dose toxicology studies. The recommendations and perspectives set forth represent input from the industrial, academic, and regulatory scientists who comprise our HESI working group.
Cardiovascular Testing Requirements
An extensive review of the evolution and content of cardiovascular testing requirements is available elsewhere 5 and will not be repeated in detail here. Concerns over delayed ventricular repolarization associated with noncardiovascular drugs led to the first guidelines regarding QT interval prolongation. 2,6 Similar concerns prompted the International Conference on Harmonization to publish the ICH S7A 7 and S7B guidelines, 8 which required the in vivo assessment of blood pressure, heart rate, and ECG, as well as the in vitro hERG voltage clamp assay. 5 Other functional cardiovascular parameters, such as cardiac output, ventricular contractility, and vascular resistance, were classified as followup studies.
While avoiding specific recommendations about statistical data analysis or study design, ICH S7A encouraged investigators to meet the sensitivity requirements necessary to detect a biologically important change. The guidelines advised investigators to utilize enough animals (or isolated biological preparations) to produce meaningful scientific data in order to clearly demonstrate or rule out a significant effect of the test substance. Finally, ICH S7A advised that sample size be guided by the magnitude of the biological effect of concern in humans. 7
Study Designs
It is useful to understand the various study designs, their limitations, and their impact on study sensitivity and specificity to fully appreciate the issues and opportunities facing cardiovascular safety assessment scientists today. The most popular formats for nonclinical in vivo pharmacological studies are Latin square, parallel, and escalating dose designs. The Latin square crossover is commonly used in nonclinical cardiovascular safety pharmacology studies testing small molecule compounds in conscious animals to fulfill ICH S7A and S7B core battery requirements. Originally developed with one endpoint for use in agriculture, the Latin square can be adapted to time series data within each treatment day. By embedding a repeated measures analysis within each cell, the magnitude, onset, and duration of an effect can be evaluated.
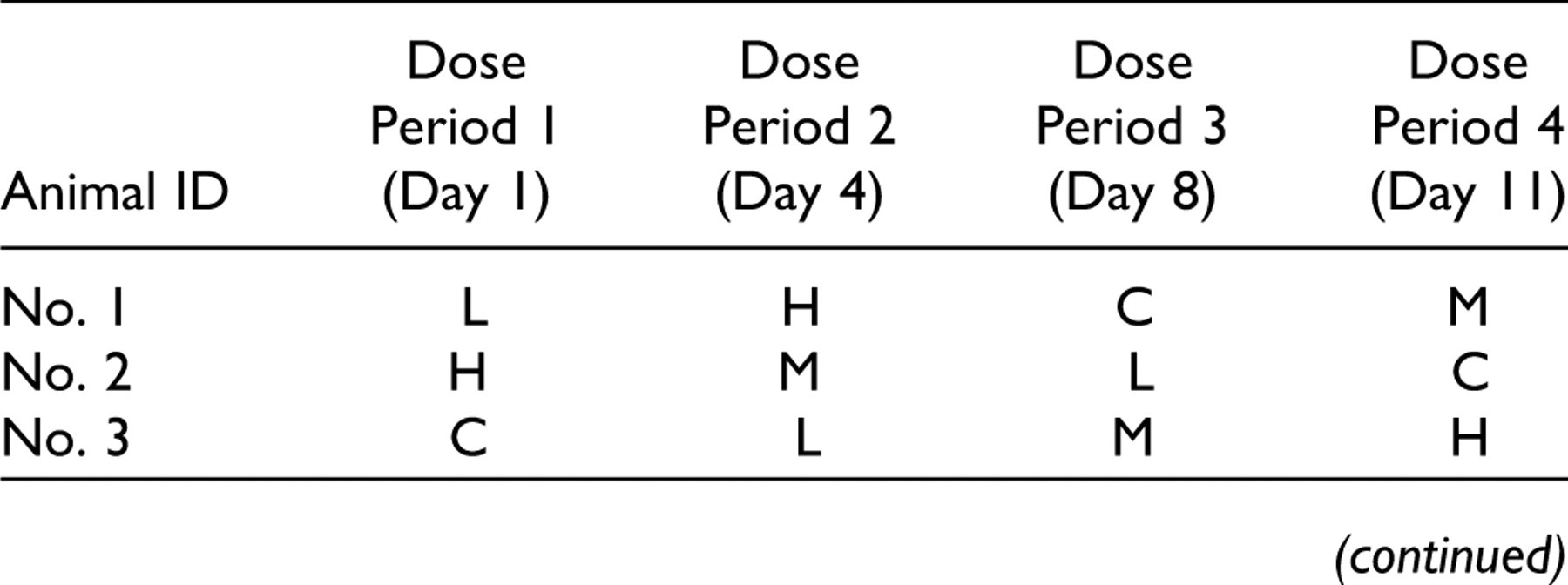
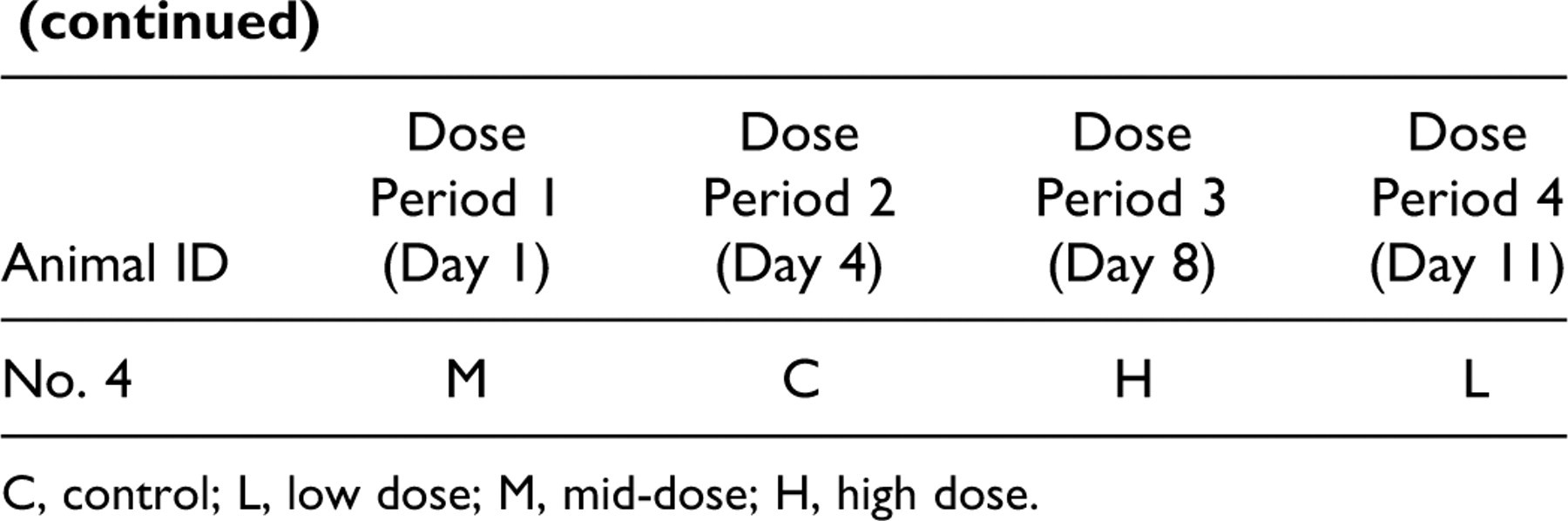
C, control; L, low dose; M, mid-dose; H, high dose.
There are many advantages to a Latin square design. Since each animal provides information for each treatment group including the vehicle treatment (ie, control), the number of animals required to detect the change of a specified magnitude is reduced. By removing interanimal variability from the mean square error term in the statistical analysis, statistical power is substantially increased, with fewer animals, compared to parallel group designs.
