
Abstract
Many immunosuppressive drugs are associated with an increased risk of B-cell lymphoma, squamous cell carcinoma, and Kaposi sarcoma. Thirteen immunosuppressive drugs have been tested in 2-year carcinogenicity studies (abatacept; azathioprine; busulfan; cyclophosphamide; cyclosporine; dexamethasone; everolimus; leflunomide; methotrexate; mycophenolate mofetil; prednisone; sirolimus; and tacrolimus) and in additional models including neonatal and genetically modified mice; chemical, viral, ultraviolet, and ionizing radiation co-carcinogenesis, and in models with transplanted tumor cells. The purpose of this review is to outline the mechanisms by which immunosuppressive drugs can influence neoplasia, to summarize the available preclinical data on the 13 drugs, and to critically review the performance of the models. A combination of primary tumor and metastasis assays conducted with transplanted cells may provide the highest value for hazard identification and can be applied on a case-by-case basis. However, for both small molecules and therapeutic proteins, determining the relative risk to patients from preclinical data remains problematic. Classifying immunosuppressive drugs based on their mechanism of action and hazard identification from preclinical studies and a prospective pharmacovigilance program to monitor carcinogenic risk may be a feasible way to manage patient safety during the clinical development program and postmarketing.
Introduction
Since the pioneering work in the 1950s, 1 the use of immunosuppressive drugs in transplantation has grown dramatically. Originally, transplantation was a treatment of last resort and was limited to closely matched donors with a similar pattern of major histocompatibility (MHC) antigens. In these early days, the pharmacopea of immunosuppressive drugs was largely limited to “spin-off” application of cytotoxic anti-neoplastics and glucocorticoids. The introduction of calcineurin antagonists in the late 1970s (cyclosporine 2 ) has revolutionized the field. Similarly, as our knowledge of immunosuppressive cocktails and our skill in patient management has grown, transplantation has become almost routine. In the 21st century, transplantation is no longer limited to life-saving procedures and transplantation of a wide variety of tissues and organs to address quality-of-life issues is becoming more common. In addition to their use in transplantation, immunosuppressive agents are now used in a variety of autoimmune diseases, for example, rheumatoid arthritis (RA), 3 systemic lupus erythematosis (SLE), 4 and psoriasis. 5 Thus, an ever-growing number of patients are being exposed to immunosuppressive drugs.
It has long been appreciated that immunosuppressive drugs increase the risk of infections and essentially all immunosuppressive drugs carry a warning concerning infectious disease (see Table 1). Similarly, from the first reports of cyclosporine’s benefits in renal transplantation, it was realized that it was associated with lymphomas. 6 With the rekindling of interest in immune surveillance against neoplasia 7 –9 and the growing experience with the newer immunosuppressive agents, there is an increased awareness that immunosuppressive drugs can increase the risk of neoplasia. 10
Human Neoplasms Associated With Immunosuppressive Drugs
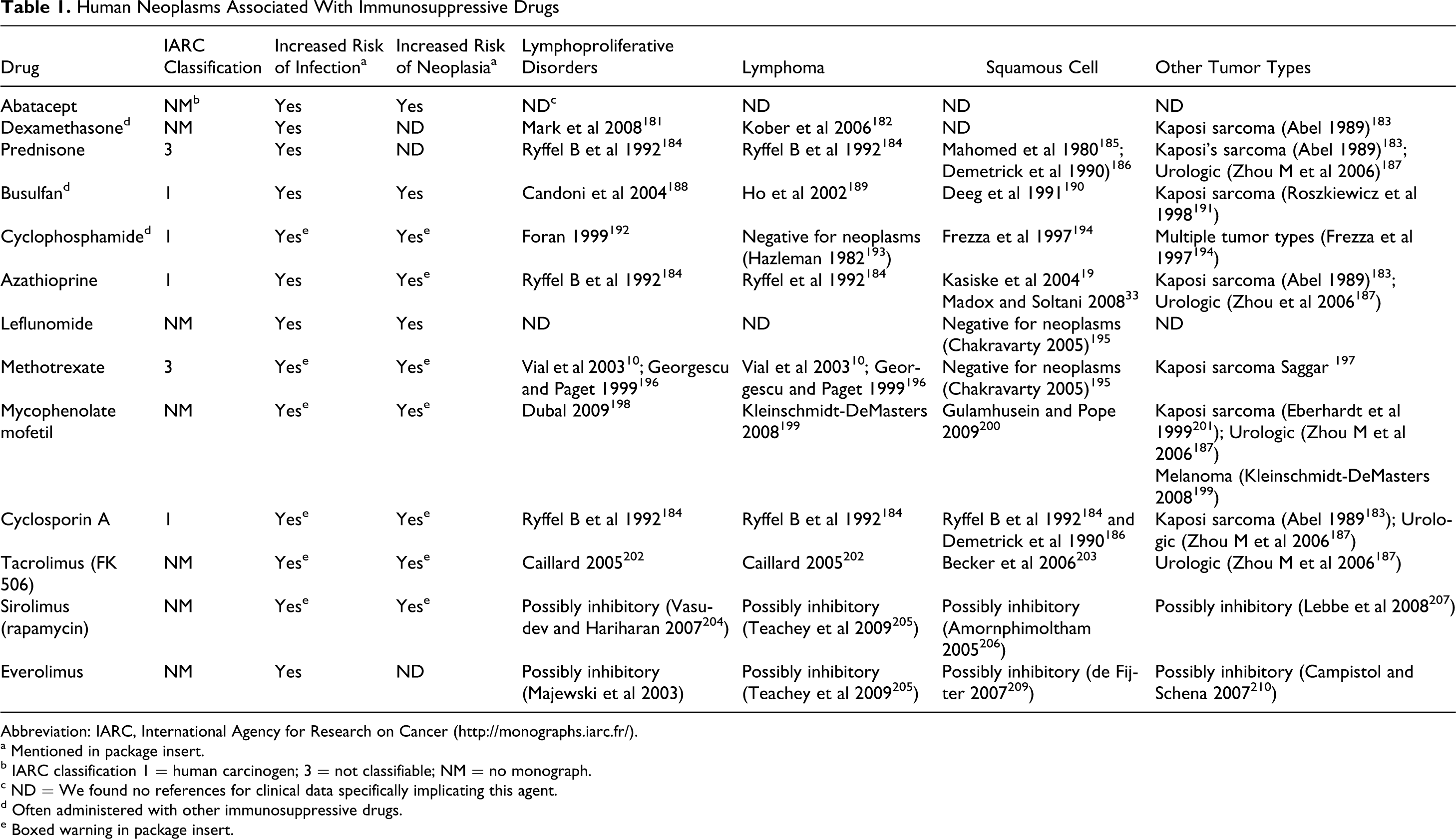
Abbreviation: IARC, International Agency for Research on Cancer (http://monographs.iarc.fr/).
a Mentioned in package insert.
b IARC classification 1 = human carcinogen; 3 = not classifiable; NM = no monograph.
c ND = We found no references for clinical data specifically implicating this agent.
d Often administered with other immunosuppressive drugs.
e Boxed warning in package insert.
The purpose of this review is to provide a synthesis of the predictive value of preclinical testing for the effects of immunosuppressive agents used in organ transplantation and the treatment of autoimmune disease on the risk of neoplasia in patients. For more than 40 years, the 2-year bioassay in rodents has been used to support development of small molecule drugs in the attempt to gain insight into the risk of neoplasia posed to patients receiving therapeutic doses. As shown in Table 2, as listed in the carcinogenicity potency database, 11,12 13 immunosuppressive drugs (including abatacept, an antibody fusion protein) have been tested in rodent 2-year bioassays. These drugs will be the focus of this review.
Effects of Immunosuppressive Drugs on Mutagenesis and in 2-year Bioassays
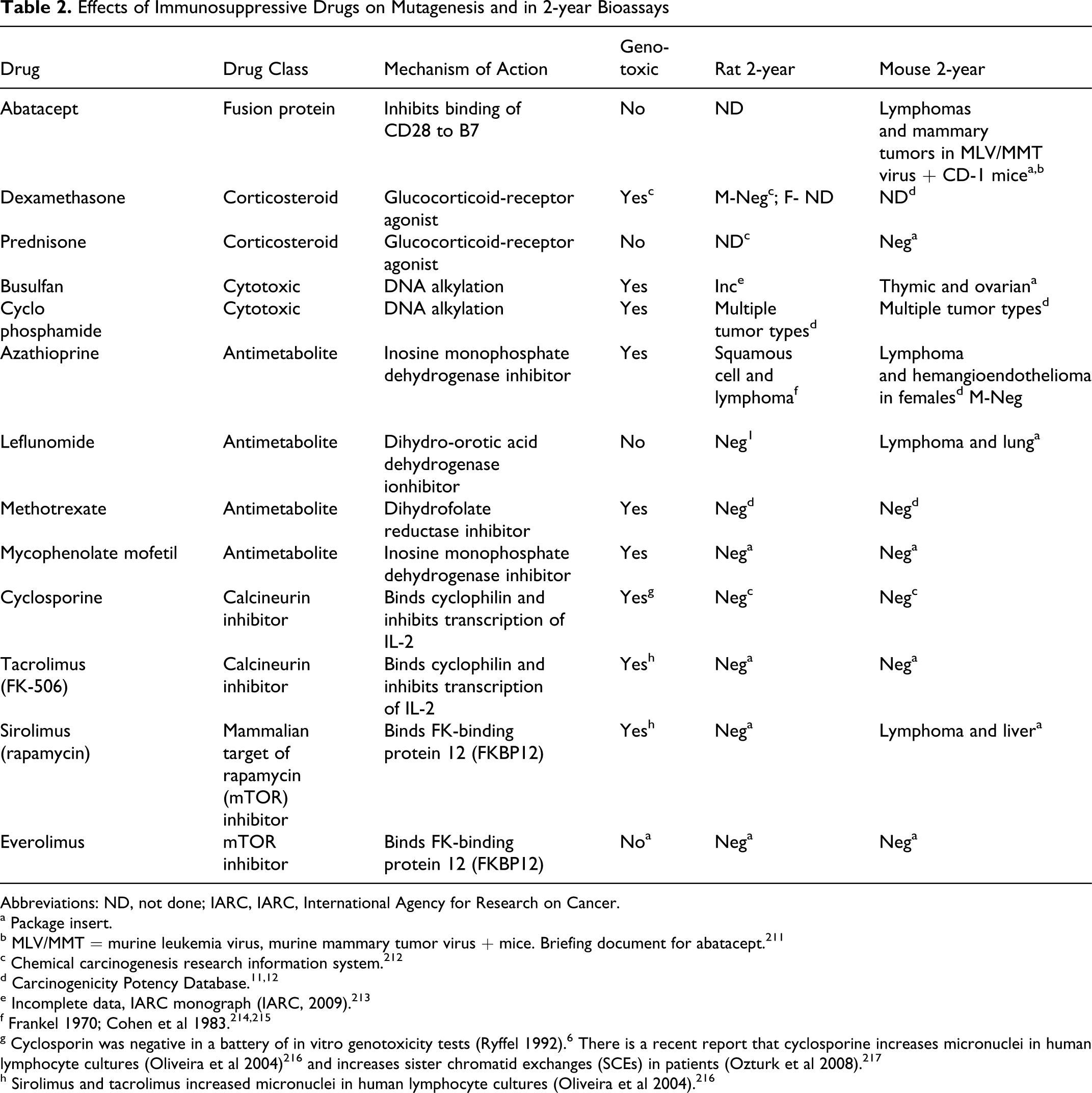
Abbreviations: ND, not done; IARC, IARC, International Agency for Research on Cancer.
a Package insert.
b MLV/MMT = murine leukemia virus, murine mammary tumor virus + mice. Briefing document for abatacept. 211
c Chemical carcinogenesis research information system. 212
e Incomplete data, IARC monograph (IARC, 2009). 213
g Cyclosporin was negative in a battery of in vitro genotoxicity tests (Ryffel 1992). 6 There is a recent report that cyclosporine increases micronuclei in human lymphocyte cultures (Oliveira et al 2004) 216 and increases sister chromatid exchanges (SCEs) in patients (Ozturk et al 2008). 217
h Sirolimus and tacrolimus increased micronuclei in human lymphocyte cultures (Oliveira et al 2004). 216
As an aid to understanding, we will first survey the putative roles of the immune system in carcinogenesis and review the clinical data on the 13 immunosuppressive agents that have been tested in the 2-year bioassays and the state of knowledge on the mechanisms of carcinogenesis associated with these neoplasms. We will then explore the data on these agents from alternative experimental approaches: neonatal and genetically modified mice; models of initiation and promotion; and models that rely on transplanted tumor cells. Finally, we will critically evaluate the predictive value of the various models and provide recommendations on potential alternatives to the 2-year bioassay that may be appropriate for immunosuppressive drugs.
