
Abstract
Ketorolac tromethamine is a potent analgesic and moderately effective anti-inflammatory drug approved for treatment of moderately severe acute pain as an intravenous/intramuscular injectable solution and an oral tablet. ROXRO PHARMA, Inc has developed an intranasal formulation, SPRIX, that delivers the drug with a similar pharmacokinetic profile to that obtained with intramuscular administration. Local tolerance and systemic toxicology studies were performed in rats and rabbits and showed that intranasal administration of SPRIX exhibits toxicity similar to that of other routes of administration and does not result in any adverse effects on the nasal passage and upper and lower respiratory system.
Introduction
Ketorolac tromethamine [(±)-5-benzoyl-2,3-dihydro-1H-pyrrolizine-1-carboxylic acid, 2-amino-2-(hydroxymethyl)-1,3-propanediol (1:1)] is a nonsteroidal anti-inflammatory drug (NSAID) that is approved for the short-term management of severe acute pain that requires analgesia at the opioid level. 1 The CAS registry number of ketorolac tromethamine is 74103-07-4 and its molecular structure is presented in Figure 1.
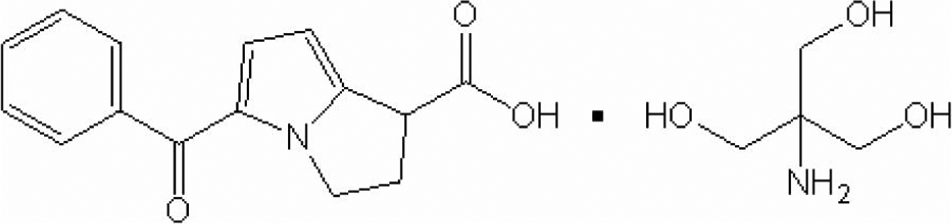
Molecular structure of ketorolac tromethamine.
Ketorolac exhibits anti-inflammatory, analgesic, and antipyretic activity. Like other NSAIDs, its therapeutic activities are produced through inhibition of cyclooxygenase (COX) enzymes, the enzymes that metabolize arachidonic acid to prostaglandins, prostacyclin, and thromboxane. When prostaglandins are released from injured tissues, they increase the sensitization of afferent nerve endings that originate at sites of injury and inflammation. Nonsteroidal anti-inflammatory drugs decrease the hyperalgesic settings of these nerve endings, and thus diminish pain. All NSAIDs share analgesic, anti-inflammatory, and antipyretic activities, but each agent has its own efficacy and safety profile; some NSAIDs have predominantly analgesic activity, whereas others have stronger anti-inflammatory effects.
Ketorolac tromethamine is a potent analgesic and a moderately effective anti-inflammatory drug. It is used for the symptomatic relief of moderate-to-severe postoperative pain, including that associated with abdominal, gynecologic, oral, orthopedic, or urologic surgery. It has also been used for relief of acute renal colic pain associated with trauma and visceral pain associated with cancer. 2 The most common side effects associated with short-term administration of ketorolac tromethamine are gastrointestinal effects similar to those seen with other prostaglandin inhibitors, which are usually mild to moderate and occur in approximately 39% of patients. 2,3 Ketorolac tromethamine does not appear to cause physical dependence or tolerance, unlike opiate analgesics. Additionally, ketorolac tromethamine lacks the respiratory depressant effects of opiate analgesics. 2
Ketorolac tromethamine was first approved for marketing in the United States in 1989 as an intravenous/intramuscular injection formulation at doses up to 120 mg/d. It is the only NSAID available for parenteral administration in the United States. An oral tablet formulation was approved in 1991 at doses up to 40 mg/day for use after parenteral treatment.
Ketorolac tromethamine is highly water soluble, and its solubility profile, unique among NSAIDs, permits the preparation of a solution for intranasal use. ROXRO PHARMA, Inc (Menlo Park, CA) has developed a ketorolac tromethamine nasal spray, SPRIX (tested as ROX-888), with a formulation that delivers the drug with a similar pharmacokinetic profile to that obtained with intramuscular administration without the need for painful injections or intravenous access. SPRIX also avoids the oral route, benefiting patients who are nauseated or unable to take oral medications. In addition, because administration by intravenous and intramuscular injection cannot be continued after the patient is discharged, SPRIX provides a more convenient form of drug delivery in ambulatory settings. Single and multiple doses of SPRIX are well tolerated by patients receiving a dose of 31.5 mg following major surgical procedures and can reduce opioid use and related side effects. 4 –6 The primary adverse event reported by both healthy volunteers and surgery patients was mild, transient nasal irritation. 4 –7
The nonclinical safety profile of intranasally administered ketorolac tromethamine (SPRIX) is described in this article. Information on the toxicity profile of ketorolac tromethamine via the intranasal route of administration has not been published previously and was the subject of nonclinical testing to support the safe use of this route of administration in the clinic. The nonclinical testing strategy for SPRIX included local tolerance and systemic toxicology studies using intranasal administration for 5 and 28 days in rats and 14 days in rabbits. Results of these studies are reported here for the first time.
Materials and Methods
The rabbit study was conducted at SRI International (Menlo Park, California). SRI International is fully accredited with the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. The study was conducted under US Food and Drug Administration (FDA) Good Laboratory Practice standards (21 CFR 58) for preclinical laboratory studies. All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). The rat studies were conducted at Charles River Laboratories (Edinburgh, UK). The facility operates under a Certificate of Designation from the Home Office, the relevant governing body for the United Kingdom, under the terms and controls of the Animals (Scientific Procedures) Act 1986. As part of the certification process, the facility is inspected regularly by the Home Office to standards that are equivalent to AAALAC accreditation. All procedures involving animals were reviewed and approved by the Animal Welfare and Ethics Committee and the Named Animal Care and Welfare Officer, under regulations relevant to the United Kingdom and comparable to the IACUC in the United States.