Certain assumptions must be met before using a Latin square. There can be no treatment carryover effects that would influence response to a subsequent treatment with the same drug or with the vehicle. Carryover effects could be due to slow elimination of the compound, the presence of an active metabolite, or to an alteration in treatment responsiveness for other reasons (eg, tissue retention, receptor desensitization). Drugs with extremely slow elimination (t ½ > 24 hours) and those that permanently alter an animal’s physiology (possibly through a toxic effect) are generally not suitable candidates for this design. Although one could consider extending the period between treatment days for compounds with long half-lives, this approach is impractical and risky due to the possibility of animal or instrumentation failures before study completion. Users of a Latin square design must also consider that few degrees of freedom remain in the error term. Missing data from the loss of an animal or a physiological signal can be detrimental to assay sensitivity.
The parallel design is commonly used in toxicology studies and is sometimes necessary for safety pharmacology telemetry studies. In this design, a group of animals is permanently assigned to each treatment group, including the vehicle control. The study can be either a single dose (acute) or a repeat-dose study. Data accumulation and analysis can continue for as long as desired after treatment cessation (referred to as the recovery period). The analysis of variance is conducted across groups, thus including interanimal variability in the error term. Parallel studies require more animals and are usually less sensitive than Latin square studies. However, compounds with very slow elimination (eg, many antibodies) must be studied via parallel design.
With an escalating dose design, all animals receive all doses in the same order of increasing magnitude, from vehicle to the highest dose. This design allows for dose modification as the study evolves, making it preferable when there are minimal preliminary pharmacokinetic, pharmacological, and toxicological data available. One concern with this design is the possibility of initial doses causing desensitization of the pharmacological effect, leading to the inaccurate conclusion that a single high dose of the drug has minimal cardiovascular effects. This observation may instead be the result of a cumulative pharmacodynamic effect associated with repeat dosing. Since the treatment day is confounded by prior exposure to the test substance, it is impossible to separate true dose effects from the number of exposures without repeating the highest administered dose in a naive group of animals.
Hemodynamics
Background
During the June 2009 HESI workshop, multiple speakers cited a 2002 meta-analysis of blood pressure data from 61 prospective studies in adult human participants. 10 Its authors concluded that small changes in systemic arterial blood pressure are strongly and directly related to cardiovascular disease mortality, even in normotensive adults. Thus, we advocate the routine assessment of arterial blood pressure and heart rate in nonclinical studies due to the potential clinical significance of drug-induced effects on these parameters in patient populations.
Several marketed drugs are known to increase blood pressure. Some agents (eg, norepinephrine reuptake inhibitors) do so acutely, while others (eg, COX2 inhibitors) have this effect over time. 11,12 It is generally assumed that drugs which acutely raise blood pressure in a clinical setting generally demonstrate the same effect in nonclinical studies, as long as the site of pharmacodynamic action is conserved across species. Conversely, it is assumed that drugs which manifest this effect by indirect mechanisms (eg, fluid retention) with chronic administration will not be detected in an acute safety pharmacology study. It is also doubtful that repeat-dose toxicology studies have the sensitivity to detect these small blood pressure changes due to the lack of methodological precision of blood pressure measurement and the type of experimental design generally used. However, these are assumptions. To date, an analysis of compounds with known clinical effects on blood pressure has not been systematically performed in preclinical models, representing an excellent research opportunity.
Current Approaches
One consideration in performing nonclinical studies to assess drug effects on hemodynamics is whether to use anesthetized or conscious animals. Drug-induced cardiovascular effects can appear to be quite different in the presence versus absence of fully functional homeostatic mechanisms. If a negative inotropic drug directly reduces cardiac output in a conscious animal, this effect may be compensated for through sympathetic activation, including peripheral vasoconstriction and/or increased heart rate, resulting in no significant change in arterial blood pressure. It would appear, based only on the arterial pressure signal, that the drug simply increased heart rate in the conscious dog and caused a drop in blood pressure in the anesthetized dog. One might then make the potentially false assumption that the drug was working through different mechanisms in the different models. This example underscores the important point that initial studies measuring blood pressure and heart rate can only be utilized to conclude that a drug is having an effect on the cardiovascular system. Study data cannot be utilized to make conclusions on the mechanism of the effect unless other parameters (eg, cardiac output and inotropy) are also measured.
Anesthetized animal models (primarily canine) have been used for decades to evaluate the functional effects of drugs on the cardiovascular system. When evaluating potentially toxic parenteral drugs (eg, oncological agents), animals must be anesthetized to eliminate cardiovascular responses secondary to other drug side effects. Using anesthetized animals offers some theoretical advantages over using conscious instrumented animals. Anesthetized animals do not react to environmental stimuli, thus eliminating one source of variability. If a drug is a direct vasoconstrictor or direct vasodilator, anesthetized animals may be more sensitive due to the absence of response to external stimuli. 13 Another advantage of anesthesia is the ability to closely control and measure plasma concentration with intravenous infusions. Anesthesia provides a greater opportunity to measure a comprehensive array of parameters that may not be easily measured in conscious animals (eg, indices of cardiac contractile function, cardiac output, pulmonary arterial pressure, and systemic vascular resistance).
However, there are disadvantages to using anesthetized animals. Indirect cardiovascular effects due to neurohumoral responses that would be present in a conscious telemetry model may be missed. 14 Furthermore, cardiovascular responses in the absence of normal hemodynamic reflexes do not emulate the physiological experience of the clinical trial participant or patient.
Conscious animal models with fully functioning neurohumoral homeostatic mechanisms are more likely to mimic effects seen clinically. Since 1965, the technology to remotely monitor blood pressure and pulse rate by telemetry in conscious animals has been available. 15,16 The use of instrumented conscious animals shifted from academic laboratories to human pharmaceutical safety assessment in the 1990s. 17,18
Chronically instrumented (conscious) animals offer some advantages for arterial pressure measurement. Implantable pressure transducers with telemetry capability provide a sensitive and robust system for monitoring arterial blood pressure in undisturbed animals over a prolonged period. These devices reliably reconstruct arterial pressure signals. Compared to ECG and left ventricular pressure, the analysis of a blood pressure signal is straight forward. Newer devices have improved stability with respect to long-term drift. Software algorithms can automatically determine systolic and diastolic pressures and calculate the difference (arterial pulse pressure), as well as mean arterial pressure and heart rate.
One limitation of chronically instrumented animal models for arterial pressure measurement is that tissue reaction following surgical implantation of the transducer may confound toxicology studies. Because toxicological endpoints include gross morphologic changes and histopathology, effects due to chronic instrumentation may be pathologically indistinguishable from those due to the test substance. To avoid this limitation, attempts have historically been made to collect arterial blood pressure noninvasively in repeat-dose toxicology studies.
Mechanistic Interpretation
With the high bandwidth of modern implantable devices, the two basic components of the blood pressure signal—tonic and phasic—can be measured accurately. The phasic component contains information on frequency (pulse rate), systolic and diastolic arterial pressure, and pulse pressure (ie, systolic minus diastolic pressure). The tonic aspect of the signal is mean arterial pressure. Obtained by applying Ohm’s law to fluid mechanics, mean arterial pressure is the mathematical product of cardiac output (flow) and total peripheral vascular resistance. Thus, a drop in mean arterial pressure could result from either a decrease in cardiac output and/or a decrease in vascular resistance (vasodilation). The converse is true for an increase in pressure.
Without measuring blood flow (ie, cardiac output), it may be unclear which mechanism is responsible for a change in mean arterial pressure. In addition, it is possible to have decreased cardiac output due to cardiac depression in concert with increased vascular resistance due to vasoconstriction (or vice versa) and have no detectable change in blood pressure at all. Thus, evaluation of cardiovascular drug effects using only arterial pressure can be misleading. A significant change in arterial pressure clearly indicates that the cardiovascular system has been perturbed in some way. However, unchanged blood pressure does not rule out a significant effect on cardiac output and vascular resistance (ie, false negatives).