Review of the Role of the Immune System in Neoplasia
It is widely accepted that carcinogenesis is a multistep process that involves initiation, promotion, and progression. 13 As outlined, in Table 3, in considering the role of the immune system in neoplasia, it is useful to consider additional steps in the carcinogenic process. The boundaries of these steps are somewhat arbitrary as is the assignment of acquiring a specific phenotype in sequential manner. For example, it is likely that genomic instability of transformed cells facilitates changes (damage) in genomic DNA that accumulates over the life of a neoplasm rather than being restricted to just the earliest phase. 14 Similarly, the order of acquisition of specific phenotypes, for example, unregulated cell growth and invasiveness, may not fit nicely as discreet sequential steps in the model. Regardless of the shortcomings of the model, as will be described in detail later, it has been possible to dissect these phases experimentally and thus to study the effects of immunosuppressive drugs on the different phases.
Putative Roles of Immune System in Tumor Initiation, Promotion, and Progression
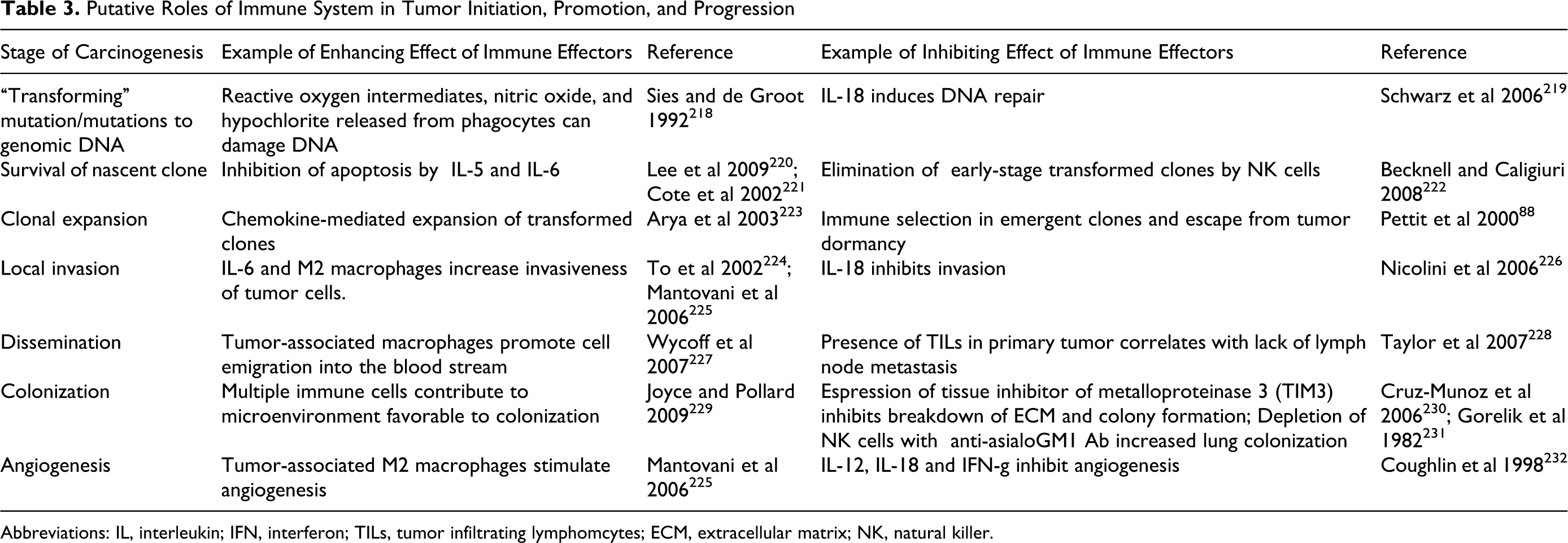
Abbreviations: IL, interleukin; IFN, interferon; TILs, tumor infiltrating lymphomcytes; ECM, extracellular matrix; NK, natural killer.
There is also an emerging understanding that the immune system can play multiple roles in oncogenesis; potentially both inhibiting and potentiating neoplasia. 15,16 As shown in Table 3, at each stage of carcinogenesis the immune system can either inhibit or facilitate the process. A further complicating factor is the observation that tumor growth itself can cause immunosuppression. 17,18 Thus, when studying the effects of immunosuppression in neoplasia, one must be prepared to consider both inhibition and potentiation of each step. This has important implications on interpreting the results of the various test systems. As will be developed in the later sections, despite the clear association between immunosuppressive drugs and an increased risk of neoplasia in transplant patients, many preclinical models fail to predict this increased risk.
Review of the Clinical Literature on the Impact of Immunosuppressive Drugs on the Risk of Neoplasia
As shown in Table 1, the list of neoplasms associated with the use of immunosuppressive drugs is rather short. Lymphomas, nonmelanoma skin cancers, and Kaposi sarcoma (KS) are increased by more than 20-fold in renal transplant patients. 19 In contrast, only a 2-fold increase in risk of developing the neoplasms that account for the majority of human tumors, that is, colon, breast, prostate, and lung, has been reported. This suggests that we are dealing with a special case.
Lymphoproliferative Disorders and Lymphomas
Lymphoproliferative disorders are characterized by the abnormal proliferation of lymphocytes into a monoclonal lymphocytosis, ultimately resulting in an increased frequency of malignant transformation and an inability to eliminate transformed cells. 20 The 2 major types of lymphocytes are B and T cells, which are derived from pleuripotent hematopoetic stem cells in the bone marrow. 21 Individuals who receive immunosuppressive drugs are susceptible to developing a lymphoproliferative disorder due to deregulation of these lymphocytes. The incidence of disease is significantly higher in individuals who have drug-induced immunodeficiency compared with primary immunodeficiencies. 21 The immunodeficiency-associated lymphoproliferative disorders are clinically and pathogenically heterogeneous, vary in clonal composition, and differ according to the immunodeficiency involved. 21 Posttransplant lymphoproliferative disorder (PTLD) occurred in up to 11% of patients, increasing with age and time of posttransplant and duration of immunosuppressive treatment. 22 There is a greater risk for men than women to develop PTLD. 23 Among pediatric transplant recipients, PTLD is recognized as the most common neoplasm with associated risk factors of patient age of <18 years and male gender. 22 Early lesions in children and young adults have been found to appear within the first year of transplantation, with a high occurrence after heart and lung transplant. 22 The overall risk 21,22 of developing PTLD over a 10-year period is approximately 12%.
B-Cell Lymphomas
B-cell lymphomas are a type of non-Hodgkin lymphoma (NHL) that are derived from lymphocytes of the B-cell lineage. They occurred most often after solid organ and bone marrow transplant and can range from indolent malignancies with low-grade histologies to rapidly growing and highly aggressive tumors with high-grade histologies. 24 Approximately 90% of PTLD lymphomas are of B-cell origin and are Epstein-Barr virus (EBV) positive. 25 They also share features, including origination or involvement of extranodal sites, aggressive histology, and rapid clinical progression. 21,26 –28 Between 1% and 5% of these lymphomas involve the kidney and liver, 5% and 15% the heart and lung, and 20% the small bowel. 21,22,25,28 –30 The median age for B-cell lymphoma presentation in rapidly growing and high-grade histologies is 42 years, and the incidence increases with advancing age and degree of immune suppression. 24 B-cell lymphoma is a potentially life-threatening complication in transplant recipients; the median survival time for B-cell lymphoma is approximately 6 years. 24,31
Squamous Cell Carcinomas
Nonmelanoma skin cancer (NMSC) is another common malignancy to occur as a result of immune suppression and develops after solid organ transplantation. The incidence 32 of NMSC after transplantation is steadily increasing and varies in the United States and Western Europe from 5% at 2 years from transplantation, 10% to 27% at 10 years from transplantation, and 40% to 60% at 20 years from transplantation; higher figures are observed in Australia, where the 20 years incidence reaches 70% to 82%. Half of the patients who develop NMSC develop a second NMSC within 3.5 years. 22 Squamous cell carcinoma (SCC) is the most common posttransplant NMSC. Squamous cell carcinoma is an aggressive carcinoma that disseminates to form multiple metastases. 22,33 –35 Squamous cell carcinoma is found most often in patients with greater sun exposure and fair skin types, above 40 years of age, with a genetic predisposition, and in men, who have a significantly higher incidence than women. 33,35 –37 The incidence of SCC has been found to increase with cumulative exposure to immunosuppressive agents and is found in 84% head and neck, 13% extensor upper extremities, 10% ear, and 30% lip. 22,33 Squamous cell carcinoma is associated with a poor prognosis, especially in patients with poorly differentiated extracutaneous tumors, tumor thickness >5 mm, and invasion of underlying tissue. 22
Kaposi Sarcoma
In addition to its prevalence as a malignancy in HIV-infected patients, KS is also a leading tumor type resulting from immunosuppression after solid organ transplantation. 30,38 Approximately 6% of post solid organ transplant patients develop KS, with a median time from transplantation to diagnosis of KS of approximately 1.5 years. 22 Cutaneous lesions account for 80% of KS and visceral lesions for approximately 20%. 22 Kaposi sarcoma is more prevalent in males and has been found to be more aggressive in older men of Mediterranean descent; however, there has been an increased incidence in white, African American, and Hispanic women. 39 Previous studies have demonstrated a 400- to 500-fold increase in KS in transplant patients compared with nontransplant controls of the same ethnic origin. Kaposi sarcoma has also been associated with NHLs and invasive cervical cancer related to human papillomavirus (HPV). 30,38
Carcinogenesis in Immunosuppression
Viral Oncogenesis
Viruses are believed to contribute to 20% to 25% of cancer deaths in developing countries and to 7% to 10% in industrialized countries. 40 Viruses have oncogenic potential at multiple organ sites, including lymphoid tissues and nasopharynx (EBV), uterine cervix (HPV), liver (hepatitis B virus [HBV]), hepatitis C virus (HCV), and KS (human herpes virus 8 [HHV-8]). 22,40 In drug-induced immuosuppression, EBV and HHV-8 are the most important oncogenic viruses.
Epstein-Barr virus is a member of the HHV family that was originally isolated from a cultured Burkitt lymphoma cell line that has been linked to numerous neoplasms, including hematopoietic, epithelial, and mesenchymal tumors. 26 Epstein-Barr virus has been associated with nasopharyngeal carcinoma, gastric carcinomas, lymphoepitheliomas of the foregut, inflammatory pseudotumors of the liver and spleen, and HIV-associated smooth muscle neoplasms, as well as lymphoproliferative disorders of B-cell, T/NK-cell, and HIV-related origin. 26 Most EBV-related lymphoid malignancies are of B-cell origin, where EBV has been thought to infect naive B-cells and induce their proliferation, which subsequently undergo a germinal center (GC) reaction and differentiate into memory B-cells, where the virus gains access to the memory B-cell pool (the site of viral persistence). 26,41 In PTLD, EBV has been suggested to interfere with normal B-cell differentiation and selection processes. 41 Expression of the latent EBV-positive encoded proteins has suggested a role in the oncogenic process by somatic hypermutation of GC cells. 26 Of note, a number of EBV-positive lymphoproliferative disorders have been observed to spontaneously regress on cessation or modification of the immuosuppressive regimen. 27 Epstein-Barr virus-negative PTLDs constitute only 20% of all transplant cases and have an unknown etiology. 26
The pathogenesis of KS is likely related to viral infection with HHV-8, also called KS-associated herpes virus (KSHV). 22,40 Susceptibility to KS has been related to a lack of host defense against HHV-8, which can result in the reactivation of a latent HHV-8 infection. This has been evidenced by the detection of viral DNA in patient blood after receiving immunosuppressive therapy. 42 In addition to KS, KSHV is also associated with lymphoproliferative disorders in persons with HIV infection/AIDS, following organ transplantation or following immunosuppressive therapy. In some cases, such as renal transplantation, KS has been found to regress depending on the type and duration of immunosuppressive therapy used. Highly immunosuppressed patients can develop KS within 6 months of initiation of therapy. 30,38,43 In 70% of cases, KSHV occurred in conjunction with EBV, suggesting that there may be a role for both viruses in tumorigenesis, first in transformation of B-cells and then in driving the growth of the tumor. 26
Ultraviolet Radiation
In contrast to the viral origin of lymphoid neoplasms and KS, SCC is believed to result primarily from exposure to ultraviolet radiation. 44 Ultraviolet radiation in sunlight that is relevant to human exposure consists of UVA (320-400 nm) and UVB (290-320 nm). UVB irradiation accounts for most of the harmful effects of sunlight because most of the UVB radiation is absorbed by the epidermis. UV irradiation induces DNA damage (primarily pyrimidine dimers via free radicals and reaction oxygen intermediates). 45 If not repaired, UV-induced pyrimidine dimers can lead to permanent mutations in the DNA sequences. UVB is able to cause skin cancer without additional initiators or promoters and is thus a complete carcinogen. UV also increases cellular proliferation, damages collagen fibers, and causes inflammation and erythema, and may activate latent viruses, such as herpes simplex, resulting in viral recrudescence and the induction of local and/or systemic immune suppression. 44 UV irradiation also leads to immune suppression. This is in part due to the inhibition of antigen presentation by Langerhans cell and due to the release of immunosuppressive cytokines. 46
The cells of the skin contain mechanisms to prevent DNA damage from leading to skin carcinogenesis. One of these mechanisms is growth arrest followed by DNA repair and the other is cell death by apoptosis. 47 Mutations of the p53 tumor-suppressor gene, which result in failure of apoptosis to occur, appear to be a key event for skin tumor development as UV-induced skin tumors develop much sooner in p53-deficient mice. 48 UV irradiation-induced inflammation may also contribute to tumor progression, whereas inhibition of UV-induced inflammation has been shown to inhibit tumor progression. 49
The risk of developing skin cancer in immunosuppressed patients is increased up to 70% in regions with high sun exposure. 30 In addition to its direct effects, research also indicates that UV radiation has immunosuppressive effects that can contribute to skin cancer induction by suppressing cell-mediated immune reactions that normally serve to keep developing skin cancers latent. 44 Although not well understood, UV-induced immunosuppression may involve disruption of cytokine cascades and inflammatory T helper type 1 (Th1) cell-driven immune reactions.