Test Article
Rat studies
Two local tolerance studies were conducted in rats; in one study, the dosing duration was 5 days and in the other study, the dosing duration was 28 days.
For the 5-day study, the intranasal ketorolac formulation was manufactured by West Drug Delivery and supplied by West Pharmaceutical Services (Nottingham, UK) as ready-to-use 10% (batch no ROX2/021/I) and 22.5% (batch no ROX2/022/I) solutions. A vehicle-only solution (batch no ROX2/023/I) was also manufactured by West Drug Delivery and supplied by West Pharmaceutical Services as a placebo control. The vehicle contained disodium EDTA, potassium dihydrogen phosphate, and water for injection. The pH of SPRIX and vehicle control formulations was adjusted to 7.2 using 2 mol/L sodium hydroxide. All test material solutions were stored at 2°C to 8°C and protected from light. All test material solutions were analyzed to confirm the intranasal ketorolac formulation content using standard analytical techniques.
For the 28-day study, the intranasal ketorolac formulation was supplied by Archimedes Development Limited (Nottingham, UK) as a white crystalline powder (batch no 10880410007) and was stored at ambient temperature, protected from light. Test solutions of SPRIX were formulated weekly. The vehicle contained disodium EDTA, 0.2 mol/L potassium dihydrogen phosphate, and water for injection. The pH of SPRIX and vehicle control formulations was adjusted to 7.2 using 2 mol/L sodium hydroxide. Standard analytical techniques were used to analyze samples of each formulated solution for ketorolac tromethamine concentration and to determine the stability under conditions of use.
Rabbit study
The intranasal ketorolac formulation was manufactured by Spectrum Laboratory Products, Inc, (Gardena, CA) as a white powder (lot no QFO345) and was stored at room temperature, protected from light. The vehicle was a 0.02% solution of disodium EDTA in phosphate buffered saline with pH adjusted to 6.5 with 0.1 N phosphoric acid. A stock solution of the vehicle was used to prepare 7.5%, 15%, and 22% active ingredient dosing solutions. Dosing solutions were prepared 3 days prior to dose administration. The pH of the dosing solutions was measured to be between 6.15 and 6.51. Concentration verification was performed on samples of the dosing solutions prior to the start of dosing and following the end of dosing. The dosing solutions remained within ±5% of the nominal concentration during the course of the study.
Test Systems
Rats
For the 5-day study, young adult male Sprague Dawley (Crl:CD [SD] IGS BR) rats were supplied by Charles River (UK) Limited, Margate, UK. The animals were assigned to 2 phases of study: Phase 1 was designed to identify the maximum tolerated dose (MTD) and phase 2 was designed to assess toxicity after 5 days of repeat dosing. The animals were 9 to 11 weeks old at the initiation of dosing. Animals assigned to phase 1 weighed 232 to 321 g, and animals assigned to phase 2 weighed 275 to 382 g. Within 24 hours of arrival, all animals were given a clinical examination to ensure that each animal assigned to the study was healthy and fit for use on the study. Following arrival at the testing facility, the animals were acclimated for a period of at least 2 weeks before the initiation of dosing.
Rats were housed 2 or 3 per cage in suspended polypropylene cages with solid bottoms and stainless steel mesh tops. All cages contained wood shavings. Animal rooms were set to maintain the temperature at 20°C ± 2°C, relative humidity at 50% ± 15%, a minimum of 15 air changes per hour, and light hours normally from 0700 to 1900. The animals had access to food (Rat and Mouse [modified] No 1 Diet SQC Expanded, supplied by Special Diets Services) and water ad libitum, except during dosing.
For the 28-day study, male and female young adult Sprague Dawley (Crl:CD [SD] IGS BR) rats (64/gender) were supplied by Charles River (UK) Limited, Margate, UK. At the initiation of dosing, the males weighed 211 to 332 g and the females weighed 170 to 232 g. Within 24 hours of arrival, all animals were given a clinical examination to ensure that each animal assigned to the study was considered healthy and fit for use on the study. Following arrival at the testing facility, the animals were acclimated for a period of at least 2 weeks before initiation of dosing. Rats were acclimated to the dosing procedure by handling to simulate the dosing procedure on days −5 and −4, and then sham dosed with 10 μL saline per nostril once per day on days −3, −2, and −1.
Rats were housed up to 5 per cage in suspended polypropylene cages with solid bottoms and stainless steel mesh tops. All cages contained wood shavings. Animal rooms were set to maintain the temperature at 20°C ± 2°C, relative humidity at 50% ± 15%, a minimum of 15 air changes per hour, and light hours normally from 0700 to 1900. The animals had access to food (Rat and Mouse [modified] No 1 Diet SQC Expanded, supplied by Special Diets Services) and water ad libitum, except during dosing and urine collection.
Rabbits
Male and female young adult New Zealand white rabbits were supplied by Covance Research Products. Twenty-five animals per gender were assigned to the study. At the initiation of dosing, the animals weighed 2.1 to 3.0 kg. Following arrival at the testing facility, animals were quarantined for 6 days.