An example of this scenario is illustrated in Figure 1 . In this study, the effects of compound A on blood pressure, cardiac output, and vascular resistance in an anesthetized dog were investigated during three 30-minute infusions. During the 3rd infusion, cardiac output decreased while vascular resistance increased (vasoconstriction), resulting in unchanged blood pressure. If only blood pressure and heart rate had been measured, one might have concluded that compound A had no effect on the cardiovascular system. This demonstrates a limitation of the cardiovascular safety pharmacology assessment as defined in the ICH S7A core battery. 7
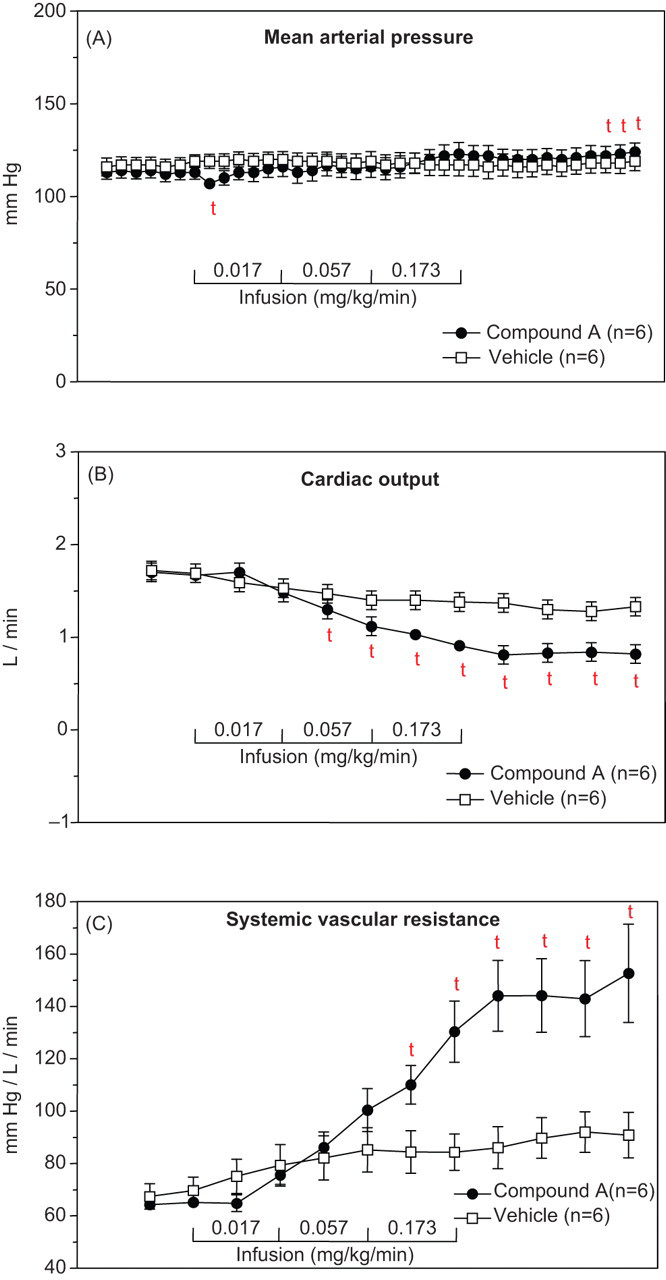
After three 30-minute intravenous infusions in pentobarbital anesthetized dogs, Compound A had no clear drug-related effects on mean arterial blood pressure (A) yet caused significant decreases in cardiac output (B) and increases in vascular resistance (C). Cardiac output was measured by thermodilution and aortic blood pressure was measured in the right carotid artery with a micromanometer catheter. Systemic vascular resistance was calculated as (MAP−central venous)/cardiac output. For each parameter at each posttreatment time point, 2-sample, 2-sided, t tests were used to compare the change from baseline for compound A-treated dogs and vehicle-treated dogs. Mean ± SE; n = 6. t = P < .05 versus vehicle.
Measuring cardiac output in conscious animals with a fully implantable telemetry system is not currently possible, but available technology can measure cardiac output in large animals wearing a backpack containing an external telemetry system. 19 Furthermore, investigators can indirectly assess potential effects on cardiac output in unanesthetized animals by implanting a pressure catheter in the left ventricle to obtain indices of left ventricular function. The advantages and disadvantages of left ventricular pressure measurements will be discussed later in this article.
Besides cardiac output and vascular resistance, the phasic component of the arterial pressure signal is affected by blood vessel compliance (capacitance) and the dynamics of moving blood (kinetic energy and waves). Arterial conductance vessels convert the pulsatile ejection of blood from the left ventricle into continuous flow through the microcirculation, accomplishing this feat by virtue of the elastic properties of the aorta and other large conductance vessels (Windkessel function). About half the blood ejected from the left ventricle may be retained in the dilating arterial conductance vessels during systole and flow through the peripheral circulation during diastole. 20
Although pulse pressure is a sensitive indicator of the elastance of the arterial distribution network, changes in elastance are rarely associated with acute pharmacology effects. Assessing blood vessel elastance offers little value to safety pharmacology studies but can serve as an indicator of chronic disease or aging. In repeat-dose toxicology studies, pulse pressure could potentially be utilized to measure chronic drug effects on vascular elastance. If a given drug does not impact elastance, pulse pressure can provide useful indirect information on cardiac stroke volume changes when no direct measure of cardiac function is available.
The final phasic parameter in an arterial pressure signal is pulse rate, which must be distinguished from heart rate. Although uncommon, it is possible for the 2 parameters to differ, especially in the presence of cardiac arrhythmias. In pulsus alternans, where cardiac contraction occurs without aortic valve opening 50% of the time, interpretation of pulse rate may underestimate true heart rate by one half.
Discussion
Implantable pressure transducers with telemetry capability provide a robust assessment of blood pressure changes with a fairly low bandwidth requirement. These devices have changed little since their introduction in the 1980s. One serious limitation of a commonly used system is its inability to simultaneously study multiple animals in the same cage, due to cross-talk of the transmitter-receiver pairs. In recent years, investigators have devised effective solutions to this problem, including the use of multiple discrete frequencies 19 or repeater collars with discrete frequencies. 21 Recent technological advances have occurred in noninvasive and minimally invasive blood pressure measurement systems often used in repeat-dose toxicology studies. These technologies are discussed later in this article.
Chronic implanted devices are limited by their inability to measure blood flow, which would allow the assessment of drug effects on cardiac output and vascular resistance. Ultrasound technology (ie, Doppler and differential transit time) is currently the only readily available means of blood flow measurement. 16,17 Because ultrasound is extremely energy intensive, these devices require cumbersome, external power sources placed in jackets worn by the animals. Externalized wires, prone to migrating tract infections from skin bacteria, limit the lifespan of these models. Since the ICH S7A core battery does not require hemodynamic parameters beyond blood pressure and heart rate, drug companies, contract research organizations, and equipment providers have had little motivation to invest resources into improving this technology.
Recommendations
Cross-talk of transmitter-receiver pairs in many of the devices used today makes studying multiple animals in proximity with implanted pressure transducers difficult. While creative means of overcoming this problem exist, there is a need for new approaches to minimize or eliminate cross-talk. Considering the recent advances in other aspects of digital telemetry, it seems that more sophisticated technology could be applied to surmount this problem. Another opportunity for technological advancement lies in developing chronic fully implantable devices that can reliably measure cardiac output (blood flow). Such devices could garner valuable information about cardiovascular drug effects.
Clearly, the blood pressure effects of drugs are vitally important in the clinical setting. Hence, we propose that all drug candidates undergo a more thorough preclinical evaluation for potential effects on blood pressure than is often done currently. This nonclinical assessment must be sensitive enough to yield practical information that can translate to clinical medicine. Further research is warranted to better understand how blood pressure changes in experimental animals translate to human therapeutic drug usage. Can a drug associated with acute blood pressure changes in laboratory animals always be expected to produce similar changes in humans? If not, could problematic blood pressure changes develop over time, with chronic administration?
Cardiac Contractility (Inotropy)
Background
The body’s varying metabolic needs are met through dynamic changes in cardiac output, which adjust oxygen delivery as needed. This is accomplished through a close relationship between local vasodilatory mechanisms (which control the delivery of oxygenated blood to tissues) and tight regulation of myocardial pump function (which helps maintain tissue perfusion pressures). Left ventricular pump function is affected by neurohumoral input from the autonomic nervous system. This neurohumoral input controls both heart rate and intrinsic myocardial contractile function, referred to as “contractility” or “inotropic state.” Physiologically, this occurs via activation of cardiac β-adrenergic receptors by sympathetic nervous inputs and/or circulating catecholamines.