Review of Preclinical Approaches to Evaluate the Impact of Immunosuppressive Drugs on Carcinogenesis
2-Year Bioassays
The current regulatory approach to the evaluation of human carcinogenicity risk is based on a series of in vitro and in vivo nonclinical assays. The multistep model of carcinogenesis identifies damage to genomic DNA as the initiating step in the oncogenic process. Accordingly, nonclinical safety evaluation of human pharmaceuticals involves a battery of in vitro and in vivo tests that identify potential drug-induced gene mutations, clastogenic damage, and direct DNA damage. 50 For most pharmaceuticals that are intended to be used in patients for durations exceeding 6 months, lifetime carcinogenicity studies are generally conducted in 2 rodent species. 51 –53 The 2-year bioassay has been the standard approach to testing the carcinogenic potential of chemical and small molecule drugs (xenobiotics) for over 30 years. 54 Although some modifications have been explored, for example, group size, number of groups, additional end points, housing conditions, dose selection, and duration of the assay, the design of the 2-year bioassay is basically unchanged 55 and can be summarized as follows: conducted in rats and mice; ≈50/gender per group, 3 dose levels and control, high-dose set at the maximum tolerated dose (MTD), lower doses set at a fraction of the MTD. The purpose of the assay is “to determine the carcinogenicity of the test agent in both sexes of 2 species and is designed to cover the greater part of the animals' life spans” 54 and the primary end points evaluated are the incidence of tumors and time to death due to tumor. 56
Over, 400 drugs have been tested in the 2-year bioassay. 57 However, for more than 25 years, the validity and usefulness of this assay for pharmaceuticals have been called into question. 58,59 Approximately 40% of marketed pharmaceuticals are positive in the assay 57 and overall, approximately 50% of all compounds tested are positive. 59 Moreover, the reproducibility of the results of the 2-year bioassays is also questionable. As reviewed by Gottmann et al, 60 comparing the results from 121 replicate bioassays revealed a concordance of only 57% indicating that “rodent carcinogenicity assays are much less reproducible than previously expected.”
Survey of the literature revealed 13 agents that are either approved or are in clinical trials as immunosuppressive agents in transplantation or for the treatment of autoimmune disease that have undergone testing in the 2-year bioassays. Some were tested in rats, others in mice, and some in both. These agents, the results of genotoxicity testing, and the results of the 2-year bioassays are listed in Table 2. For these 13 agents, a total of 26 potential (rat and mouse) bioassays were possible, however in only 21 cases were complete data available, that is, data from both genders. From these bioassays, only in 7 cases (33%) were increased numbers of neoplasms detected. Of the 5 agents that were positive in one or more bioassays, 4 are considered to be genotoxins. As described above, most of these agents are believed to pose an increased risk of neoplasia in humans (see Table 1). Thus, the 2-year bioassay not only failed to detect increased neoplasms with 66% of immunosuppressive drugs, but it even failed with some known genotoxins. These findings cast doubt on the predictive value of the 2-year bioassay for immunosuppressive agents.
Neonatal Mice
The neonatal mouse has been evaluated as a potential assay for testing of potential human carcinogens since the late 1950s, where it was first observed that administration of a single dose of a human carcinogen to newborn mice was sufficient to induce tumors. 61 Neonatal mice are thus considerably more susceptible to some carcinogens than adult mice. The typical tumors in this assay are liver and lung. Although the neonatal mouse assay has been used for chemical testing since the early 1960s, the mechanisms of the carcinogenic effect have not been fully described. Mice are born at a developmentally immature stage and their tissues are undergoing extensive cell proliferation and expansion. One theory for the high susceptibility to carcinogenic agents may be that chemicals that are genotoxic may induce transformation in cells that are more likely to survive and undergo clonal expansion than cells in the adult system. Moreover, as the immune system in neonates is poorly developed, for example, natural killer cell activity develops after 3 weeks of postnatal life and does not reach a maximum until approximately 2 months of age, 62 testing immunosuppressive drugs in neonatal mice may mask effects on immune surveillance.
In the neonatal mouse assay, mice are typically dosed by intraperitoneal injection or by oral gavage with test article on postnatal days 8 and 15 and then necropsied at 1 year of age. 61,63 Gross lesions and masses are recorded and mice are subjected to full histopathology. Typical group sizes are 24/gender per group. 61
As shown in Table 4, only 3 of the 13 drugs of interest have been tested in this assay. As was observed with the 2-year bioassay, the genotoxic carcinogens azathioprine (administered intraperitoneally to Swiss mice on days 1-4 at a dose of 40 mg/kg) and cyclophosphamide (administered orally to CD-1 mice on days 8 and 15 at doses ranging from 20 to 60 mg/kg) were positive and cyclosporine (administered orally to CD-1 mice on days 8 and 15 at doses ranging from 10 to 90 mg/kg) was negative. 61
Effects of Immunosuppressive Drugs on Carcinogenesis in Neonatal Mice
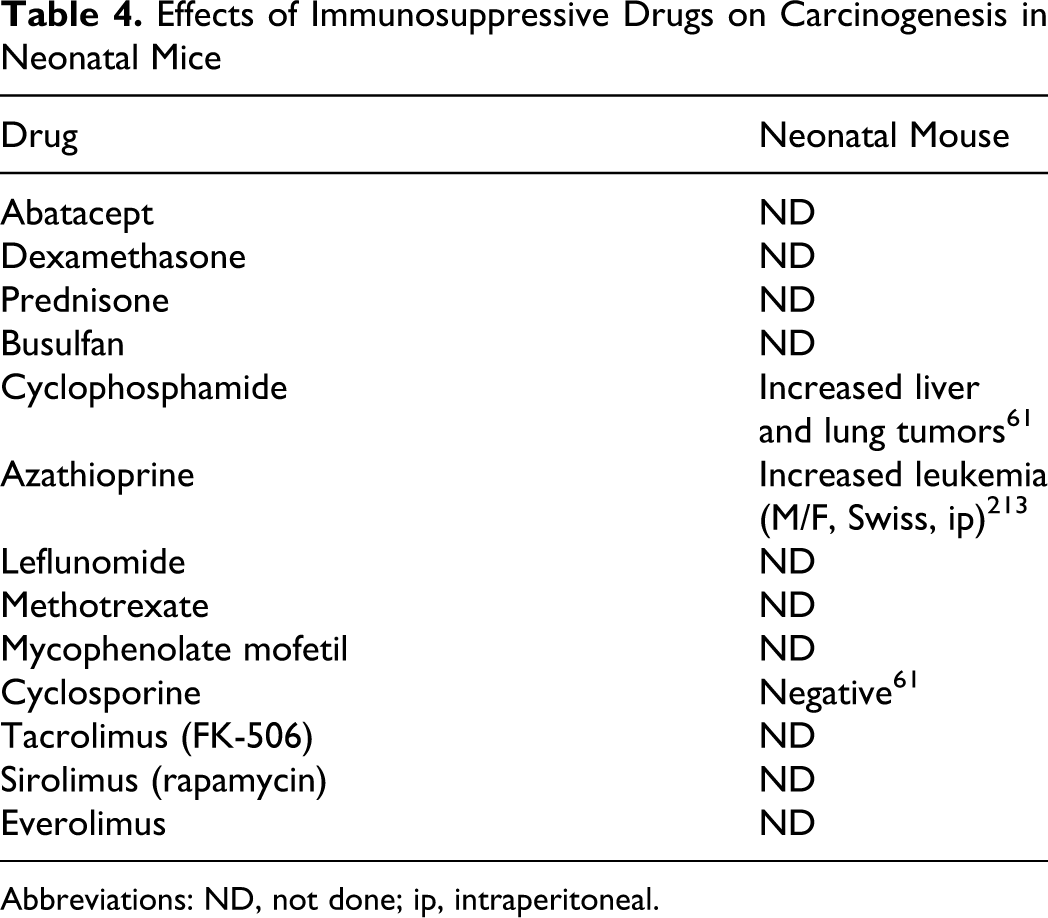
Abbreviations: ND, not done; ip, intraperitoneal.
Genetically Modified Mouse Models
A variety of genetic modifications have been made in mice in an attempt to increase the predictive value of testing chemicals for their carcinogenic potential. The genetically modified mouse models described in this section can basically be subdivided into 2 main categories, (a) genetic modifications designed to increase the detection of carcinogenic chemicals and (b) genetic modifications designed to evaluate tumor biology and antitumor therapies.
The first category of genetic modification has been evaluated as alternatives to the 2-year bioassay. These transgenic models have specific altered oncogenes or tumor-suppressor genes. According to the ICH S1B guidance document, 52 examples of some transgenic models that can be considered in place of the 2-year bioassay are p53+/−- and XPA−/−-deficient models and the Tg.AC and Ha-ras2 transgenic models. These are the models that have most widely been used for pharmaceutical nonclinical safety testing. 64 The models were developed on the basis that specific tumor-suppressor genes (p53 or XPA) or proto-oncogenes (ras) play a critical role in cancer development in humans and therefore genetic modification of these genes will alter susceptibility to carcinogenic agents. 65 The models have been designed to have a higher probability of detecting potential carcinogens within a shorter time period than the traditional 2-year bioassay.
The second category of genetic modifications includes models that show a high incidence of a specific tumor type that is relevant to clinical tumors of interest. In these models, tumors develop within a short period of time and treatments are initiated either prior to (preventative) or after (therapeutic) the tumors have developed. These studies may have little relevance to risk assessment but are included because they provide information that is relevant to understanding the spectrum of effects that can be observed with immune suppressive agents.
A typical study design for the genetically modified models is exemplified by the work of Tennant et al 66 where 8- to 9-week-old hemizygous or homozygous Tg.AC mice and the FVB/N parental strains (15/gender per group) were dosed with the test agent at doses up to the MTD by either the dermal or the oral route of administration for 26 weeks, the mice necropsied, and all tissues examined histologically. 66
As shown in Table 5, the effects of 6 of the 13 drugs of interest have been tested in one of more genetically modified mouse models with highly variable results. Because these models have been the subject of great regulatory interest, the results in each model will be described in detail below. In summary, depending on the model, the immunosuppressive drug and the dose levels administered, increased, decreased, or no effect on tumor incidence was observed. Moreover, as immune function in the genetically modified mice may be altered, for example, transgenic mice with an activated v-Ha-ras oncogene have higher serum levels of IL-1α and higher levels of IL-2 in spleen and thymus cell supernatants, 67 interpreting the results with immunosuppressive drugs may be problematic.