Animals were individually housed in suspended stainless steel cages. Animal rooms were set to maintain the temperature at 21°C to 22°C, relative humidity at 30% to 70%, at least 10 air changes per hour, and a 12-hour light/12-hour dark cycle. The animals had access to food (Purina Certified Rabbit Chow, #5325) and purified tap water ad libitum.
Five-Day Local Tolerance Study in Rats
This study was consisted of 2 phases. The objective of phase 1 was to identify the MTD of intranasally administered SPRIX in rats. The objective of phase 2 was to evaluate the potential for drug-induced toxicity in the upper and lower respiratory tract and gastrointestinal (GI) tract following repeat intranasal dosing for 5 days at doses lower than the MTD. This study was conducted according to GLP (Good Laboratory Practice) standards. The upper and lower respiratory tract were targeted because of the route of administration and the GI tract was targeted because of the known toxicity profile with ketorolac. 8 Results of this study were used to select doses for the subsequent 28-day intranasal study in rats.
A total of 6 rats each were assigned to 4 dose groups for phase 1, and 5 additional rats each were assigned to 4 dose groups for phase 2. Animals were stratified by body weight and randomly assigned to control and treated groups. For both study phases, rats were dosed intranasally with SPRIX solutions using an automatic pipette 3 times per day at 3-hour intervals. The target dose for each group was achieved by varying the dose volume (20-40 µL) per dose session each day and the concentrations of ketorolac in the formulation (10% or 22.5%).
In phase 1, the target doses were 24, 36, 54, and 108 mg/kg per d, corresponding to nasal surface area doses of 0.58, 0.87, 1.30, and 2.60 mg/cm 2 per d, respectively, based on a nasal surface area of 10.4 cm 2 for a 0.250 kg rat 9 (Table 1). The estimated achieved doses, based on test item use, were 33.6, 53.1, 66.3, and 113.0 mg/kg perday, corresponding to nasal surface area doses of 0.8, 1.3, 1.6, and 2.7 mg/cm2, respectively.
Study Design for 5-Day Study in Rats: Phase 1
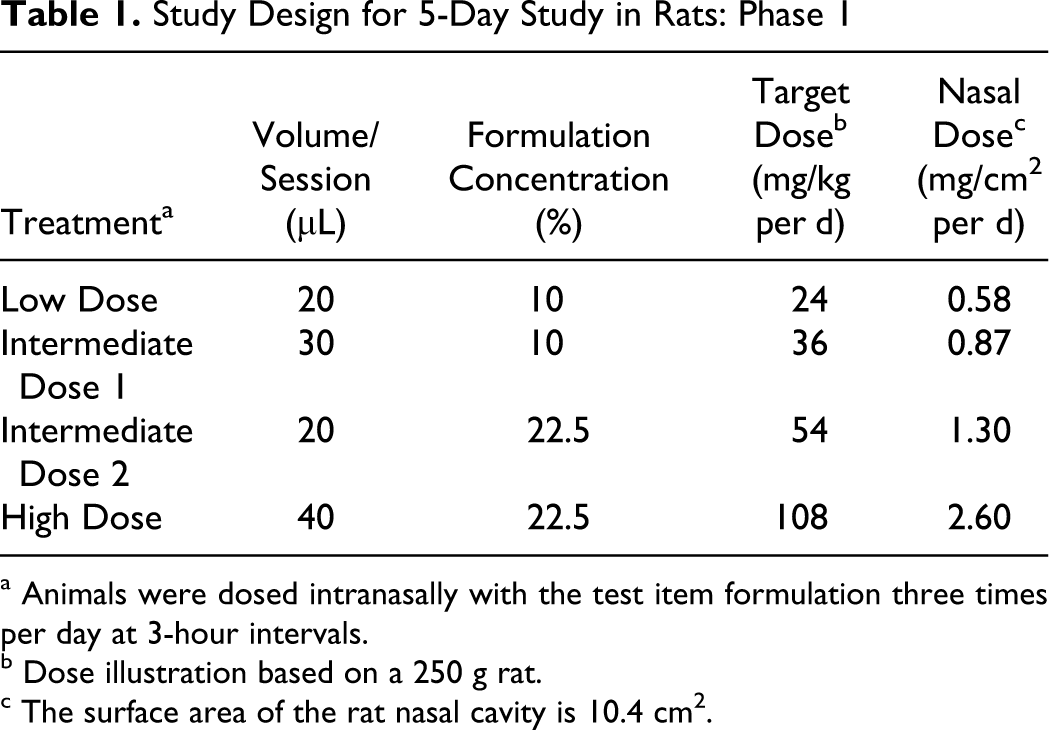
a Animals were dosed intranasally with the test item formulation three times per day at 3-hour intervals.
b Dose illustration based on a 250 g rat.
c The surface area of the rat nasal cavity is 10.4 cm2.
In phase 2, the target doses were 24, 36, and 54 mg/kg per d for the test-article treated groups, corresponding to nasal surface area doses of 0.58, 0.87, and 1.30 mg/cm2 per d, respectively. A vehicle control group was also included and received the same dose volume as that administered to the high-dose group (Table 2). The estimated achieved doses, based on test item use, were 0 (vehicle control), 26.7, 35.0, and 58.1 mg/kg per d, corresponding to nasal surface area doses of 0, 0.6, 0.8, and 1.4 mg/cm2 per d, respectively.