Cardiac contractility, defined as the ability of cardiac muscle to do work at constant end-diastolic fiber length, 22 is an intrinsic property of the myocardium. With increased contractility, cardiac muscle performs more work at the same diastolic fiber length. This principle is demonstrated in vivo as enhanced pump function (ie, ejection fraction or stroke volume) at a given end-diastolic volume. For a rigorous discussion of the physiology of contractility, the reader is referred to the original work of Suga et al. 23
A positive inotropic drug increases, whereas a negative inotropic drug decreases, myocardial contractility. The most familiar positive inotropic agents are the primary neurotransmitters of the sympathetic nervous system, epinephrine, and norepinephrine. Positive inotropes increase the work performed by the heart, which leads to higher cardiac output and increased myocardial oxygen consumption. This scenario can be detrimental in disease states where restricted coronary perfusion results in myocardial ischemia.
Decreased myocardial contractility results in reduced cardiac work capacity, which presents clinically as acute or chronic heart failure. Some pathophysiological causes of heart failure include hypertension, coronary artery disease, valvular dysfunction, and drug toxicity. 24 Since diminished cardiovascular reserve is not usually apparent in sedentary individuals, impaired myocardial contractility may be clinically asymptomatic, becoming evident only when increased demand is applied to the cardiopulmonary system.
Nonclinical investigators should be vigilant about discovering possible drug effects (both positive and negative) on myocardial contractility that may have clinical implications in humans. For example, the antifungal itraconazole has been shown in animal studies to be a negative inotrope. 24 This effect also occurs in humans, based on a summary of 58 clinical cases of congestive heart failure associated with itraconazole use. 25
Although the potential for drug-induced changes in myocardial contractility is recognized, its actual measurement (both preclinically and clinically) is fraught with problems. Research indices used to assess myocardial contractility are simultaneously affected by changes in preload (ie, left ventricular end-diastolic pressure [LVEDP]), afterload (ie, aortic resistance), and heart rate. Thus, a drug-induced change in any of these parameters can indirectly affect an index of contractility without actually changing intrinsic myocardial contractile function. Additionally, drug effects on lusitropy (ie, diastolic relaxation) may be significant. Both inotropic and lusitropic effects can be measured in the experimental models described below.
Current Approaches
Because contractility is an intrinsic property of the myocardium, drug-induced effects on contractility can be demonstrated using in vitro models of myocardial function, commonly utilized in cardiovascular drug safety and efficacy pharmacology studies. Perfused with an artificial physiologic buffer, in vitro models can assess direct drug effects on the myocardium, independent of secondary effects from cardiac sympathetic input. Isolated from the influences of an intact biologic system, in vitro models are not subject to neurohumoral homeostatic mechanisms. The most rigorous in vitro approach for assessing myocardial contractility utilizes isolated, perfused (ie, Langendorff) hearts. 23 The use of isolated myocardial contractile tissue (eg, papillary muscle or cardiac myocytes) allows investigators to examine both contractile and electrophysiological properties of cardiac tissue using one preparation. In contrast, noncontractile Purkinje fibers (used for electrophysiological assessments) cannot also be used to evaluate inotropic effects. While in vitro findings can help prioritize among lead drug candidates, they always require in vivo confirmation.
A high-fidelity pressure transducer placed in the left ventricle is the most common in vivo approach for evaluating drug-induced changes in cardiac contractility. 17,26 Other techniques include noninvasive echocardiography (EC) and ECG calculation of the QA interval, the period between Q wave onset and aortic pressure signal upstroke. 27 Although QA interval roughly approximates the duration of excitation-contraction coupling plus isovolumic systole, it is affected by several factors besides myocardial contractility. Its use should be limited to early compound-screening studies followed by more direct measures in definitive safety studies. 28
The peak positive value of the first derivative of left ventricular pressure (LV dP/dt max) is a widely used index of cardiac contractility, which relies upon measurements from a left ventricular pressure transducer. This parameter provides an in vivo index that closely approximates in vitro unloaded muscle shortening velocity. LV dP/dt max represents isometric contraction of cardiac muscle fibers during the isovolumic phase of systole. This rise in muscle tension causes increased chamber pressure. If preload, afterload, and heart rate remain constant, LV dP/dt max provides a sensitive index of changes in ventricular contractility.
Because left ventricular preload cannot truly be measured without including cardiac chamber dimensions, LVEDP provides a very good approximation, assuming diastolic compliance is constant. LV dP/dt max data should always be interpreted in context with concurrent LVEDP values to rule out an indirect effect of drug-induced preload changes. Changes in LVEDP can also independently support the observation of a strong negative inotropic effect. Impaired systolic function leads to reduced stroke volume, resulting in higher residual left ventricular diastolic blood volume and increased LVEDP. Decreased LV dP/dt max in conjunction with increased LVEDP is a strong indicator of drug-induced heart failure.
Measuring instantaneous dP/dt at a predetermined pressure (eg, LV dP/dt at 40 mm Hg) rather than at its maximum value (ie, LV dP/dt max) is an alternative index of contractility. Most data analysis software systems calculate these parameters automatically. While this approach may minimize sensitivity to variations in preload and afterload, LV dP/dt max remains the most commonly used index of contractility.
Besides left ventricular preload and afterload, heart rate also substantially modulates LV dP/dt max. This was originally observed in an isolated myocardial preparation where contractile force was found to be directly related to tissue stimulation rate (ie, the force-frequency relationship). This in vitro characteristic translates to the in vivo setting where a direct, nonlinear relationship exists between contractility (ie, LV dP/dt max) and heart rate. This relationship is more important at higher heart rates or under sympathetic stimulation. 29 The force-frequency relationship also differs somewhat between species (Figure 2 ). Because LV dP/dt max is directly related to heart rate, drug-induced effects on heart rate will have implications on contractility. Because heart rate effects on LV dP/dt max are analogous to heart rate effects on the QT interval, they can be addressed via similar data analysis and correction factors.
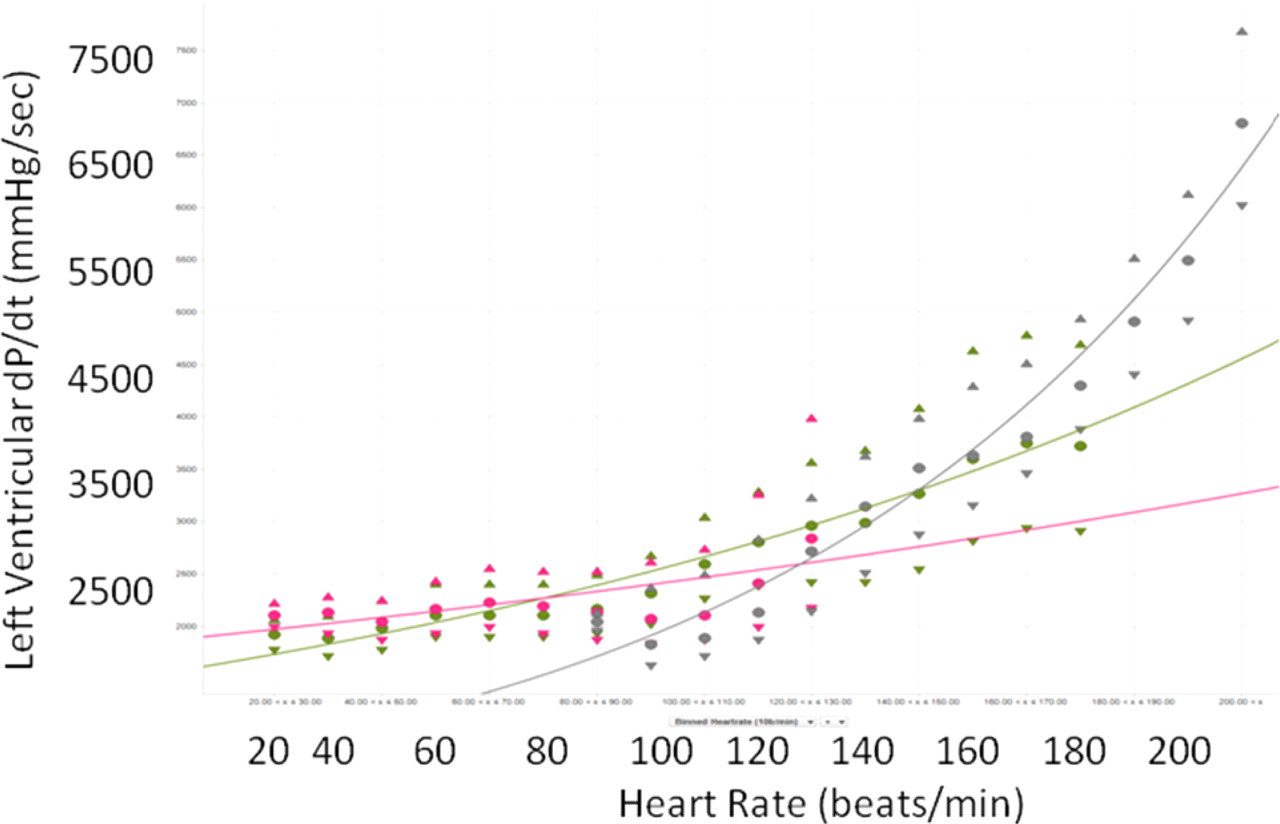
The relationship between heart rate (beats/min, X-axis) and LV dP/dt (mm Hg/s, Y-axis) for the conscious dog (blue), mini-pig (red), and cynomulgus monkey (green). Data were collected over 24 hours from animals chronically instrumented with a micromanometer in the left ventricle without any treatment. Thus, the heart rates reflect the normal range seen under control conditions with normal activity. The data were fit using a second-order polynomial equation (Graphic provided courtesy of Michael Markert, Boehringer Ingelheim Pharma GmbH & Co KG).