Effects of Immunosuppressive Drugs on Carcinogenesis in Genetically Modified Mice
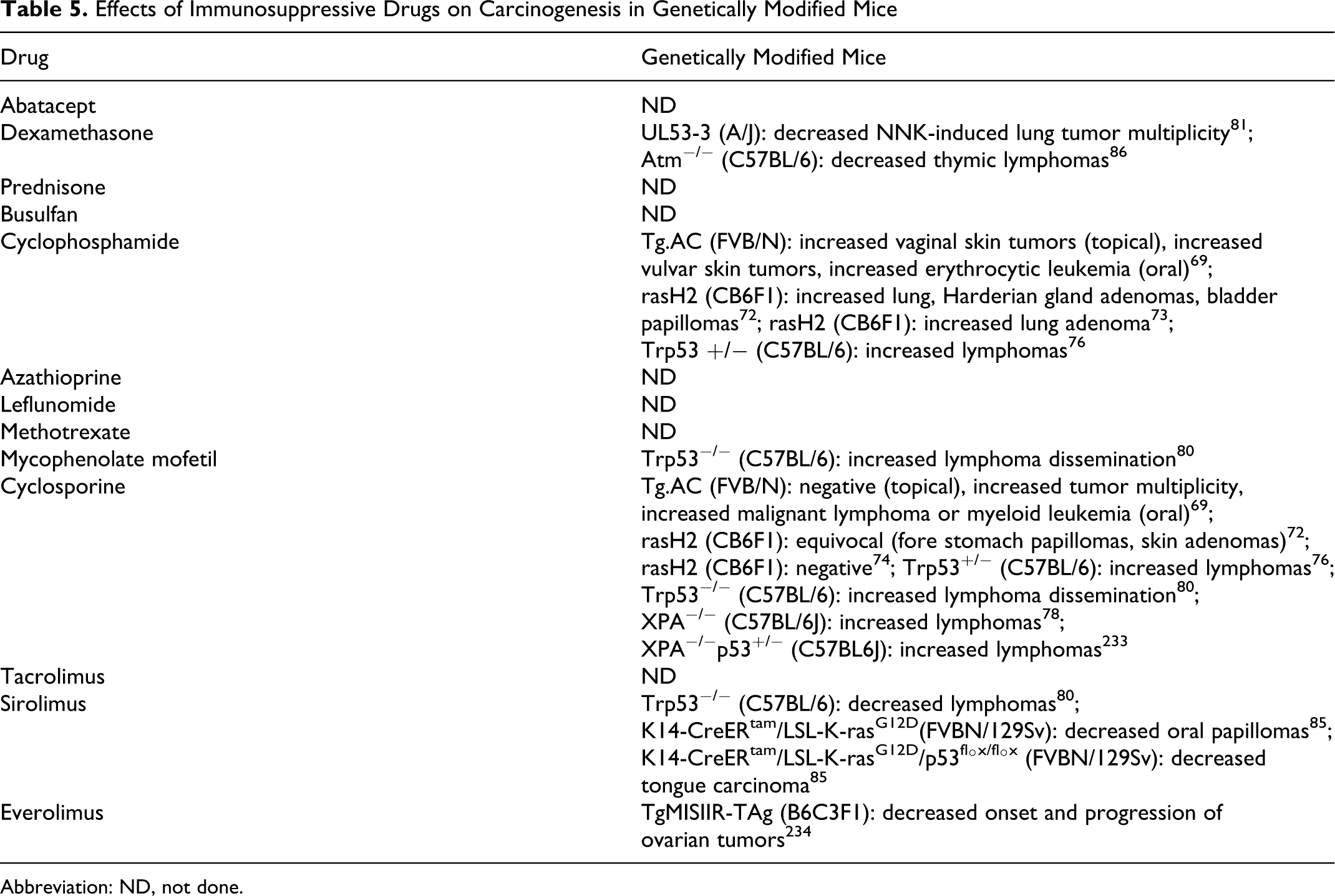
Abbreviation: ND, not done.
Tg-AC (v-Ha-ras)
Based on the knowledge that the c-Ha-ras proto-oncogene is a major player in the initiation of papillomas, a mouse model was developed in which the Ha-ras gene was mutated to be preactivated. 68 In the Tg.AC mouse model (FVB/N strain), the transgene is transcriptionally silent until activated by wounding, UV light, or chemical exposure. Topical or oral administration of carcinogens leads to papillomas or carcinomas of skin or fore stomach, respectively. Typical group sizes for these studies are 15/gender per group and the duration of the study is 6 months. An important feature of the Tg.AC mouse model is that the transgene is not constitutively expressed but appears normal when compared with that of the parental strain. The spontaneous incidence of tumors in this model is very low. 66
As shown in Table 5, only 2 of the drugs of interest, cyclophosphamide and cyclosporine, have been tested in this assay. 69 When administered orally, cyclophosphamide (administered by gavage at doses of 10-60 mg/kg 2×/week starting at 8-9 weeks of age) increased the incidence of skin tumors in females, mostly squamous cell papillomas of the vulva. The mid-dose (30 mg/kg) was associated with an increase in erythrocytic leukemia. Cyclosporine when administered topically (at doses ranging from 0.05 to 0.8 mg/kg per day starting at 8-9 weeks of age) did not increase the number of animals with skin tumors but did produce a slight increase in tumor multiplicity in the high dose (0.8 mg/kg per day) female group only. When administered orally, cyclosporine (at doses ranging from 7.5 to 30 mg/kg per day starting at 8-9 weeks of age) was negative for fore stomach tumors. Interestingly cyclosporine at the highest dose tested (30 mg/kg) did result in an increase in lymphoma and myeloid leukemia in females. This may be serendipitous as these are not the expected tumor types for this model.
rasH2 Mice
The rasH2 mice are hemizygous transgenic mice (C57BL/6J) containing the human prototype c-Ha-ras gene with its own promoter region. 70,71 The transgene encodes a c-H-ras protein, p21, that is integral to cell proliferation. Overexpression of the ras gene can also induce cellular transformations. The ras protein has the potential to act as a potent carcinogen when the ras gene expressing the protein has undergone mutations in certain critical domains. In the rasH2 model, the transgene is constructed by ligating human activated c-H-ras genes with single-point mutations at codons 12 and 61. In this model the life span of the animals is reduced relative to that of wild-type animals due to spontaneous tumor development starting at 6 months of age. The tumor spectrum includes lung adenomas/adenocarcinomas, fore stomach, and skin papillomas, Harderian gland adenomas, liver proliferative lesions, splenic hemangiomas/sarcomas, and lymphomas. The rasH2 model was demonstrated to be particularly useful at detecting genotoxic carcinogens and showed fewer false positives than the 2-year bioassay. 72
Immunosuppressant drugs that have been tested in the rasH2 model include cyclophosphamide and cyclosporine. Mice treated with cyclophosphamide (by oral gavage at doses between 1 and 10 mg/kg 5×/week or 50 and 150 mg/kg 1×/week) developed an increased incidence of lung and Harderian gland adenomas and bladder papillomas. 72 At doses of 10 to 30 mg/kg 2×/week cyclophosphamide produced lung tumors at an incidence similar to that of the nontransgenic CB6Fl mice. 73 Cyclosporine (administered by oral gavage at doses of 10 or 25 mg/kg 5×/week) has been categorized as either negative 74 or equivocal 72 in this assay. The equivocal designation was based on a higher incidence of skin and fore stomach papillomas and skin adenomas in the rasH2 mice versus wild-type mice but not versus vehicle-treated rasH2 mice.
Trp 53 Heterozygous Null (+/−) Mouse
The p53 tumor-suppressor protein functions in normal cells as a transcription factor. Its levels rise in response to DNA damage and/or other stresses and it triggers cellular responses, including cell cycle arrest, senescence, and apoptosis. Mutations in the gene encoding the p53 protein are found in more than half of human cancers. The Trp53 heterozygous null (+/−) mouse model has one copy of the wild-type allele of the p53 tumor-suppressor gene and one copy of a null allele that is not transcribed or translated. 75 Trp53 heterozygotes (+/−) on a C57BL/6 background have a low spontaneous tumor incidence up to 9 months of age but have increased spontaneous tumor rates thereafter, with approximately 50% survival at 18 months. The 3 most common spontaneous tumors in this model are subcutaneous sarcomas, lymphomas, and osteosarcomas. The p53 model appears to be particularly useful as an in vivo test for mutagenic carcinogens. 76
Cyclophosphamide (administered orally at doses of 8-32 mg/kg 2×/week for 26 weeks starting at 6-10 weeks of age) was positive in this model and produced an increased incidence of lymphomas. 76 Cyclosporine was negative when administered orally at doses up to 25 mg/kg 5×/week. 69 When administered orally in the diet at doses ranging from 5 to 60 mg/kg per day cyclosporine was reported to be positive at the highest doses tested (50-60 mg/kg). 76 Interestingly wild-type mice showed a similar pattern of results suggesting that the lymphoma response was independent of the p53 genotype. 76 Also the location of several of the lymphomas in the high-dose cyclosporine treated mice, to small intestine and lung, differed from that of the spontaneous lymphomas which were generally thymic.
XPA−/− and XPA/p 53 +/− Mice
The XPA-deficient mouse model was developed on the basis that patients that lack the XPA protein are highly susceptible to UV-induced skin tumors. The XPA protein is essential for nucleotide excision repair and therefore in the absence of XPA DNA damage is not removed. Homozygous XPA−/− mice have a low background incidence of tumors up to 11 months of age but thereafter develop an increased incidence of lymphomas, bronchiole-alveolar adenomas, and adrenal cortical adenomas. 77 After 18 months, these mice developed hepatocellular adenomas. XPA−/− mice are also highly susceptible to UV-induced SCCs. In an attempt to further increase the selectivity and sensitivity of the model, XPA−/− mice were crossed with heterozygous p53+/− mice. These mice, therefore, have a defect in cell cycle control and apoptosis and a defect in DNA repair. 77 The synergistic effects of these 2 genetic deletions are only apparent after exposure to carcinogens.
Of the immunosuppressive drugs, only cyclosporine (administered orally in the diet at doses of 3-80 mg/kg per day for 9 months starting at 6-8 weeks of age) was tested in the XPA and XPAp53 models. Although these models are intended to detect the carcinogenic effects of genotoxic carcinogens, cyclosporine was positive in both the XPA−/− and XPA/p53+/− models, but only at doses that were at (30 mg/kg per day) or exceeded (80 mg/kg per day) the MTDs. 78 Malignant lymphomas were the major treatment-related tumors. Lymphomas were also present in the wild-type C57BL/6J mice.
Trp 53 Homozygous Null (−/−) Mouse
The Trp53 homozygous null (−/−) mice on a C57BL/6 background that are completely deficient in the p53 protein are not generally used in hazard-identification studies because these mice develop spontaneous tumors, mostly lymphomas and sarcomas, in the first 3 to 6 months of life. 79 Therefore, it is difficult to differentiate treatment-related effects on tumor incidence from background tumor incidence.
A side-by-side comparison of 3 immunosuppressive agents, rapamycin, mycophenolate mofetil, and cyclosporine (administered in the diet starting at 9 weeks of age and continuing through week 29) was conducted. 80 The actual doses administered in this study were not specified but the serum concentrations obtained were reported to be within the immunosuppressive range. This study showed no effect on tumor incidence with cyclosporine or mycophenolate mofetil but showed a reduction in tumor incidence with serolimus. One other interesting finding from this study is that with all 3 immunosuppressive treatments tumors, when they did occur, were manifested systemically more often than in the controls, suggesting that the immune system may play a role in inhibiting the spread of tumors.
UL 53-3 Crossed With A/J Mice
A/J mice that are susceptible to chemically induced lung tumors were crossed with UL53-3 mice that carry 3 copies of a transgene containing a 135Val p53 mutation. Two weekly injections of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) resulted in lung tumors in all of these mice. Treatment of mice with a combination of dexamethasone (0.5 mg/kg, orally) and myo-inositol beginning 7 days before NNK administration and for 20 weeks after NNK administration resulted in a reduction in tumor multiplicity. 81 This inhibition occurred in both p53-deficient and in the wild-type mice. The rationale for combining dexamethasone treatment with myo-inositol was based on previous studies that showed that both dexamethasone and myo-inositol reduced chemical-initiated lung tumor multiplicity in A/J mice and that the effects of the 2 agents were additive. 82 Interestingly in that study dexamethasone reduced tumor multiplicity if administered prior to chemical-induced initiation as well as after chemical-induced initiation.