All animals were observed at least once daily during the pretrial period. During the treatment period, each animal was examined once before dosing, continuously during dosing, and at approximately 1 hour after dosing. Animals were observed for clinical signs; onset, intensity, and duration of any signs observed were recorded and reported for each individual animal. In phase 1, gross pathology was performed on 3 animals per group approximately 24 hours after the start of dosing. Gross pathology was performed on the remaining animals approximately 48 hours after the start of dosing. In phase 2, gross pathology was performed on all animals at the end of 5 consecutive days of treatment, that is, day 6, for surviving animals. Gross pathology was also performed on animals that died prematurely. The necropsy consisted of a complete external and internal examination including body orifices (ears, nostrils, mouth, anus) and cranial, thoracic, and abdominal organs and tissues. All macroscopic findings were recorded in descriptive terms, including location/locations, size (in mm), shape, color, consistency, and number. The respiratory and gastrointestinal tracts were closely examined for signs of gross irritation.
Twenty-eight-Day Local Tolerance Study in Rats
SPRIX was administered to rats for 28 days by the intranasal route, and the reversibility of any effects was evaluated after a 28-day recovery period. This study was conducted according to GLP standards. Three groups received SPRIX and a fourth group served as a vehicle control. Each group was consisted of 16 animals/gender per group; 10 animals/gender per group for the main study, and 6 animals/gender per group for toxicokinetic evaluations. The 6 animals designated for toxicokinetic evaluations were also designated as recovery animals in the vehicle control, 18.0, and 27.0 mg/kg per d dose groups. Animals were stratified by body weight and randomly assigned to treatment and control groups. Animals were dosed with the vehicle or test item formulation (7.5%, 15%, or 22.5% ketorolac) 5 times per day at 3-hour intervals with 10 μL to 1 nostril using a calibrated automatic pipette. Nostrils were alternated for dosing for each dose session. Recovery animals were dosed in the same manner for 28 consecutive days and then observed for a 28-day treatment-free period.
The targeted doses were 0 (vehicle control), 9.0, 18.0, and 27.0 mg/kg per d. These doses equated to nasal surface area doses of approximately 0, 0.21, 0.43, and 0.65 mg/cm2, respectively, based on an assumed nasal surface area of 10.4 cm2 for a 0.250 kg rat 9 (Table 3). Due to the gain in body weight over the course of the study and the marked difference between male and female body weight, the body-weight-adjusted dose levels were higher in week 1 compared to the subsequent weeks for both genders; females in the treated groups received higher doses on a body weight basis than males. For males, the achieved mean weekly dose levels were in the range of 7.53 to 5.73, 14.80 to 10.91, and 22.90 to 17.53 mg/kg per d for the targeted 9.0, 18.0, and 27.9 mg/kg per d dose groups, respectively, over the course of the 28-day study. For females, the achieved mean weekly dose levels were in the range of 10.54 to 8.80 and 21.61 to 18.08 mg/kg per d for the targeted 9.0 and 18.0 mg/kg per d dose groups, respectively, over the course of the 28-day study. Females in the targeted 27.0 mg/kg per d dose group received a dose of 33.61 mg/kg per d for week 1. By the end of day 8, all females in the 27.0 mg/kg per d dose group had either died or were euthanized due to adverse clinical signs. Accordingly, males and females in the 18.0 mg/kg per d dose group originally designated for toxicokinetic investigations were reassigned to the recovery phase.
Study Design for 5-Day Study in Rats: Phase 2
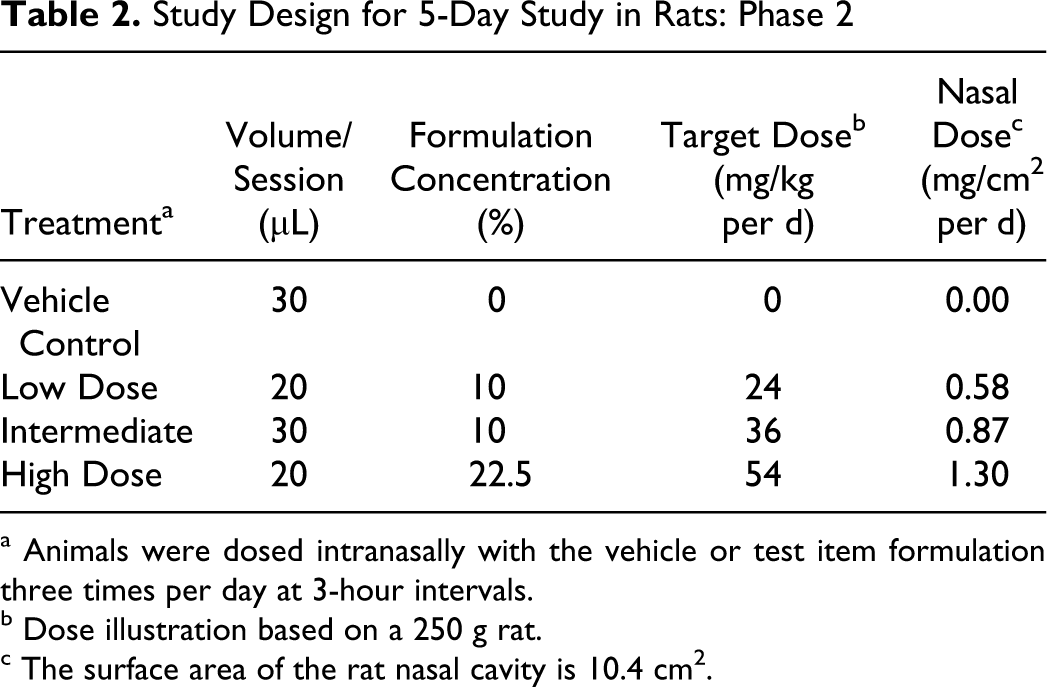
a Animals were dosed intranasally with the vehicle or test item formulation three times per day at 3-hour intervals.
b Dose illustration based on a 250 g rat.
c The surface area of the rat nasal cavity is 10.4 cm2.