Regarding left ventricular pressure data, 2 critical technical factors to consider are bandwidth (ie, frequency response) and sampling rate of the transducer and data acquisition systems. This is especially important if increases in LV dP/dt max are expected. Upstroke of the LV pressure signal during systole contains its highest frequency component when dP/dt max occurs. If the data acquisition rate is too slow or the system bandwidth is too low (upper limit < ~100 Hz), dP/dt max will be affected. When dP/dt max values are expected to exceed ×7000 mm Hg/s for any reason (eg, positive inotropic drug and exercise), the sampling rate should be at least 500/s. 30
Mechanistic Interpretation
Although inotropic state is an important physiological parameter, it is rarely assessed in either safety pharmacology or toxicology studies. Due to its clinical relevance, we contend that cardiac contractility should be more frequently measured in nonclinical studies. 31 There are numerous examples of heart failure cases induced by noncardiac drugs. Some drugs intended for noncardiac pharmacological targets affect myocardial contractility. 24 Whenever a pharmacological mechanism suggests potential cardiac effects, the preclinical assessment of drug effects on myocardial contractility is clearly indicated.
However, consensus is lacking on whether preclinical evaluation of drug effects on cardiac inotropy should be routine or conducted only in cases where there is obvious cause for concern. Data assessing a drug candidate’s potential effects on cardiac contractility are not routinely required to be compliant with the ICH guidelines.
Little information exists on how inotropic drug effects in animals translate to human therapeutic drug usage. One well-known exception is the antifungal drug, itraconazole, a proven negative inotrope in nonclinical studies associated with numerous clinical cases of heart failure. 24,25 Recognizing the potential for adverse cardiac effects early in the drug development process would enable pharmaceutical companies to focus attention and resources on identifying alternative agents without such undesirable effects.
One reason that myocardial contractility is not routinely assessed in the preclinical studies is the uncertainty surrounding the translation of preclinical findings to the clinic. Since the invasive methods utilized in cardiovascular safety pharmacology studies are more sensitive than the noninvasive techniques available to clinicians, it is difficult to demonstrate that an effect does not also occur in humans. These sensitivity differences contribute to the lack of consensus regarding interpretation of nonclinical measures of myocardial contractility. Whether these changes are clinically meaningful relies on the precision of the methodologies available to the clinical pharmacologist. Nonclinical data can, however, alert clinicians to a possible concern. Available clinical techniques are generally adequate to rule out significant effects in both healthy volunteers and patients, but this requires additional monitoring and cost.
Discussion
Chronically instrumented animal models with high-fidelity left ventricular pressure transducers have been used in drug safety assessment for over 20 years. 17,32 Telemetry systems have recently been improved to allow continuous monitoring of cardiac contractility in numerous conscious, unrestrained animals.
The direct application of epicardial ECG electrodes to research animals has become increasingly popular. 33 Since the heart is already surgically exposed for electrode placement, the additional burden of inserting a pressure transducer (or catheter) into the left ventricle is relatively small. Modern data acquisition and analysis systems use simple algorithms that allow real-time analysis of cardiac contractility indices. Thus, newer technology and surgical approaches have reduced the difficulty of the collection and analysis of left ventricular function in safety pharmacology telemetry studies. In fact, the difficulty of these measures is now comparable to measuring blood pressure or performing an ECG.
One possible drawback of routine left ventricular pressure measurement is an increased potential for spontaneous ventricular arrhythmias due to local irritation at the transducer insertion site in the left ventricular apex. This recent criticism arises from a preliminary report citing an increased rate of spontaneous ventricular arrhythmias in dogs and nonhuman primates instrumented with left ventricular pressure devices. However, this differs from the experience of the authors during our combined decades of laboratory experience using chronically implanted left ventricular pressure transducers. Following carefully performed surgeries and an adequate postoperative recovery period, we have not found an increased incidence of spontaneous ventricular arrhythmias. Furthermore, despite the widespread use of instrumented, conscious animal models in physiology and pharmacology research since the mid-1960s, we are unaware of any problems with spontaneous ventricular arrhythmias being cited in the literature, until the aforementioned preliminary report.
Using a balanced Latin square design, there is little possibility to confound surgery-related spontaneous arrhythmias with drug effects since all animals are treated both as a control group and with the drug. Agents known to increase sensitivity to spontaneous ventricular arrhythmias (eg, catecholamines and some anesthetics) will cause an increased rate of spontaneous arrhythmias in treated groups. However, this is a pharmacological consequence that is related to human safety. As with surgically attached epicardial ECG electrodes, careful surgical technique should preclude an increased incidence of spontaneous ventricular arrhythmias in animals with implanted left ventricular pressure transducers. These devices have the capability to provide cardiac contractility information in safety pharmacology telemetry studies with very little increased cost or effort. 17,18 Whether this approach is justified in all cases, however, remains debatable.
Although this section has been limited to the evaluation of cardiac contractility in acute studies with chronically instrumented animals, the described methodology may also be applied to subchronic or chronic investigations. Measurement of left ventricular pressure and LV dP/dt max can also be incorporated into models using anesthetized animals in the acute setting. Despite possible influences from anesthetic and analgesic agents used, these models are sensitive to the acute administration of inotropic agents and may help detect effects on myocardial contractility. Alternatively, noninvasive methodology is available for chronic applications.
Echocardiography (EC) is a noninvasive method that provides valuable information about cardiac morphology and contractile function. Hanton et al provided an excellent review of EC in preclinical drug toxicology and safety pharmacology. 34 There are 3 modes of EC: (1) 2-dimensional (2-D), (2) time-motion (M-mode), and (3) pulsed Doppler. They offer complementary data, resulting in a comprehensive assessment of the cardiac structures, their movements during the cardiac cycle and blood flow. 2-D EC allows for measurement of the cardiac cavities in systole and diastole. Based on left ventricular systolic and diastolic volumes, standard equations are applied to determine stroke volume and ejection fraction, revealing much about cardiac contractility. M-mode EC links the changes in cardiac morphology over time to the cardiac cycle. Pulsed Doppler EC measures blood flow velocity in vessels and cardiac cavities, highlighting flow variations during the cardiac cycle. This reveals blood flow patterns, pressure gradients or changes, vascular resistance, and cardiac function during both systole and diastole.