TgMISIIR-Tag Model
The TgMISIIR-Tag model is a transgenic mouse model of spontaneous epithelial ovarian cancer that was developed by expression of SV40 Tag/tag under transcriptional control of the MISIIR promoter. 83 These mice developed bilateral ovarian carcinoma with complete penetrance surviving approximately 20 weeks. 84
In TgMSIIR-TAg mice administered everolimus (administered orally at a dose of 5 mg/kg for 20 weeks starting at 5 weeks of age), there was a delay in the tumor onset and progression. In addition to the inhibition of mTOR, everolimus inhibited expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinase 2 and inhibited angiogenesis that may have contributed to the observed antitumor effect in this model. 84
K 14-CreERtam/LSL-K-rasG12D and K 14-CreERtam/LSL-K-rasG12D/p53fl°x/fl°x Models
The K14-CreERtam/LSL-K-rasG12D and K14-CreERtam/LSL-K-rasG12D/p53fl°x/fl°x transgenic models were developed as models for head and neck cancer. Mice that express a tamoxifen-inducible Cre recombinase under the control of the cytokeratin 14 (K14) promoter (K14-CreERtam and the ras oncogene) from its own promoter after Cre excision of a stop signal (LSL-K-rasG12D) develop papillomas exclusively in the oral cavity and hyperplasia in the tongue within 1 month of tamoxifen induction. 85 When these mice are crossed with floxed p53 conditional knockout mice, the resulting K14-CreERtam/LSL-K-rasG12D/p53fl°x/fl°x mice, which have combined inactivation of p53 and activation of ras, develop carcinoma of the tongue as early as 2 weeks after tamoxifen induction. In these models, tamoxifen was administered orally daily for 5 days to 1-month-old mice to induce the tumors.
In the K14-CreERtam/LSL-K-rasG12D model, serolimus (administered intraperitoneally [ip] every other day at 10 mg/kg per day starting on day 45 and continuing through day 100-150) prevented the development of oral papillomas and increased their life span. Similarly, in the K14-CreERtam/LSL-K-rasG12D/p53fl°x/fl°x model, serolimus inhibited tongue carcinoma and increased their life span relative to controls. 85
Atm(−/−) Model
Ataxia telangiectasia is a genetic disorder with a mutation (Atm) that results in a number of pathologies and an increased incidence of lymphoreticular tumors of T-cell origin. Atm(−/−) mice recapitulate some of the features of human ataxia telangiectasia including the development of lethal thymic lymphomas by 4 to 5 months of age. 86 Patients and Atm(−/−) mice exhibit elevated DNA synthesis.
Treatment of Atm(−/−) mice with dexamethasone (administered by the ip route at a doses of 1.25 to 10 mg/kg per day for 7-day cycles at 2 to 4 week intervals starting at 2 to 4 weeks of age and continuing for 4 months) showed a reduction in tumor incidence and an increase in survival. 86 In this model, dexamethasone also inhibited DNA synthesis, which is a likely mechanism for the inhibition of the tumor development.
Chemical Co-Carcinogenesis
As described above, carcinogenesis can be considered in 3 steps, initiation, promotion, and progression. In this model, initiation is believed to be the result of heritable changes (mutations) in the genome of a cell. Initiators (or their metabolites) must gain access to the genome and through chemical interactions with DNA result in mutations. 87 In the process of neoplastic transformation, it is considered unlikely that a single mutation can result in the multiplicity of phenotypic changes associated with malignancy. 88,89 For full malignant transformation to occur, a cell must survive in its native milieu, accumulate a number of mutations, and expand its numbers as a nascent neoplastic clone. At this stage, the nascent clone can be considered neoplastic but is not yet malignant. For a malignancy to occur, the nascent clone must continue to accumulate transformed phenotypes so as to include the ability to invade, disseminate, and escape destruction outside its native milieu (ie, in the interstitial space, the lymphatic system, or the blood stream). Genotoxic carcinogens, for example, azathioprine 90 can mediate all 3 steps.
In an experimental setting, chemical carcinogenesis can usually be accomplished only after repeated exposure to the carcinogen, for example, acetylaminofluorine. 91 With some chemical carcinogens, a single exposure is sufficient to cause tumors, for example, vinyl-carbamate. 92 Typically, rodents are exposed to a dose level of carcinogen sufficient to cause tumors and coexposed to the agent to be tested. The assay is conducted over several months with variable numbers (up to 24) of animals per group and the number and size of the resultant tumors measured. 93
As complete carcinogens are tested and the end point measured is tumor incidence, this assay provides a global measure of carcinogenesis, and it is unlikely that the assay will provide insight into the mechanisms by which an immunotoxicant has affected carcinogenesis. Moreover, because many chemical carcinogens are immunosuppressive, for example, dimethylbenz[a]anthracene (DMBA), 94 understanding the effects of an immunotoxicant could be problematic.
As shown in Table 6, there is experience with 9 of the 13 immunosuppressive compounds of interest in chemical carcinogenesis assays. In the interest of space, these results for all 9 drugs will not be described in detail. Examples include cyclosporine, azathioprine, and tacrolimus.
Effects of Immunosuppressive Drugs on Chemical Carcinogenesis
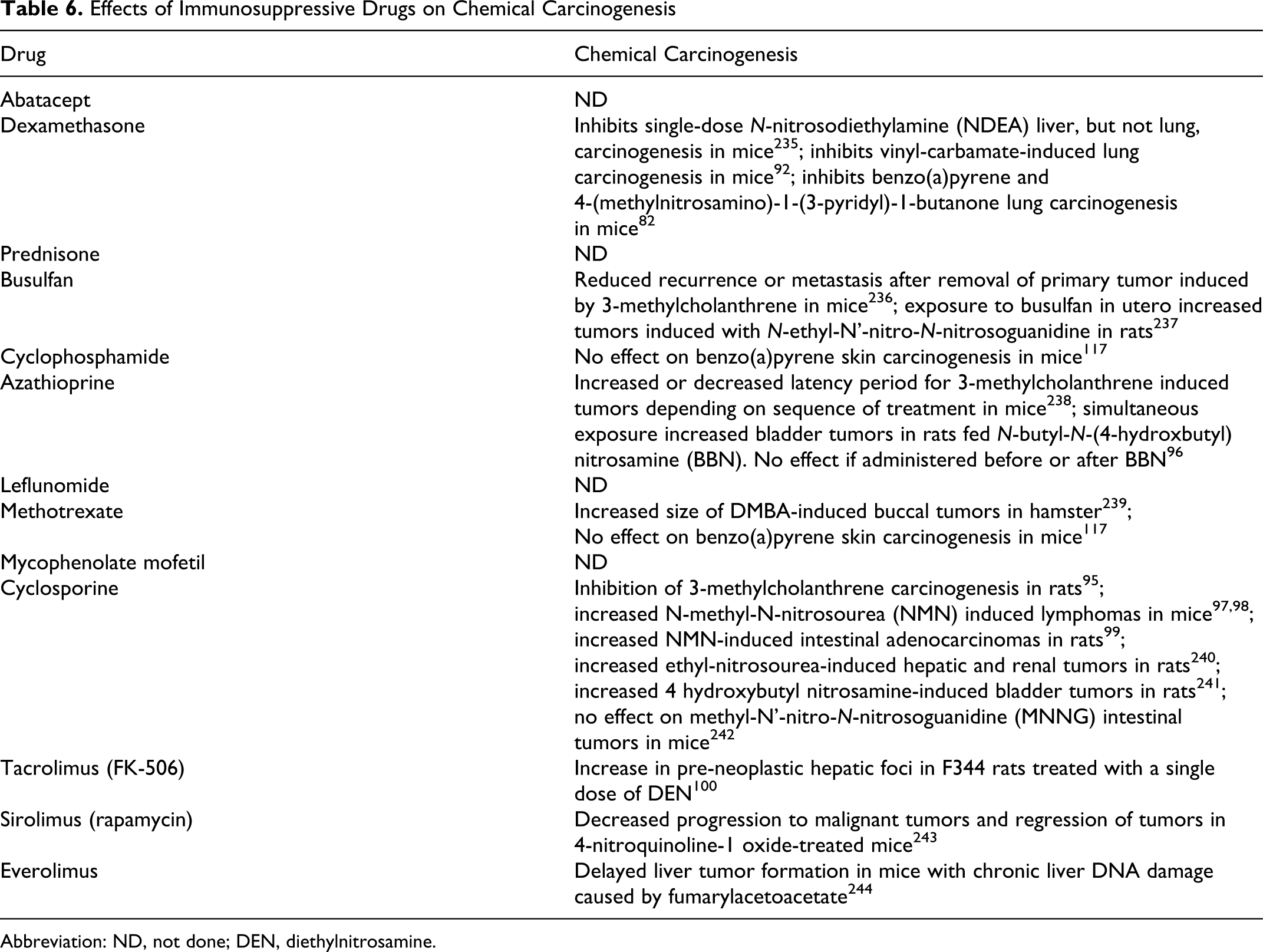
Abbreviation: ND, not done; DEN, diethylnitrosamine.
When cyclosporine was administered subcutaneously to male Sprague Dawley rats at a dose of 2.5 or 5 mg/kg for 2 days prior to and again at 2 weeks (2.5 mg/kg group) or twice weekly for 10 weeks (5 mg/kg group) following a single 5 mg/rat subcutaneous (sc) injection of 3-methycholanthrene (MCA), tumor development was inhibited. In contrast, when treatment with cyclosporine followed exposure to MCA, no effect on tumor incidence was observed. 95 Similarly, simultaneous exposure of male Fischer rats to N-butyl-N-(4-hydroxbutyl) nitrosamine (BBN) at a dose of 0.01% in the drinking water and azathioprine administered by the diet at a concentration of 0.02% for 8 weeks increased bladder tumors, but there was no effect if azathioprine was administered before or after BBN. 96
In other experiments, Swiss Webster mice and Sprague Dawley rats were given the direct-acting carcinogen N-methyl-N-nitrosourea (MNU) ip at a dose of 25 mg/kg followed by treatment with cyclosporine (administered by the diet at a dose up to 0.015% for 20 weeks [mice] or up to 34 weeks [rats]) starting 1 week after MNU treatment. The mice exhibited an increase in the incidence of thymic lymphomas 97,98 and the rats an increased incidence of intestinal adenocarcinomas. 99 Similarly, when the macrolide antibiotic tacrolimus was administered via the diet to F344 rats (calculated dose range of 2.0-2.5 mg/kg for 8 weeks that started after completion of 7 weeks on a choline-deficient diet) following a single exposure to diethyl-nitrosamine, an increase in the number and size of pre-neoplastic hepatic foci was observed. 100
In summary, testing immunosuppressive drugs in chemical carcinogenesis assays gave highly variable results. Depending on the carcinogen, the immunosuppressive drug, the dose levels administered, and the dosing schedule of the carcinogen and immunosuppressive drug, increased, decreased, or no effect on tumor incidence was observed.
Initiation/Promotion Models
As described above, carcinogenesis can be considered in 3 steps, initiation, promotion, and progression. Initiation/promotion models have been developed in an attempt to dissect these stages of carcinogenesis. In these models, promoters inhibit the intracellular mechanisms that can eliminate the nascent (initiated) clone, facilitate accumulation of the mutations necessary for full malignant transformation, or disrupt intercellular signaling. 101 An example is 12-O-tetradecanoylphorbol-13-acetate (TPA, also referred to as phorbol myristate acetate [PMA]).
In an experimental setting, initiation/promotion is usually accomplished by a single exposure to the initiator, for example, DMBA followed by repeated exposure to the promoter, for example, TPA 102 and thereafter co-exposed to the drug to be tested. As many initiators are complete carcinogens, rodents are typically exposed to a dose level insufficient to cause tumors. The assay is conducted over a period up to 6 months using 20 to 30 animals per group (most often), and the number and size of the resultant tumors are measured. 103
As shown in Table 7, there is experience with 5 of the 13 compounds of interest in initiation/promotion assays. In the interest of space, these results will not be described in detail. Examples include dexamethasone and cyclosporine.