All animals were observed at least once daily for clinical signs. Body weights were generally recorded once weekly except for the 18.0 and 27.0 mg/kg per d dose groups, for which the appearance of adverse clinical signs prompted more frequent monitoring. Food consumption was recorded weekly. Laboratory investigations of blood hematology, coagulation, and clinical chemistry were conducted during week 4 (day 23) for main study animals and toward the end of the recovery period (day 50 for males and day 49 for females) for recovery animals. Urine was collected for analysis during week 4 (day 28 for main study males, day 27 for main study females) and toward the end of the recovery period (day 51 for males and day 50 for females) for recovery animals. Blood samples for bioanalysis of SPRIX and subsequent toxicokinetic analysis were obtained from the designated animals on days 1 and 28.
Animals were sacrificed by carbon dioxide asphyxiation, followed by exsanguination, and were subjected to a detailed necropsy and organ weight analysis. A complete histopathology evaluation, including 4 transverse sections of the nasal cavity, was carried out in males and females in the vehicle control group, males in the 27.0 mg/kg per d dose group, and females in the 18.0 mg/kg per d dose group. Females in the 27.0 mg/kg per d dose group were not evaluated due to early deaths and moribund sacrifices.
Fourteen-Day Local Tolerance Study in Rabbits
The local tolerance of the SPRIX formulation administered via the intranasal route by spray pump for 14 days was determined in rabbits. This study was conducted according to GLP standards. Three groups received SPRIX, a fourth served as a saline control group, and a fifth served as a vehicle control group. Each group consisted of 5 animals/gender per group. Three animals/gender per group were designated for the main study and 2 animals/gender per group were designated for a 14-day recovery period. Animals received 100 μL saline, vehicle or test item formulation to the left nostril 3 times per day at 4-hour intervals for 14 consecutive days.
The target doses were 9.0, 18.0, and 27.0 mg/kg/d, corresponding to nasal surface area doses of 0.75, 1.5, and 2.25 mg/cm2/d, respectively, based on a nasal surface area of 60 cm2 (30 cm2 for the single treated nostril) for a 2.5 kg rabbit 10 (Table 4).
Study Design for 28-Day Study in Rats
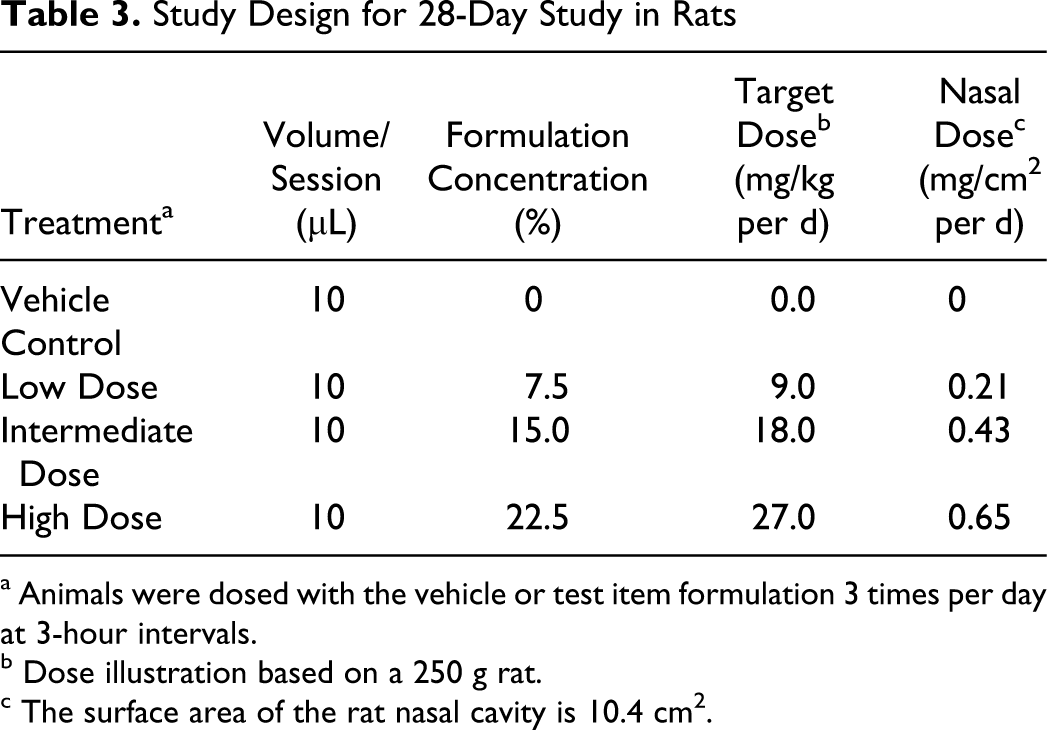
a Animals were dosed with the vehicle or test item formulation 3 times per day at 3-hour intervals.
b Dose illustration based on a 250 g rat.
c The surface area of the rat nasal cavity is 10.4 cm2.