Widely used in humans, EC also has numerous applications in laboratory animals, especially dogs. Echocardiography has been used to evaluate cardiac function in canine models of heart disease (eg, valvulopathy, congestive heart failure, cardiac ischemia, and cardiomyopathy) and to assess the drug effects in these groups. It is considered an important method of investigation in preclinical toxicology using canine models. 34 Echocardiography has been used in monkeys to evaluate the cardiac sequelae of several diseases and to assess the effects of cardiovascular agents. In rats and mice, EC has been used to evaluate experimental models of heart disease and the effects of drug interventions. Although well-established in dogs, monkeys, rats, and mice, EC has been less frequently used in other laboratory animal species.
Recommendations
The measurement of cardiac contractility, in both acute animal models and chronically instrumented animals, is technically robust with the use of in-dwelling micromanometers for the measurement of left ventricular pressure and then deriving the parameter LV dP/dt. Digital data acquisition and analysis systems can therefore provide accurate, real-time information on cardiac contractile function. The further miniaturization of implanted manometers is desirable and the selection of a system allowing simultaneous monitoring of animals housed in groups is strongly recommended. While the systems currently available work well, there is room for improvement.
The clinical relevance of contractility changes detected in nonclinical studies remains uncertain. Some pharmaceutical companies routinely screen drug candidates for the effects on cardiac contractility, discarding any candidate with such effects in favor of an analog having lesser effects. Because little data exist regarding translation of these effects to clinical use, further studies of compounds with known clinical impact on myocardial contractility are needed to establish a relationship between preclinical results and clinical relevance.
Presently, there is no consensus on the role of routine nonclinical evaluation of cardiac contractility. However, the authors believe drug effects on the inotropic state are so important that they should be viewed in the same category as drug-induced changes in blood pressure and heart rate. Despite the current guidelines, we recommend that investigators consider performing measurements of cardiac contractility in nonclinical studies. In determining whether to evaluate inotropic function in the preclinical setting, reasonable questions include: Are other members of the same therapeutic or chemical class associated with cardiac function effects (eg, antifungals similar to itraconazole)? Does the drug’s therapeutic mechanism have a high risk of cardiac interaction? Sympatho- and parasympathomimetic, sympatho- and parasympatholytic drugs; drugs indirectly affecting autonomic function. Drugs that may affect mitochondrial function. Drugs that may affect surface sodium and calcium channels and/or intracellular sodium or calcium handling. Drugs having a novel mechanism of action that exists in a pathway known to affect myocardial function. Have cardiovascular effects been discovered in prior preclinical studies with the same or a similar compound? This is the ICH S7A definition of a “follow-up” study. Was a blood pressure change detected? If so, was it due to a change in cardiac output or in peripheral vascular resistance? This includes toxicology studies demonstrating evidence of myocardial morphological changes that might be preceded by changes in inotropic state. Are there any additional risk factors to be considered for drugs that have met the above criteria (eg, diabetic or elderly patient populations)?
Functional CV Assessment in Toxicology Studies
Background
While acute cardiovascular telemetry studies in chronically instrumented conscious animals are typically conducted to meet ICH guidelines for drug development, in some cases safety pharmacology endpoints (eg, cardiovascular studies) should be incorporated into general toxicology studies. When accumulation of small molecule drugs (or actions of their metabolites) may have cardiovascular effects, repeat-dose studies can evaluate for these potential effects. Repeat-dose studies may also be appropriate for biological agents. ICH S7A advises: “For biotechnology-derived products that achieve highly specific receptor targeting, it is often sufficient to evaluate safety pharmacology endpoints as a part of toxicology and/or pharmacodynamic studies.” 7 Regarding oncology drugs, ICH S9 states: “An assessment of the pharmaceutical’s effect on vital organ functions (including cardiovascular, respiratory and central nervous systems) should be available before the initiation of clinical studies; such parameters could be included in general toxicology studies.” 35
Adequately sensitive and predictive measurement of cardiovascular drug effects via repeat-dose toxicology studies could save time, money, and resources in the drug development process. An important purpose of preclinical safety studies is to lead to safer, more appropriate, and efficient clinical studies. This is critical with large molecule drugs (not often studied in traditional acute safety pharmacology studies), which are ideal candidates for repeat-dose toxicology studies to evaluate cardiovascular safety. 31
Many HESI workshop participants voiced concerns about the sensitivity and scientific robustness of functional cardiovascular evaluations extracted from general toxicology studies. Although cardiovascular safety pharmacology endpoints can be incorporated into general toxicology studies, the high doses necessary for toxicology studies may produce adverse effects that interfere with cardiovascular assessment. In contrast, doses administered in safety pharmacology studies are controlled to avoid confounding effects. If restrained animals are used in general toxicology studies, ECG and blood pressure measurements may be compromised. Differences in experimental design (parallel vs Latin square) can also limit sensitivity.
Current Approaches
The ECG has an important role in toxicology studies. The standard 10-lead ECG on a restrained canine is obtained in right lateral recumbency with surface electrodes attached directly to the skin, including 3 limb leads, 3 augmented limb leads, and up to 4 precordial leads. 36 Qualified personnel may obtain high-quality ECGs, which may then be analyzed for basic arrhythmias and drug effects on standard ECG intervals.
Until recently, a significant limitation of ECGs in toxicology studies was the nature of the equipment. Traditional ECG machines designed for clinical use were heavily filtered and required manual analysis and measurement from paper records. Digital ECG systems introduced over the past decade have adequate bandwidth to record laboratory animal ECGs and allow semi-automated digital analysis. A consequence of improved bandwidth is higher performance expectations from ECG technicians who strive to collect artifact-free ECG signals.
Unfortunately, a sensitive assessment of drug effects on heart rate is not possible in physically restrained dogs, as significant variability exists in the heart rate data. Obtaining ECGs in nonhuman primates is much more difficult than in dogs. Restraining primates to perform ECG results in high sympathetic activation and movement artifacts (due to physical struggling), both of which compromise ECG quality. Sedation has drawbacks, such as possible drug interaction between the test agent and the sedative. Sedation also limits the number of ECG samples that can be collected over time.
Noninvasive blood pressure measurements can be made during nonrodent toxicology studies utilizing cuff techniques (similar to sphygmomanometry in humans) around an animal’s limb or tail. 37 The most common devices, developed for veterinary anesthesia monitoring, utilize an ultrasonic Doppler probe to detect arterial wall motion distal to the inflatable cuff. Carefully used, these systems offer reasonable estimates of systolic arterial blood pressure in anesthetized cats and dogs. However, minimal evidence exists to support using these systems for reliable blood pressure measurement in conscious dogs or nonhuman primates in toxicology studies.
Although indirect blood pressure measurement has been available in human medicine for a very long time, 38 its reliability in veterinary medicine is considerably lower. This is due to the need for restraint (physical or chemical) in uncooperative animals, as well as anatomic differences from humans. Positioned around an animal’s tail or limb, conventional oscillometric cuff systems have limited sensitivity. 37 Recently, oscillometric cuff systems for nonclinical studies have been enhanced by the high-definition oscillometry (HDO) principle. 37,39
An alternative approach is to include chronically instrumented animals in a toxicology study and evaluate blood pressure (and other parameters) using standard safety pharmacology telemetry. These animals can be added as a satellite group and avoid necropsy, or be included as part of the regular study group and undergo necropsy. In the latter case, necropsy may reveal instrumentation-related artifacts such as fibrosis and/or emboli. If instrumented animals are included in both the vehicle control and the treated groups, these artifacts can be distinguished from drug-related abnormalities observed on necropsy or histopathology. Pilot investigations may be appropriate before initiating such procedures in a research laboratory.
Currently, the only viable noninvasive means to assess cardiac contractility in a nonrodent toxicology study is the use of EC. 34 Used in humans for several decades, EC has become more sophisticated in veterinary medicine in recent years. This modality provides noninvasive assessment of structural heart disease (eg, hypertrophy and dilated cardiomyopathy). Since animals in toxicology studies ultimately undergo necropsy where structural changes can be definitively measured, performing EC to evaluate for structural pathology may be less valuable in the research laboratory than in the clinic. Yet, EC may still be useful for monitoring structural cardiac changes during a study.