Effects of Immunosuppressive Drugs on Initiation/Promotion
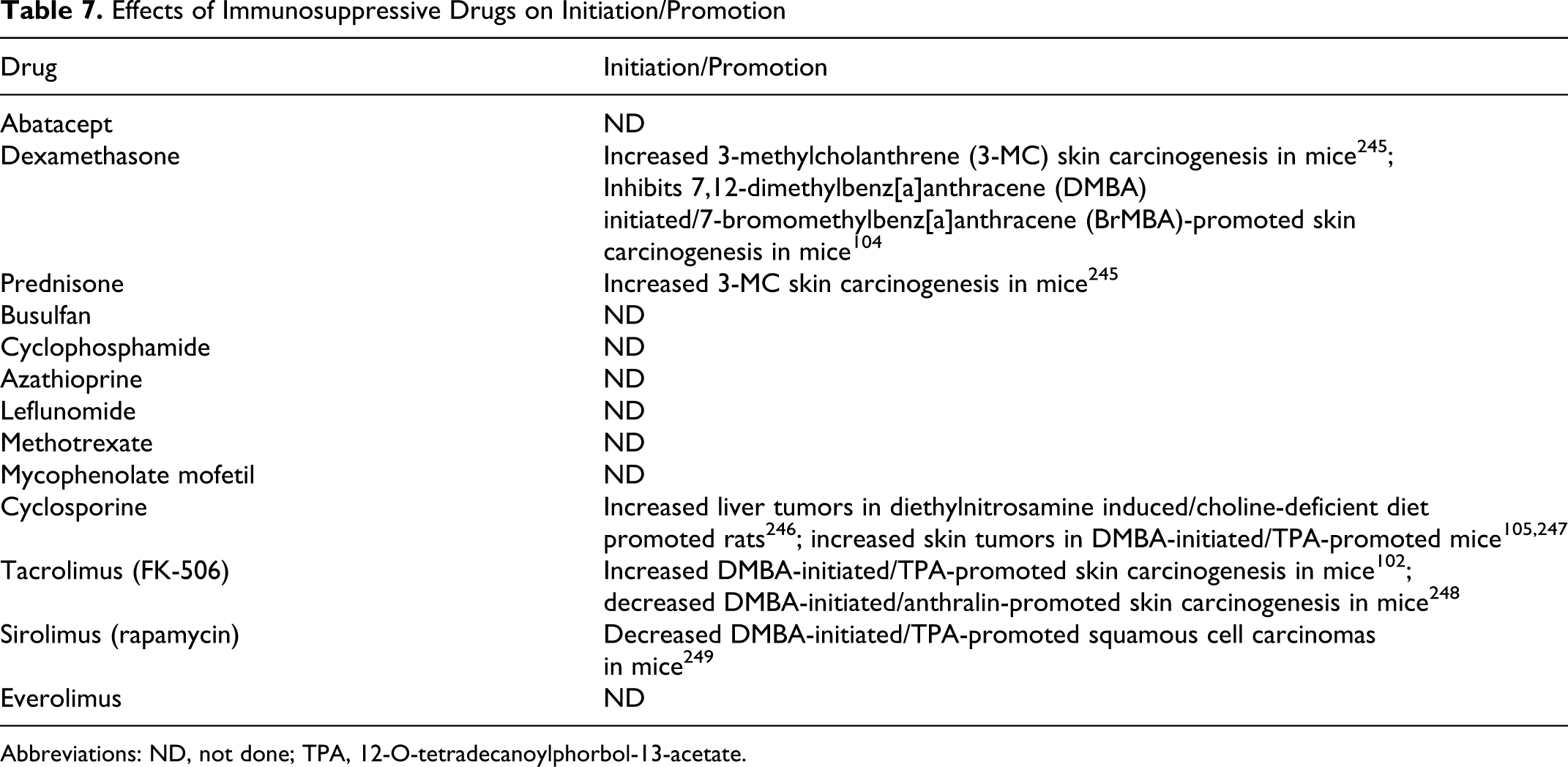
Abbreviations: ND, not done; TPA, 12-O-tetradecanoylphorbol-13-acetate.
In a 7,12-dimethylbenz[a]anthracene (DMBA)-initiated/7-bromomethyl-benz[a]anthracene (BrMBA)-promoted skin carcinogenesis model in CD-1 (Swiss) mice, dexamethasone (administered dermally twice weekly at a dose of 76 nmol starting 14 days after tumor initiation) inhibited tumor formation. 104 In contrast, in a DMBA-initiated/TPA-promoted skin carcinogenesis model in Swiss Webster mice, cyclosporine (administered in the diet at 0.015% for 35 weeks starting on day 3) had no effect on papillomas but increased squamous cell skin tumors. 105
In summary, testing these drugs gave variable results. Depending on the initiator, the promoter, the immunosuppressive drug, the dose levels administered and the dosing schedules, increased, decreased, or no effect on tumor incidence was observed.
Viral Co-Carcinogenesis
As described above, viral carcinogenesis with DNA viruses is believed to play a role in many of the neoplasms associated with immunosuppressive drugs. The mechanisms of viral carcinogenesis with DNA viruses have been described above. In most murine systems, however, retroviruses are the causative agents. With these viruses, viral RNA is reverse transcribed to DNA which becomes incorporated into the genome, disrupting host cell genes and encoding viral proteins that ultimately lead to malignant transformation. 106
In an experimental setting, viral carcinogenesis is accomplished by exposure to the virus followed by repeated exposure to the immunosuppressant drug. Viral exposure can be experimental, that is, a deliberate, timed infection, or the mice can congenitally harbor the virus through vertical transmission, for example, mouse mammary tumor virus. 107 The assay is conducted over many months with 20 animals per group and the number and size of the resultant tumors measured. 108 Because many oncogenic viruses are immunosuppressive, 109 understanding the effects of an immunotoxicant could be problematic.
As shown in Table 8, there is experience with 7 of the 13 compounds of interest in viral carcinogenesis assays. In the interest of space, these results will not be described in detail. Examples include cyclophosphamide, dexamethasone, and cyclosporine.
Effects of Immunosuppressive Drugs on Viral Carcinogenesis

Abbreviation: ND, not done; LDH, lactate dehydrogenase.
In a murine sarcoma virus inoculation model, treatment of B6DF1 mice with cyclophosphamide (administered as a single ip dose of 300 mg/kg) accelerated tumor growth when administered 24 hours prior to virus. 110 Cyclosporine gave conflicting results. Although cyclosporine (administered in the diet at a concentration of 0.015% for up to 35 weeks) increased lymphomas in murine leukemia virus + (AKR) mice, 111 it delayed the onset of leukemia in radiation leukemia virus (RadLV)-inoculated C57Bl/6 mice (administered ip at a dose of 50 mg/kg for 2 weeks starting on week 3 after viral inoculation). 112
In summary, testing these drugs gave variable results. Depending on the virus, the immunosuppressive drug, the dose levels administered, and the dosing schedules, increased, decreased, or no effect on tumor incidence was observed.
Photo Co-Carcinogenesis
UV irradiation is believed to be the primary cause of skin cancers associated with immunosuppressive drugs. The mechanisms of UV carcinogenesis have been described above. In an experimental setting, UV carcinogenesis is accomplished by repeated exposure of hairless or shaved mice to UVA, UVB, or a combination of wavelengths. 113 The assay is typically conducted over an interval spanning 11 to 26 weeks with 20 to 30 animals per group and the number and size of the resultant tumors are measured.
Most of the tumors that arise in rodents following exposure to UV are papillomas rather than the SCCs of relevance to patients receiving immunosuppressive drugs. 113 Although some papillomas can progress to squamous cell tumors, it is necessary to conduct a histologic examination to accurately type the neoplasm. Also, because exposure to UV is immunosuppressive 114 and can enhance tumor growth and metastasis, 115 understanding the effects of an immunosuppressive drug could be problematic.
As shown in Table 9, there is experience with 9 of the 13 compounds of interest in UV carcinogenesis assays. In the interest of space, these results will not be described in detail. Examples include cyclosporine, methotrexate, and sirolimus.
Effects of Immunosuppressive Drugs on Photocarcinogenesis
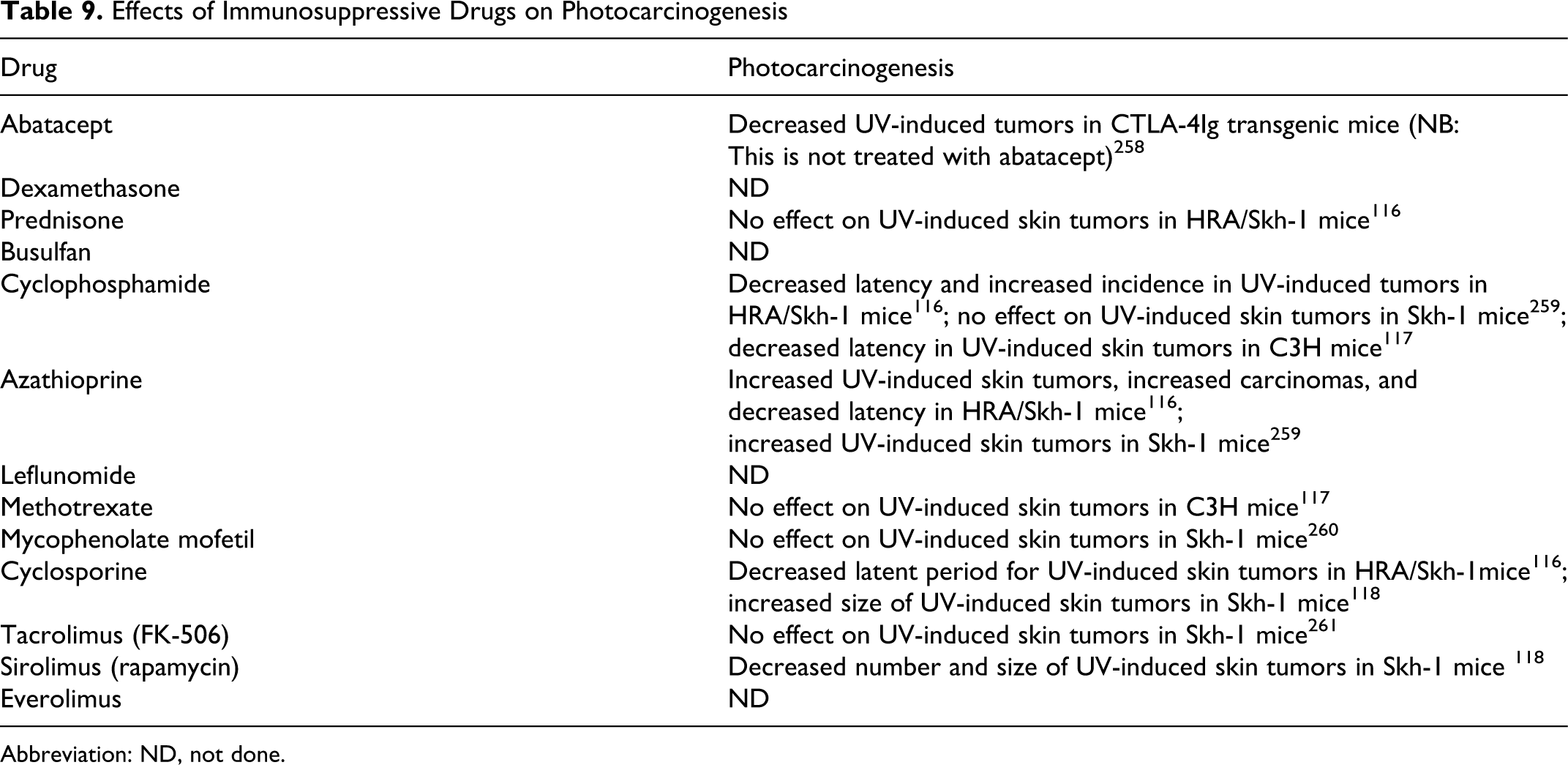
Abbreviation: ND, not done.
In outbred albino HRA/skh-1 mice exposed to 1.72 mW/cm2 UVA and 0.2 mW/cm2 UVB initially for 4.5 minutes, increasing by 20% weekly with a final exposure time of 14 minutes, treatment with cyclosporine (administered by gavage at a dose of 60 mg/kg, 3 times weekly for 10 weeks starting at week 2) decreased latent period for skin tumors. 116 In contrast, in C3Hf/HeN mice exposed to 1.79 × 103 erg/cm2 per UV for 10 minutes, 3 times per week, treatment with methotrexate (administered ip at a dose of 2 mg/kg for 23 weeks starting on day 0) had no effect on UV carcinogenesis. 117 And, in Skh-1 mice exposed to 2240 J/m2 UVB, treatment with sirolimus (administered ip at a dose of 2 mg/kg daily for 9 weeks starting at 15 weeks) decreased the number and size of skin tumors. 118
In summary, testing these drugs gave variable results. Depending on the type of UV, the immunosuppressive drug, the dose levels administered, and the dosing schedules, increased, decreased, or no effect on tumor incidence was observed.