Each animal was observed at least once daily for mortality. Clinical observations were performed prior to dosing, daily after each dose administration, and weekly during the recovery period. Body weights were recorded prior to initiation of treatment and weekly thereafter. Food consumption was measured for a 24-hour period once per week during the study. Main study animals were sacrificed on day 15 and recovery animals were sacrificed on day 30 after a 14-day treatment-free period. All animals were subject to complete necropsies on days 15 (main study) and 30 (recovery). The nasal septum and left (treated) and right (untreated) turbinates from all animals were microscopically examined.
Results
Five-Day Local Tolerance Study in Rats
In the single-dose range finding study (phase 1), there were no unscheduled deaths and no adverse clinical signs. Gastrointestinal irritation was evidenced by a decrease in body weight and intestinal reddening at target doses of 54 mg/kg per d and above. At the 108 mg/kg per d dose, 2 animals also had stomach lesions that appeared to be small ulcers.
In the 5-day repeat dose study (phase 2), there were 3 unscheduled deaths: 1 animal in the 36 mg/kg per d dose group was euthanized on day 5 due to piloerection, labored respiration, and a dark nasal discharge, and 1 animal in each of the 36 and 54 mg/kg per d dose groups was found dead on day 6. Clinical signs associated with exposure to SPRIX included hunching behavior in animals receiving doses of 36 mg/kg per d and above (Table 5). No signs of nasal irritation were observed on clinical or gross examination. Mean overall body weight gain was over 10% lower in all treated groups compared to controls. As in phase 1, there was gross evidence of GI irritation in some animals in the 54 mg/kg per d dose group, including a mass associated with the jejunum and thickening of the intestinal wall. It was suspected that the jejunum had ruptured in the animal in the 54 mg/kg per d dose group that was found dead.
Study Design for 14-day Study in Rabbits
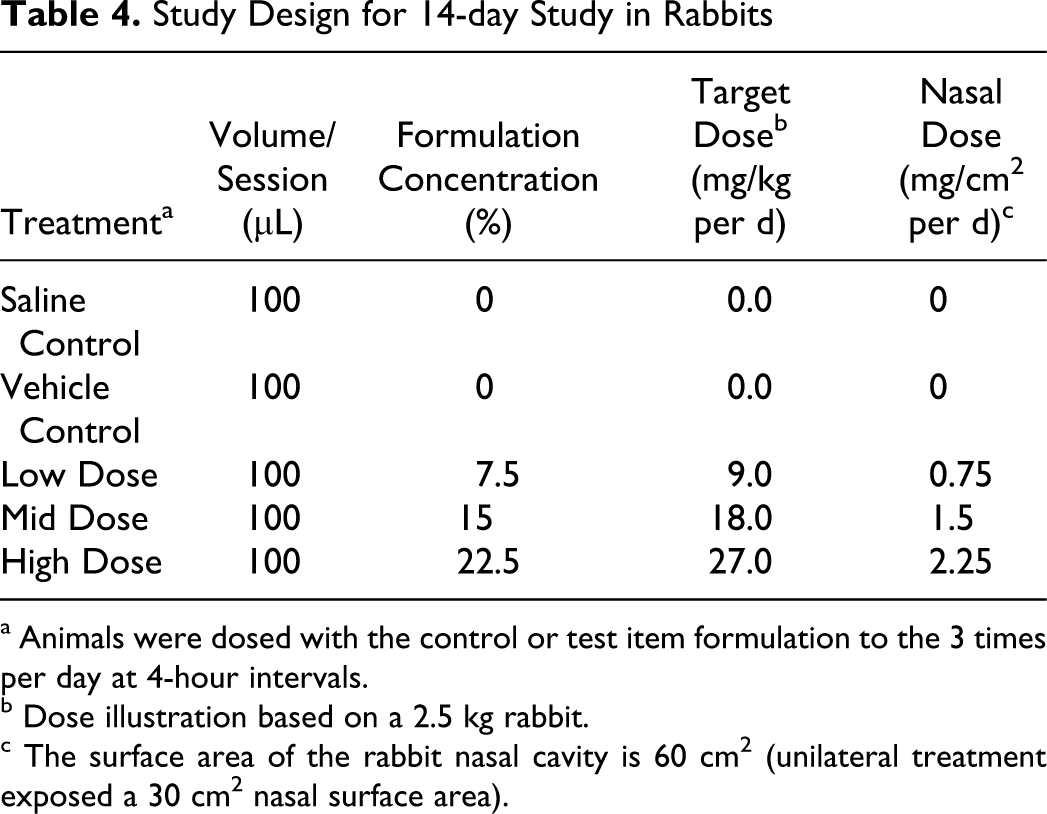
a Animals were dosed with the control or test item formulation to the 3 times per day at 4-hour intervals.
b Dose illustration based on a 2.5 kg rabbit.
c The surface area of the rabbit nasal cavity is 60 cm2 (unilateral treatment exposed a 30 cm2 nasal surface area).
Twenty-eight-Day Local Tolerance Study in Rats
Clinical Observations
In general, females were more severely affected by treatment with SPRIX than males. Mortality was noted in females in the 18.0 and 27.0 mg/kg per d dose groups. A moribund female in the 18.0 mg/kg per d dose group was euthanized on day 27. Of the 16 females in the 27.0 mg/kg per d dose group, 7 were found either dead or euthanized in a moribund condition by day 6, and the remaining females in the group were sacrificed on day 8. Additional clinical signs were observed in females in the 18.0 and 27.0 mg/kg per d dose groups, including hunched posture, swollen abdomen, shallow and labored breathing, thin appearance, marked weight loss, pale eyes, subdued behavior, dark nasal discharge, and piloerection. By week 3, all females in the 18.0 mg/kg per d dose group had markedly hunched body posture.