While little quantitative data exist on the sensitivity or reproducibility of EC in nonclinical safety studies, it appears to have an emerging role in preclinical safety pharmacology and toxicology. It has already proven to have several useful applications. 2-D EC allows for the evaluation of drug-induced morphological changes (eg, myocardial hypertrophy and cardiac chamber dilatation). M-mode and Doppler EC have the capacity to detect the changes in hemodynamic parameters and indicators of cardiac contractility consistent with drug-induced cardiac effects. Additionally, EC can assess adverse effects of cardiovascular drugs, which are often due to exaggerated pharmacological effects.
While EC can provide a noninvasive assessment of cardiovascular function in laboratory animals, its adoption in drug safety assessment has been slow. This may be due to technical constraints and/or low demand by pharmaceutical companies for such data. It may also reflect the fact that proper technique requires a high level of training and proficiency. While EC in veterinary medicine is often performed by board-certified veterinary cardiologists, it can be mastered by technical staff with proper training and experience. It requires only gentle restraint and/or sedation or light anesthesia (in some species). It is neither painful nor stressful for the animals. The examination is easily repeatable for follow-up studies.
Despite its benefits, EC is rarely incorporated in standard nonrodent toxicology studies. Yet, several experiments have shown EC to be a valuable method for assessing cardiovascular consequences of adverse drug effects. Data obtained from EC generally correlate well with invasive measurements. Echocardiography complements other cardiovascular evaluations (eg, ECG) and may have the potential to replace some invasive cardiovascular measurement methods.
Mechanistic Interpretation
Functional cardiovascular assessments in repeat-dose toxicology studies provide valuable nonclinical information on the chronic effects of drug treatment. This can be important for small molecule drugs that may either accumulate in tissues over time or produce metabolites that cause functional cardiovascular effects. One important delayed pharmacological effect is the inhibition of hERG channel trafficking. This effect would probably not alter the QT interval during a single-dose study but, rather, reveal itself in a repeat-dose study. 40 Repeat-dose toxicology studies may cause structural changes due to cumulative drug toxicity.
One major disadvantage of functional cardiovascular assessments in repeat-dose toxicology studies is that study design and environment are not optimized for the collection of these endpoints. Toxicology studies are parallel, not Latin square, designs. The doses are typically higher (ie, toxic) than those in safety pharmacology studies. The research animals are frequently disturbed during toxicology studies. Unlike the controlled environment used for safety pharmacology studies, the rooms used for toxicology studies are not specifically designed for auditory and visual isolation. Little quantitative data exist on how study environment affects outcomes. However, it would require 32 animals under typical toxicology study conditions to achieve the same sensitivity achieved with only 8 animals under acute safety pharmacology study conditions using the Latin square crossover design within a tightly controlled environment. 41
While functional cardiovascular endpoints in toxicology studies are useful to evaluate chronic dosing with small molecules, they are essential in the development of biologics. These data may be the only nonclinical in vivo cardiovascular function data available for these large molecules. They typically cannot be studied in Latin square crossover designs due to their potential for immunogenicity and exceptionally slow elimination (long half-life). Biologics can be evaluated in acute safety pharmacology telemetry studies applying a parallel experimental design. However, due to its lower sensitivity, a parallel study design requires more study animals to achieve similar statistical power to a Latin square study design.
Some researchers contend that acute cardiovascular safety studies are inappropriate for biologics.
Depending on the dosing regimen, a significant time commitment is required to achieve steady-state concentrations due to long half-life and drug accumulation. Another drawback is the lack of animal models that react pharmacologically to biologics (eg, humanized antibodies).
Debate exists as to whether highly specific large molecules (eg, monoclonal antibodies) developed for noncardiovascular targets will produce adverse cardiovascular effects and, thus, whether safety pharmacology studies are indicated at all. Although this seems reasonable with respect to QT interval prolongation via inhibition of hERG channels, 42 the evidence is less clear for other cardiovascular effects which may occur via direct or indirect effects of the biological product. This is particularly true for novel biologic agents, where biological mechanism and antibody-related effects are incompletely understood.
Discussion
Noninvasive ECG telemetry systems have been used in humans for decades, typically in cardiac care units where patients are centrally monitored. Though rarely used in animal research, Holter monitoring systems are clinical ambulatory devices that record ECG data for subsequent playback. One drawback of these traditional devices in animals is inadequate bandwidth for quantitative diagnostic ECGs. However, recent technological advances have improved capability to measure functional cardiovascular indices like ECGs.
For example, jacketed ECG telemetry systems for use in nonclinical studies are now widely available with adequate bandwidth to allow simultaneous monitoring of up to 36 animals (ie, dogs or nonhuman primates) in the same room for over 24 hours. 43 Residing in the animal’s jacket, these battery-powered systems use digital technology (eg, BluetoothTM) to eliminate signal crosstalk. This technology can continuously collect high-quality ECG signals, even in a toxicology setting. While the use of these systems (along with efforts to minimize stimuli during data collection) can significantly improve the sensitivity of ECG endpoints in toxicology studies, achieving sensitivity comparable to that of dedicated safety pharmacology studies is unlikely due to the many conflicting goals of these multipurpose studies.
High-definition oscillometry technology allows more accurate peripheral blood pressure measurement (including systolic, diastolic and mean arterial pressure) from dogs and nonhuman primates. With immediate graphical feedback provided to the user, inaccurate samples can be quickly discarded and measurements repeated when animals have been struggling. Currently available HDO systems have some limitations. For instance, they can only collect isolated data samples. They also require the physical or chemical restraint of participants, which affects the endpoints being measured (especially in nonhuman primates).
Although direct measurement by implantable telemetry is the most sensitive method for evaluating blood pressure effects, implanted foreign bodies (telemetry transducers) may confound the pathology endpoints in a toxicology study. This is not an issue in safety pharmacology studies in which the animals are meticulously monitored and avoid necropsy. A satellite group of chronically instrumented animals can be included in repeat-dose toxicology studies (under the same experimental conditions as the animals destined for necropsy). Alternatively, these animals could be studied in the controlled environment of a parallel safety pharmacology study, possibly at more appropriate doses.
The ideal solution to these challenges would combine the advantages of direct blood pressure instrumentation with those of noninvasive, jacketed telemetry. Such an approach has recently been developed and tested successfully in multiple laboratories. 43 In this system, a small pressure-sensing catheter is inserted unilaterally into an iliac artery via the adjacent femoral artery. A tiny blood pressure transducer (originally used in mice) is then implanted in the inguinal region. Thus, the distribution of any potential pathology associated with the indwelling catheter is limited to 1 hind limb. To compensate for limited transmission range, the signal is received by an antenna in the animal’s jacket. It is then electronically combined with the surface ECG signal and digitally transmitted via Bluetooth to a receiver in the room. Up to 36 animals in the same room can undergo simultaneous and continuous monitoring. In fact, nonhuman primates have been successfully monitored for up to 15 weeks, with side effects limited to slight endothelial inflammation at the catheter insertion site. 43
Recommendations
Toxicologists, veterinary pathologists, and safety pharmacologists should work closely to improve the integration of functional cardiovascular evaluations into nonclinical repeat-dose toxicology studies. While it may be challenging to collect functional cardiovascular endpoints from toxicology studies with a sensitivity similar to that achieved in acute safety pharmacology studies, the potential importance of this nonclinical information on the cumulative effects of drug treatment should encourage us to make the attempt. Performing functional cardiovascular assessment in repeat-dose toxicology studies is particularly worthwhile in the case of chronic dosing with small molecules (prone to tissue accumulation), with drugs whose metabolites may have cardiovascular effects and in the development of large molecules (eg, biologics).
We recommend quantification of the assay sensitivity of cardiovascular parameters generated within a general toxicology study. Scientists and regulatory agencies may then incorporate this quantification into data interpretation (see next section). The adoption of different approaches to enhance test sensitivity should be considered. Further research and development of new methods and technologies is strongly encouraged to address the challenges described in this article.