Ionizing Radiation Co-Carcinogenesis
The effects of ionizing radiation (IR) are an extensively studied form of human and experimental carcinogenesis. The primary mechanism of IR carcinogenesis is believed to be mutagenesis through double strand breaks in DNA. However, IR also induces numerous changes in a variety of cellular processes. 119 Although dose−response relationships for low-dose IR in humans are complex, 120 it is clear that IR is a human and rodent carcinogen. Ionizing radiation acts as a complete carcinogen, mediating initiation, promotion, and progression. 121
In an experimental setting, radiation carcinogenesis can be accomplished with either external (eg, gamma rays) or an internal source (eg, by administration of a radionucleotide such as 90 Sr). 122 Typically, animals (usually rats or mice) are exposed to a dose of IR sufficient to cause tumors and co-exposed to the agent to be tested, and the time to macroscopic tumor formation and incidence of tumors at one or more sites are recorded. 123 Leukemogenesis can also be studied. 124 Typically the assay is conducted over many months, with as many as 40 animals per group. 125
As IR is a complete carcinogen and the end points measure tumor incidence, this assay provides a global measure of carcinogenesis, and it is unlikely that the standard assay will provide insight into the mechanisms by which an immunotoxicant has affected carcinogenesis. Moreover, because IR itself is immunosuppressive and can enhance tumor growth and metastasis, 126 understanding the effects of an immunotoxicant could be problematic. That being said, special studies at the molecular level and studies of IR carcinogenesis in knockout mice or transgenic mice may shed light on the mechanism/mechanisms by which an immunotoxicants affects IR carcinogenesis. Such studies are beginning to appear in the literature, 124,127 –129 but to date we have not found examples relevant to immunosuppressive drugs.
As shown in Table 10, there is only very limited experience with the 13 compounds of interest in IR carcinogenesis assays. Only cyclosporine, either alone or in combination with prednisone has been tested. In C57Bl/6 mice exposed to a single dose of 0.1-0.6 Gy, cyclosporine (administered in the diet at 0.015% for 42 weeks starting 1 week after irradiation) increased thymic lymphomas. 130 Similarly, in patched 1 tumor-suppressor heterozygous mice (Ptch1+/−) exposed to 5 Gy 137 Cs radiation and treated with prednisone (administered sc at a dose of 10 mg/kg) or cyclosporine (administered ip at a dose of 50 mg/kg) 3 to 5 times/week for up to 32 weeks increased basal cell carcinomas. 131 These latter results should be interpreted with some caution as Ptch1+/− mice are predisposed to developing basal cell carcinomas rather than squamous cell tumors 132 and Ptch1 plays a critical role in the development of T- and B-cells. 133
Effects of Immunosuppressive Drugs on Ionizing Radiation Carcinogenesis
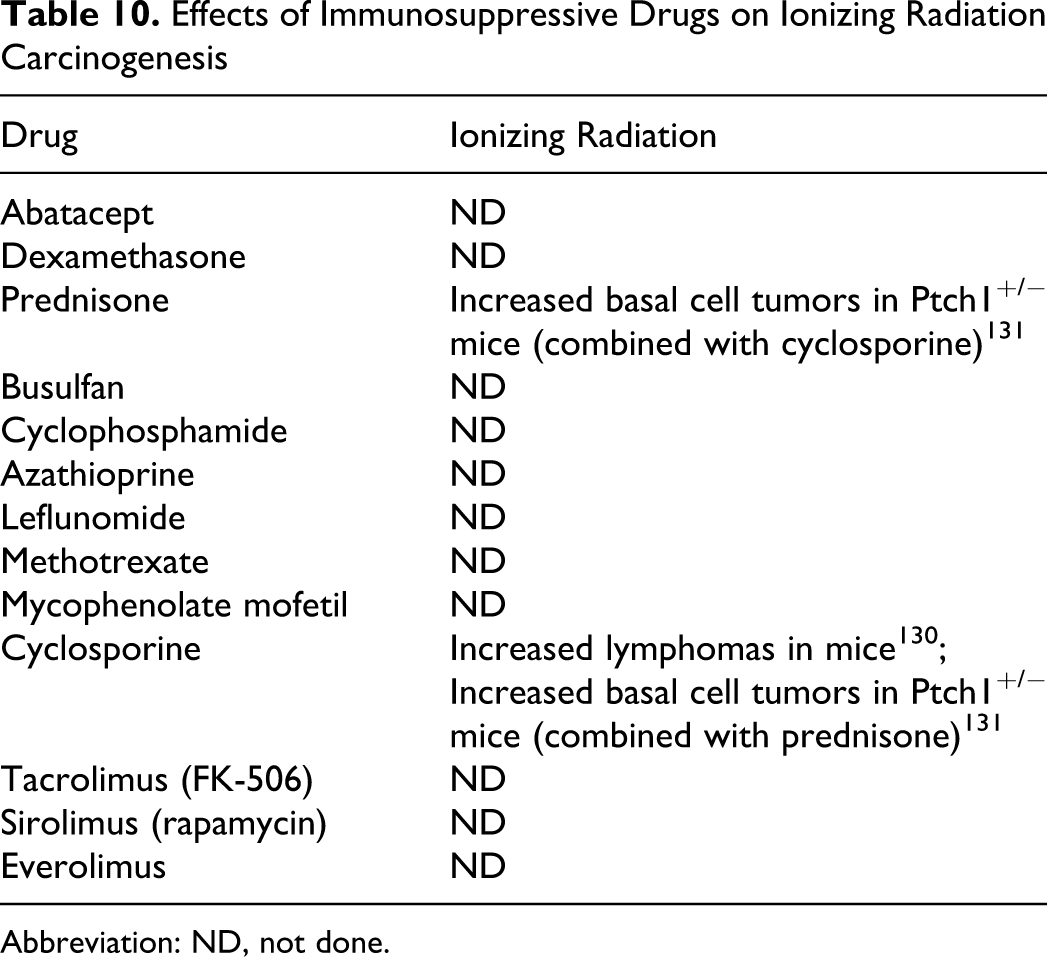
Abbreviation: ND, not done.
Review of Models Using Transplanted Tumor Cells to Evaluate the Impact of Immunosuppressive Drugs on Neoplasia
Transplantation of established tumors in rodents is one of the most widely used techniques in cancer research. A wide variety of models has been described. These include syngeneic models in mice, semi-syngeneic models in rats, and human xenografts in immunosuppressed mice. There are also limited data on human xenografts in immunosuppressed mice reconstituted with human immune cells. Most human tumor types can be modeled in this way, including the B-cell lymphomas and SCCs relevant to patients receiving immunosuppressive drugs. These models have played an invaluable role in drug discovery 134 and in particular, the B16 melanoma and EL-4 T cell lymphoma in syngeneic C57Bl/6 mice, have been used in immunotoxicology. 135
In these models, suspensions of cells or small plugs from established neoplasms (either passed from mouse to mouse or grown in culture) are injected into mice. Cells can be injected into a number of sites or routes and a number of end points measured. These include measuring local growth at the site of transplantation, counting spontaneous metastases from a local injection site, counting experimental metastases from intravenously injected tumor cells (either tail vein [usually leading to pulmonary metastases] or portal vein [leading to hepatic metastases]), and survival (following either local or intravenous injection). Complementary microscopic analysis can be used to gain insight into angiogenesis and growth fraction. Typically, group sizes are between 10 and 20 and the model runs in less than 1 to 2 months.
Because these models use cells from established tumors, they are not useful for studying effects on tumor initiation. However, there is accumulating evidence that initiated cells express the ligand recognized by the effector cells of innate immune system. 136 Moreover, by changing the conditions of the assay, for example, timing of dosing and duration of exposure to the immunosuppressant in relation to injection of tumor cells, the route of administration of the tumor cells and selection of the tumor cell line studied, drug effects on promotion and progression can be modeled. These include effects on survival of the transformed cells in the nascent tumor, local growth, invasion, metastases, angiogenesis, and survival.
As shown in Tables 11 and 12, there is experience with all the 13 drugs of interest in primary transplant models and with 7 of 13 in models of metastasis. In the interest of space, these results will not be described in detail.
Effects of Immunosuppressive Drugs on Transplanted Syngeneic Primary Tumors

Abbreviation: ND, not done.
Effects of Immunosuppressive Drugs on Transplanted Syngeneic Metastases

Abbreviation: ND, not done.
In models of primary tumors, examples include busulfan, dexamethasone, and cyclosporine. In SCH:ARS(ICR)f mice injected ip with 107 Ehrlich ascites carcinoma and treated with busulfan (administered ip, day 1 only, at a dose of 57 mg/kg), no effect on survival was observed. 137 In contrast, in C57Bl/6 mice injected sc with 105 B16 melanoma cells and treated with dexamethasone (administered sc at a dose of 1.25 mg/mouse [3/week] starting 1 day after injecting tumor cells for 30 days), decreased tumor growth was observed. 138 Again in contrast, in another experiment, C57Bl/6 were injected sc with 106 B16 melanoma and treated with cyclosporine (administered ip at a dose of 40 mg/kg per day for 28 days starting 7 days after injection of tumor cells) and increased tumor growth was observed. 139 The same group also reported increased growth of CT26 colon carcinoma tumors in BALB/c mice.
In experimental metastasis models, examples include cyclophosphamide and methotrexate. In B6C3F1 mice treated with cyclophosphamide (administered ip at a dose of 180 mg/kg
In spontaneous metastasis models examples include cyclosporine, methotrexate, and prednisone. In WAG/RIJ (RTIu) rats injected ip with a 12-15 mg plug of CC531 colon tumor and treated with cyclosporine (administered intramuscularly (im) at a dose of 20 mg/kg for 3 days starting on day 4), no effect on locoregional metastases was observed. 143 In contrast, in VM/DK mice injected sc with a suspension of small pieces of VM-M3/Fluc brain tumor and treated with methotrexate (administered ip, weekly, at a dose of 25 mg/kg for 3 weeks starting on day 7), decreased metastases to lung, liver, kidney, spleen, and brain was observed. 144 In contrast, in WHT/Ht mice injected sc with ~1 × 106 NC carcinoma cells (tumors resected on day 14) and treated with prednisone (administered orally at a dose of 0.5 mg/kg for up to 121 days starting on day 0), no effect on local, lymph node or pulmonary metastases was observed. 145
In summary, testing these drugs gave highly variable results. Depending on the type of tumor, the route of tumor cell inoculation, the immunosuppressive drug, the dose levels administered, and the dosing schedules, increased, decreased, or no effect on tumor growth or incidence of metastases was observed.
As shown in Table 13, there is experience with 12 of the 13 drugs of interest in xenograft transplant models. With the exception of cyclosporine that increased experimental metastases of T24 human transitional cell carcinoma in SCID/bg mice 146 and increased tumor growth of human BRO melanoma in normal mice, 147 the results were uniformly negative. Similarly, there is experience with 2 of the 13 drugs of interest in human-reconstituted models and again the results were uniformly negative. 148 These findings call into question the rationale for testing of immunoactive drugs in a setting of preexisting immunosuppression and cast doubt on the use of xenotransplantation models for hazard identification.
Effects of Immunosuppressive Drugs on Transplanted Xenografts in Mice

Abbreviation: ND, not done.
Critical Analysis of the Predictive Value of Preclinical Testing
As we have seen in the preceding sections, when the literature data on the 13 drugs of interest are viewed on a drug-by-drug basis, complex patterns of response are evident. It is also clear that each assay can give conflicting results for a given drug and that some assays have been used with only a few of the drugs of interest. In conducting our analysis of the relative performance of the assays, we have made the assumption that all immunosuppressive drugs should have some form of detrimental effect on immune surveillance against tumors, for example, increased incidence, growth, or metastases. Thus, a “false negative” is a result that fails to detect a detrimental effect.
To facilitate a side-by-side comparison of the performance of the various assays, listed in Table 14 are the assays, their summary performance with immunosuppressive drugs, how many of the 13 drugs of interest have been tested in the assay, and some of the advantages and disadvantages for each assay. Summary performance is defined as the percentage of compounds tested in a given assay that show a detrimental effect on carcinogenesis, tumor growth, or metastasis and thus would predict an increased risk of neoplasia. So long as a drug was shown to have a detrimental effect on tumors in at least 1 experiment, the assay was scored as positive. It should be noted that false negatives (a drug positive in 1 or more other assays or experiments that is negative in a particular experiment) are not captured in this measure of performance. Thus, the table shows each assay in its most “positive” light.