With the exception of 1 male from the high-dose recovery group that was hunched in posture from day 26 to day 30, no other treatment-related clinical signs were recorded in the males from any group or in females in the 9.0 mg/kg per d dose group (Table 6).
Summary of Clinical Observations for 5-Day Study in Rats
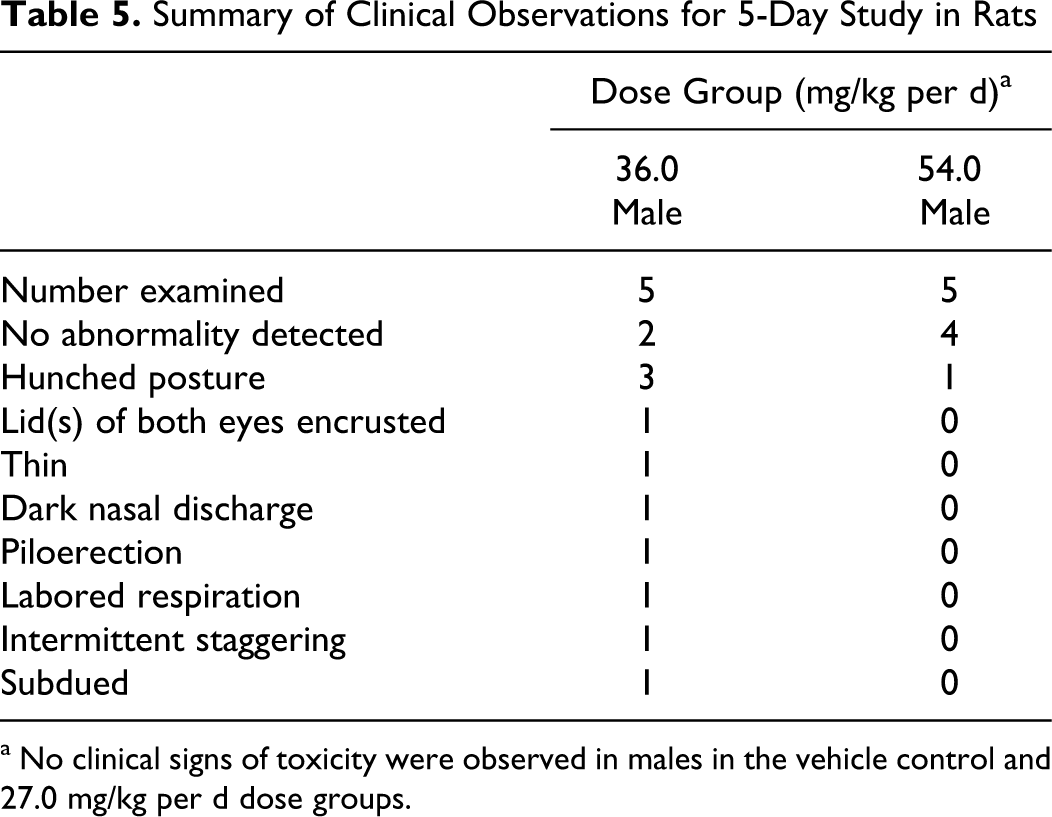
a No clinical signs of toxicity were observed in males in the vehicle control and 27.0 mg/kg per d dose groups.
During the recovery period, hunched posture persisted in females in the 18.0 mg/kg per d dose group until week 3 or 4 of recovery. By day 55 (day 27 of recovery), hunched posture was absent in these animals.
Hematology
Several hematology parameters were affected in females in the 18.0 mg/kg per d dose group: relative to controls, there were increases in leukocyte, platelet and reticulocyte numbers, and reductions in mean cell hemoglobin concentration and activated partial thromboplastin time (APTT). In males, there was a slight, but statistically significant, dose-dependent decrease in APTT values in all treated groups compared to control values during week 4. With the exception of a marked reduction in reticulocytes in females from the 18.0 mg/kg per d dose group, values for affected hematology parameters returned to control levels following the recovery period. The reported changes in hematology parameters are believed to have been secondary to GI toxicity in females.
Serum Chemistry
Serum chemistry parameters were slightly but statistically significantly different from control in several instances. In treated females, there were statistically significant decreases in aspartate aminotransferase (9.0 and 18.0 mg/kg per d dose groups), and in total protein, albumin, and albumin-globulin ratio (18.0 mg/kg per d dose group), relative to control values. Sodium levels were slightly higher in females in the 18.0 mg/kg per d dose group compared to controls. In males, sodium and chloride levels were slightly higher and albumin levels were slightly lower for the 18.0 and 27.0 mg/kg per d dose groups compared to controls. Changes in serum electrolytes have also been reported in a study with other NSAIDs (aspirin and indomethacin) in rats and rabbits. 11 Serum chemistry changes were not evident at the end of the recovery period, with the exception of a slight increase in chloride in males dosed with 27.0 mg/kg per d. All of these changes were considered slight and the biological significance of these findings, if any, was not established.
Urinalysis
In both genders, urine volume generally increased with increasing dose and, correspondingly, specific gravity decreased with dose. Urinary protein concentrations also tended to be decreased in treated groups compared to control. These effects were no longer evident following the recovery period.