Assay, Pharmacologic, and Study Sensitivities
Background
Assay sensitivity, which indicates how large of an effect can be excluded, is an important factor to consider in nonclinical safety pharmacology studies. 26 Knowledge of assay sensitivity influences interpretation of any effect (or lack thereof) observed in these studies. Much discussion within our working group has centered on the purpose of safety pharmacology studies and how this influences assay sensitivity. We contend that ICH S7A studies do not need to be highly sensitive if their main purpose is to protect Phase I human volunteers from potentially life-threatening effects. However, the risk of advancing a molecule with any degree of cardiovascular liability has challenged this perception. ICH S7B studies need to be highly sensitive if they are to detect small changes in cardiac ventricular repolarization.
Discussion
Sensitivity may be defined and described in different ways (ie, statistical, systems-based, and study-related). While each approach has its advantages and limitations, we suggest an optimal way of looking at assay sensitivity is through a combination of these approaches.
Statistical sensitivity is determined by evaluating the study power, or calculating the minimum sample size required to detect a certain amount of change between the compound tested and the control vehicle. One can calculate the detectable amount of change due to drug effects by using the knowledge of variance from the previous studies. Study power, sample size, and effect size are correlated. Normally one can be obtained if the other two are fixed based on an assumed data variability (stating Study A is more statistically sensitive than Study B could mean several things: (1) For a fixed sample size and the amount of change between study drug and placebo, Study A yields a larger statistical power [eg, 95%] than Study B [eg, 80%]; (2) For a fixed study power and the effect size to be detected between study drug and placebo, Study A needs a smaller sample size than Study B; or (3) For a fixed study power and sample size, Study A can detect a smaller difference between study drug and placebo than Study B). Since the variability in all these calculations is based on historical data, caution should be used if the study protocol or conditions are changed.
Systems' sensitivity refers to the ability of the experimental model to respond to the specific mechanism under evaluation. For example, dogs have known sensitivity to hERG inhibitors such that administration of dofetilide increases QT interval in this species. Because hERG is not the prominent repolarizing current in rats, they do not respond to this class of drugs. Consequently, one would not evaluate a new drug for hERG-related QT interval effects in rats, given this lack of pharmacologic sensitivity. Similar reasoning would apply to other experimental parameters and mechanisms, where pertinent information is known.
Using a positive control, study sensitivity intends to demonstrate that a given study design and animal population can capture an effect. Study sensitivity is determined by the inclusion of a concurrent reference compound arm in the study design. The reference arm must either have a known action at the dose tested or have established the sensitivity to detect change with a positive control agent in a previous study. This approach was advocated in ICH E14. 44
In a clinical Thorough QT/QTc Study (TQTS), assay sensitivity is established as the ability to detect a small increase in QTc, say 5 ms, induced by a positive control drug (eg, moxifloxicin). In a typical TQTS, the pharmacokinetic profile of the positive control arm is scrutinized, as well as the similarity of its effect size and time course to the established QTc effect. 45 Extrapolating to nonclinical studies, inclusion of a positive control drug at a dose that produces an effect size similar to that needed to be captured lends credibility to study results with the drug candidate. However, it is not practical to include a positive control agent for all possible cardiovascular outcomes in each animal study. In fact, this was recognized as a limitation in ICH S7A, which allowed that, “Appropriate negative and positive control groups should be included in the experimental design. In well-characterized in vivo test systems, positive controls may not be necessary.” 7
Regulatory scientists translate nonclinical safety pharmacology results to the clinic via safety margins, whereby the ratio of dose or plasma concentration at the no-observed-adverse-effect-level (NOAEL) in animals is extrapolated to the proposed human dose and exposure. Low-assay sensitivity leads to a higher “apparent” safety margin since higher doses are needed to produce an effect. For example, we can compare safety margins generated using 2 cardiovascular safety pharmacology studies with different sensitivities (or minimum effect sizes) on heart rate.
In study A, the minimum effect size is 10 beats per minute vs 50 beats per minute in Study B. Assuming the study drug increases heart rate in a dose-related manner, a higher dose would be needed to significantly increase heart rate in the less-sensitive assay. Consequently, the safety margin calculated from the less sensitive study will be higher than the safety margin from the more sensitive study. Thus, the apparent safety margin depends not only on the drug effect, but also on the sensitivity of the study used to determine it.
Another important topic concerning assay sensitivity is the relevance of an animal signal to defining a human risk. Can nonclinical findings be translated to clinical participants and human patients? This important question has been raised several times in this manuscript.
Recommendations
For nonclinical cardiovascular safety studies, either acute telemetry studies or toxicology studies that include functional endpoints, our working group achieved a reasonable consensus on recommendations for defining sensitivity.
Assay sensitivity should be calculated based on experimental design and established variability (according to a continually updated and monitored database). Information about assay sensitivity should be used prospectively by the sponsor to modify study design when indicated. Variance information used in the power calculation should be based on several historical studies whenever possible. If study modifications or changes occur, additional data should be collected and evaluated to ensure that study sensitivity has not changed. Periodic recalculation of statistical power should routinely be conducted as a means of monitoring drift in experimental procedures.
In addition to prospective evaluation of study sensitivity, we contend that a reference compound (ie, positive control) at a relevant dose (selected to affect each parameter of interest) should be evaluated to confirm assay sensitivity. The dose (concentration) should be selected to produce a submaximal response. Appreciable changes in study conditions should motivate investigators to generate additional data with the reference compound.
The HESI workshop participants recognized the importance of a concurrent positive control arm in a QT study. However, this may not be practical for safety pharmacology studies evaluating other parameters, since a control drug typically affects only one parameter. For example, moxifloxacin is used (as a control drug) at a dose that will only increase QT interval and not other parameters of interest (ie, hemodynamic measurements). To fully characterize assay sensitivity, a study would require several reference compounds, which is clearly impractical. However, it may be feasible to test a variety of reference compounds and use them as historical controls. For studies evaluating a specific parameter, investigators could include a concurrent positive control arm at a relevant dose for that parameter.
In most nonclinical studies, the combination of statistical sensitivity analysis and historical positive control data should be provided as supporting material in cardiovascular safety study submissions and should be adequate to characterize assay sensitivity. This approach will not account for differences that result when an animal population changes from study to study but is the most easily adaptable practice to follow.
Conclusion
While nonclinical evaluation of the electrophysiological effects of drugs on the heart has become quite robust, the assessment of hemodynamic and functional cardiovascular drug effects has not been as rigorously improved, despite its importance to human safety. Evaluation of cardiovascular drug effects is important not only in acute safety pharmacology studies but also in repeat-dose toxicology studies. A strong consensus was achieved that nonclinical assay sensitivity should be adequately characterized, and this information should be included when study outcomes are evaluated and translated to humans.
Within the realm of functional cardiovascular assessment in nonclinical drug trials, several points of disagreement remain. One noteworthy area of debate is the role of the routine evaluation of cardiac contractility. Consensus is lacking on whether or not surgically implanted devices to measure left ventricular function are proarrhythmic and may thus interfere with cardiac electrophysiology evaluation.
This manuscript has highlighted numerous areas in need of additional research. Several pertinent studies are either in development or already underway. Hopefully, data provided by these studies will facilitate improved nonclinical cardiovascular assessment of drug candidates, with results easily translated to human participants. Ultimately, performing sensitive yet specific evaluations of functional cardiovascular parameters in nonclinical studies will allow pharmaceutical companies to identify suspect agents early in the drug discovery and development process while allowing the promising agents to proceed into clinical development.
Footnotes
The work was conducted as part of the ongoing scientific activities managed as part of the HESI Committee on Cardiac Safety. HESI is a nonprofit, scientific organization that facilitates joint programs involving industrial, academic, and government scientists. The funding for the workshop that served as the initial source of ideas behind this manuscript came from the HESI Committee on Cardiac Safety.
Acknowledgment
Thank you to the many scientists who contributed to the Committee’s discussions over the past year and have provided input to make this manuscript possible. The authors would also like to acknowledge the editorial assistance provided by Scorr. The views in this article represent those of the authors and do not necessarily reflect the view of the U.S. Food and Drug Administration or any of the other organizations listed.
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) received no financial support for the research and/or authorship of this article.