Despite viewing the assays in their most positive light, it is clear that there is a high rate of false negatives. Reasons for this high rate include the reliance on adventitious tumor initiation or promotion in some models; the dose levels tested; the pharmacokinetic behavior of the drug and the dosing paradigm tested; and nonlinear dose−response relationships for effects on immune function. Moreover, blocking angiogenesis immunosuppressive drugs can impede development of macroscopic (or even microscopic) tumors. Furthermore, if the immunosuppressive drug exhibits cytotoxic activity, for example, methotrexate, azathioprine, and busulfan, or lympholytic activity, for example, dexamethasone and prednisone, it can have “therapeutic” activity against nascent tumors. It should, thus, not be surprising that the results of in vivo testing can be complex and sometimes contradictory.
This is particularly true for the 2-year bioassay. By experimental design, to be positive in a 2-year bioassay the test article must increase the incidence of tumors detected or shorten the “latent” period (the time to finding a mass or the time to die from a tumor). 56 For nascent autochthonous tumors, should the balance between blocking immune surveillance and blocking tumor progression favor blocking progression, an immunosuppressive drug would be expected to block or slow formation of detectable lesions and thus lead to a false negative.
As shown in Table 14, 5 of the assays (neonatal mice, genetically modified mice, initiation/promotion, IR, and human reconstituted) have been inadequately “validated” (in the context of this limited analysis), for example, <50% of the 13 drugs tested. We consider this too low to allow a meaningful comparison of their performance as predictors of an increased risk of neoplasia, and these assays will not be considered further.
Summary Performance and Critical Analysis of Preclinical Models

a Performance = Percentage of immunosuppressive drugs shown to increase tumor end point (number of immunosuppressive drugs tested).
Seven assay systems have been applied to >50% of the 13 drugs of interest: 2-year bioassays; chemical carcinogenesis; photocarcinogenesis; xenotransplantation models; primary syngeneic tumor transplantation models, and transplanted syngeneic metastasis models. Of these assays, the 2 worst performers in detecting a detrimental effect were the xenotransplantation and the photocarcinogenesis assays with only 11% and 22% accuracy, respectively. Despite their popularity, based on their performance to date, it seems unlikely that these models will prove generally useful in risk assessment for immunosuppressive drugs.
If we change our analysis to accentuate the positive, of the 5 remaining assays, the 2-year bioassay and primary syngeneic tumor assays gave similar performances of 46% and 42% accuracy and the 3 best performing assays, chemical carcinogenesis, viral carcinogenesis, and syngeneic metastases gave 56%, 71%, and 64% accuracy, respectively. Moreover, when the performance data for syngeneic primary tumors and metastases are combined, the accuracy of detecting a detrimental impact on immnosurveillance rises to 75%. It should be kept in mind, however, that this value for percentage accuracy reflects overall performance and not the predictive value of any 1 tumor cell line or model system. Importantly, the dose and dosing regimen can play a critical role in the outcome of the assay. As shown in Tables 11 and 12, there are numerous examples where assays using transplanted syngeneic tumors failed to detect an adverse effect on immune surveillance. This can be of particular importance as the test article is cytotoxic or can inhibit tumor growth and progression through noncytotoxic mechanisms. It may be necessary to use a number of tumor cell lines, doses, and dosing regimens to fully understand the potential of a novel agent to influence immunosurveillance. The need to conduct several models with transplanted tumor cells should not be viewed, however, as either too onerous or costly. Transplanted syngeneic tumors grow relatively quickly and experiments can be completed within 1 month. Moreover, robust data sets can be acquired with relatively small group sizes (≤20/group) compared with other experimental systems and the experiments can be repeated with relative ease.
Taking together the performance data, the advantages and disadvantages of the various assays, and their costs in terms of time to complete the assay, drug requirements, animal usage, and labor intensity, what emerges from our critical analysis is that a combination of transplanted primary tumor and metastasis assays conducted with several tumor types and run at doses and dosing regimens appropriate to the mechanism of action of the test article may provide the highest predictive value for an increased risk of neoplasia in patients receiving immunosuppressive drugs.
View to the Future
In developing a view to the future, we should define the purpose of preclinical testing of immunosuppressive drugs in regard to an increased risk of neoplasia. A model commonly used in risk assessment breaks the process into parts: hazard identification; dose−response relationships, exposure evaluation; risk characterization and pharmacovigilance (postmarketing drug surveillance). 149 In an ideal world, not only would preclinical testing identify and characterize the hazard, it would define the risk posed to humans receiving therapeutic doses of the drug.
Unfortunately, this is not the case. As reviewed by Olson et al, 150 preclinical testing in rodents alone identified only 43% of human toxicities identified in the course of clinical trials of 150 different compounds. Because rodents can predict only a fraction of human toxicity in general and because rodents are the only practical test system to address carcinogenesis, given the differences in the structure, development, and function 151 –153 of the immune systems among rodents and humans, it is not surprising that they have limited value in predicting a toxicity as subtle as the increase in the risk of neoplasia posed by immunosuppression. Moreover, the usual paradigm used in risk assessment includes incorporation of exposure margins and identification of a no adverse effect level (NOAEL). 154 Because the effects on risk of neoplasia are likely directly related to the desired pharmacologic activity of immunosuppressive drugs, 155,156 and because drug levels in plasma may not correlate with the degree of immunosuppression, 157 there may be little advantage in incorporating exposure margins into risk assessment for patients receiving therapeutic doses of this class of drug. The issue then is not risk avoidance. Rather, it is one of hazard identification, characterization, risk communication, and risk management. 158
As mentioned above, the original immunosuppressive drugs were antineoplastics recast as antirejection drugs. These agents can be thought of as “nonselective” immunosuppressive drugs, that is, they inhibit the activity of numerous effector arms of the immune system. More recently, interest has shifted to selective immunosuppressive drugs. Although some may be recast antineoplastic agents, for example, the use of rituximab (an anti-CD20 monoclonal antibody originally approved for treating B-cell lymphomas) in rheumatoid arthritis, 159 many have been designed and developed specifically as selective immunosuppressive agents. More recently, a variety of therapeutic proteins, especially monoclonal antibodies, have been approved for clinical use. 160 Many of these newer agents are either pharmacologically inactive or immunogenic (or both) in rodents. Thus, the traditional approach to carcinogenicity testing, that is, the rodent 2-year bioassay, is not feasible, and alternate approaches must be availed upon.
Many of the emerging selective immunosuppressive therapeutic proteins are pharmacologically active in Old World nonhuman primates (NHPs), and rhesus and cynomolgus macaques are commonly used in general toxicity and embryo-fetal development studies with these agents. 161,162 New world NHPs, for example, tamarins, marmosets, and squirrel monkeys, are evolutionarily more distant to humans, 163 and therapeutic proteins directed toward human targets are often not pharmacologically active in these species.
Macaques are not an appropriate species for carcinogenicity testing because their long life span (>20 years) makes the conduct of life-time studies impractical. Increasing the duration of a chronic toxicity study in macaques from 6 months to 9 or 12 months is an insignificant difference in the total duration of the life span of most NHPs. In addition the incidence of spontaneous tumors in NHP is quite low. Over a 15-year period at a major contract research organization, tumors were observed in <1% of necropsies of cynomolgus macaques. 164 Nonhuman primates are susceptible to PTLDs, 165,166 but the incidence is low (~5%). Similarly, NHPs are susceptible to tumor induction by IR 167,168 but the onset is delayed. Moreover, NHP show limited sensitivity to human carcinogens. Although some chemical carcinogens, for example, diethylnitrosamine 169 and cytotoxic chemotherapeutics for example, procarbazine, have been reported to cause tumors in NHP, 170 most have little or no effect. For example, over a 34-year period the US National Cancer Institute carried out an investigation of 37 chemicals in long-term studies in NHP. 171,172 Two of the immunosuppressant drugs of interest cyclophosphamide and azathioprine were included in this program. The results from the cyclophosphamide study were considered to be inconclusive. Of the 23 NHPs treated with cyclophosphamide, 2 developed malignant tumors, 1 with a small transitional cell carcinoma of the urinary bladder after 3.3 years of treatment and 1 an anaplastic carcinoma involving the heart and lungs after 13 years of treatment. Similarly, of the 29 NHPs treated with azathioprine, only 1 case of lymphocytic leukemia was detected after 15 years of treatment. These studies demonstrate that even when nonselective immune suppressant drugs (that are also genotoxic and rodent carcinogens) are administered to NHPs, tumor incidence is low, is indistinguishable from background incidence in age- and strain-matched monkeys, and prolonged dosing periods (3-15 years) are required before tumors become evident. These studies also reinforce the premise that increasing the duration of a chronic toxicity study by an additional 3 or 6 months in NHPs is unlikely to detect a carcinogenicity risk.
There are very few reports of studies with transplanted tumors in NHP. Johnson et al 173 showed that allogenic LLC-MK2 cells produced only occasional microscopic tumors in immunosuppressed NHPs, and Petricciani and Gillette showed that strong immunosuppression including anti-thymocyte globulin was required to support the growth of transplanted tumor cells. 174 Dexamethasone has been shown to have a beneficial effect on brain edema in a human choriocarcinoma xenograft implanted in the brain of rhesus macaques, 175 but this model has been shown to be unresponsive to chemotherapy. 176
Because of the resistance of NHPs to carcinogenesis, the lack of relevant tumor models, the lack of relevant validation for immunosuppressant drugs, and the long life span of NHPs, we consider that the risk assessment for carcinogenicity in NHP is not feasible.
The testing of a surrogate molecule that is the rodent homolog of the human protein in the rodent 2-year bioassay is also not an appropriate approach for the evaluation of the carcinogenicity potential of immunosuppressive drugs. This conclusion is first and most importantly based on the fact that the 2-year bioassay is a poor predictor of carcinogenicity risk associated with immunosuppression. Second, a surrogate molecule is not the same molecule as the clinical product. 177 Surrogate molecules can, however, provide useful information on mechanisms associated with desired and potentially undesired effects of stimulating or inhibiting the target pathway. 161,178 As discussed above, a combination of transplanted tumor models of primary tumors and metastases in rodents showed the greatest predictive value of an increased risk of neoplasia with immunosuppressive therapy. Thus, for selective immunosuppressive drugs that are pharmacologically active only in humans and NHPs, studies with a surrogate may be the only way of generating pharmacologic data on in vivo immunosurveillance, should such data be deemed necessary. However, creation of a surrogate is no simple task. As recently reviewed, 177,179 to be of value in hazard identification and risk assessment, such an agent must express pharmacologic activity in rodents similar to that of the human protein in humans. However, as most if not all targets for selective immunosuppression undergo study in a basic research setting, surrogates are often already available at the onset of drug development, and these same surrogates can be used to probe potential mechanisms that may affect tumorigenicity.
Conclusions
In conclusion, immunosuppressive drugs, by virtue of their intended pharmacologic activity may pose a hazard for developing certain tumor types, especially lymphomas and skin cancer, and the 2-year bioassay performs poorly in identifying this hazard. A potentially viable alternative is to conduct studies with transplanted tumors in syngeneic mice. Although conducting such studies to identify this hazard is relatively straightforward, determining the relative risk to patients is likely to remain problematic for both small molecule xenobiotics and therapeutic proteins. Moreover, because the hazard is intimately linked with the desired pharmacologic activity of immunosuppressive drugs, traditional risk assessment approaches that are used with most pharmaceuticals based on exposure margins are unlikely to be of much value. One alternative option may be to classify and label immunosuppressive drugs based on their mechanism of action and not conduct any additional experiments to specifically address immune surveillance. However, should a sponsor choose not to conduct this type of work, regulatory agencies will have to rely on the existing published literature, without access to detailed protocols, stability data on the test article, toxicokinetics, or any raw data. This literature, may be fragmentary and contradictory, and in the case of knockout mice provide no dose−response information and possibly limited information on compensatory changes in immune function (eg, see the recently issued Food and Drug Administration [FDA] pharmacology review of tocilizumab 180 ). In any event, a prospective pharmacovigilance program to monitor carcinogenic risk is likely to play a critical role in ensuring patient safety during the clinical development program and as a subsequent postmarketing commitment.
Footnotes
Notes
All authors (except Jacques Descotes) are employees of Centocor R&D and hold an equity stake in Johnson & Johnson (J&J), the parent company of Centocor R&D. J&J does not have a commercial interest in any of the drugs reviewed in this article. Jacques Descotes is professor of medical pharmacology at Claude Bernard University (Lyon, France) and head of the Poison Center and Pharmacovigilance Department from Lyon University Hospitals. He has no conflict of interest with any of the drugs reviewed in this article.
This work was supported by Centocor R&D. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.