Gross Pathological Findings
Necropsy findings in the high-dose females that died prematurely involved the GI tract and mesenteric lymph nodes. Similarly, the gross findings in the single female in the 18.0 mg/kg per d dose group sacrificed on day 27 primarily involved the GI system. Gross findings from females surviving to dosing cessation in the 18.0 and 27.0 mg/kg per d dose groups included fluid in the abdominal cavities, adhesions, and abdominal contents; intestines with abnormal contents, raised foci, abnormal shape, thickening, reddening, and adhesions; pronounced gut-associated lymphoid tissue; stomachs with distension, abnormal contents, and pale or dark foci; pallor of various organs; and skin staining. There were no notable necropsy findings in males from any group or in females in the 9.0 mg/kg per d dose group. No remarkable findings were observed after the recovery period.
Histopathology Findings
Histopathology findings were observed primarily in the GI tract, consistent with NSAID treatment. 8 Findings in the females treated at dose of 18.0 mg/kg per d included ulcerations and erosion of the stomach, jejunum, and ileum; peritonitis throughout the gastrointestinal system, peritonitis, lymphoid hyperplasia and plasmacytosis in the mesenteric lymph node; and increased hematopoiesis in the spleen. Females in the 27.0 mg/kg per d dose group were not evaluated due to the early termination of the group. In males, a gastric ulcer was noted in 1 animal in the 27.0 mg/kg per d dose group and gastric erosion was noted in 1 male in the 18.0 mg/kg per d dose group. A few animals from all treated groups had minor inflammatory reactions in the gastric lamina propria (Table 7). These findings were not observed following the recovery period (Table 8).
Summary of Clinical Observations for 28-Day Study in Rats—Main Study and Recovery
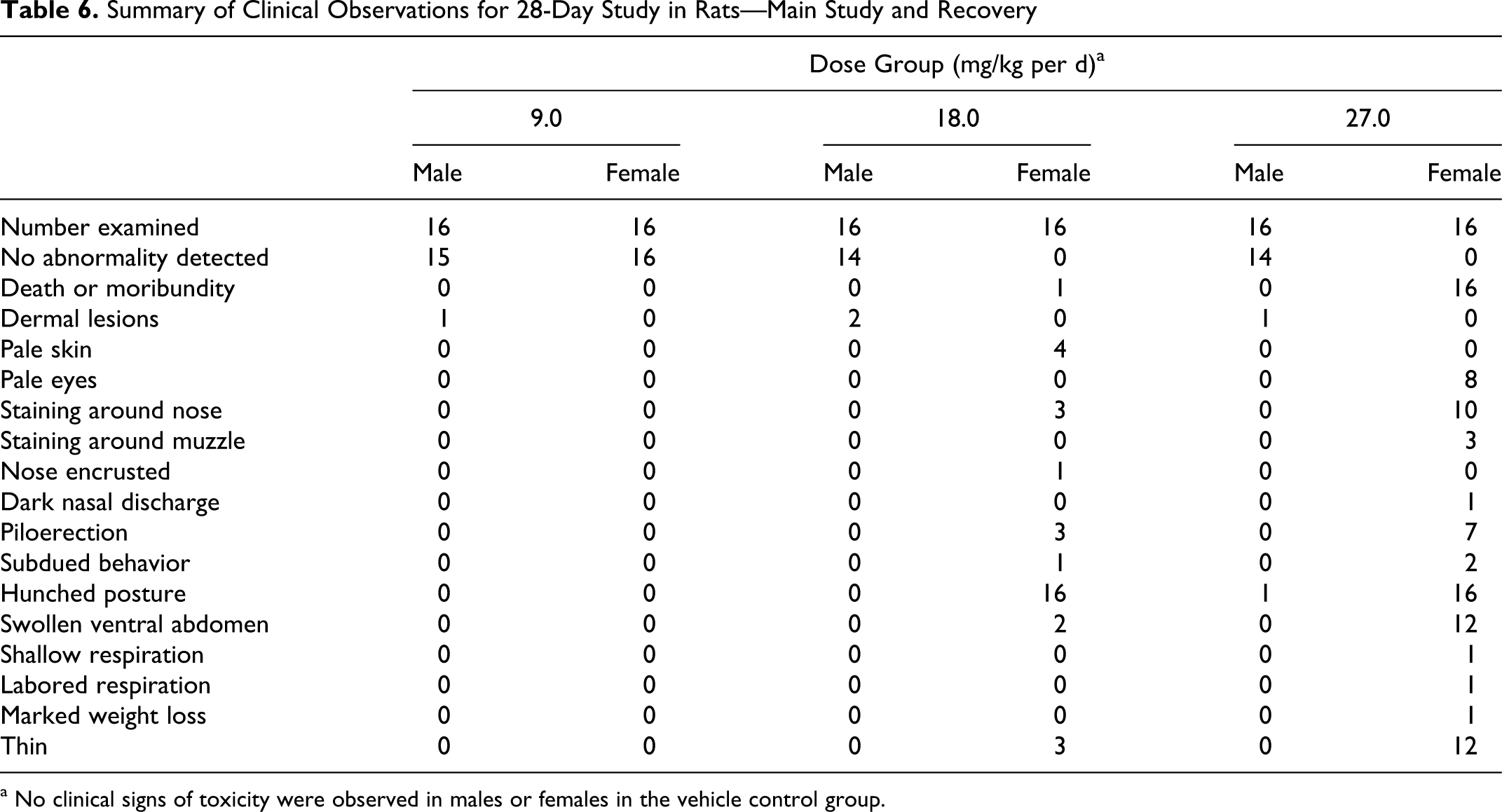
a No clinical signs of toxicity were observed in males or females in the vehicle control group.
Histopathology Findings in the Gastrointestinal Region for 28-Day Study in Rats—Main Study
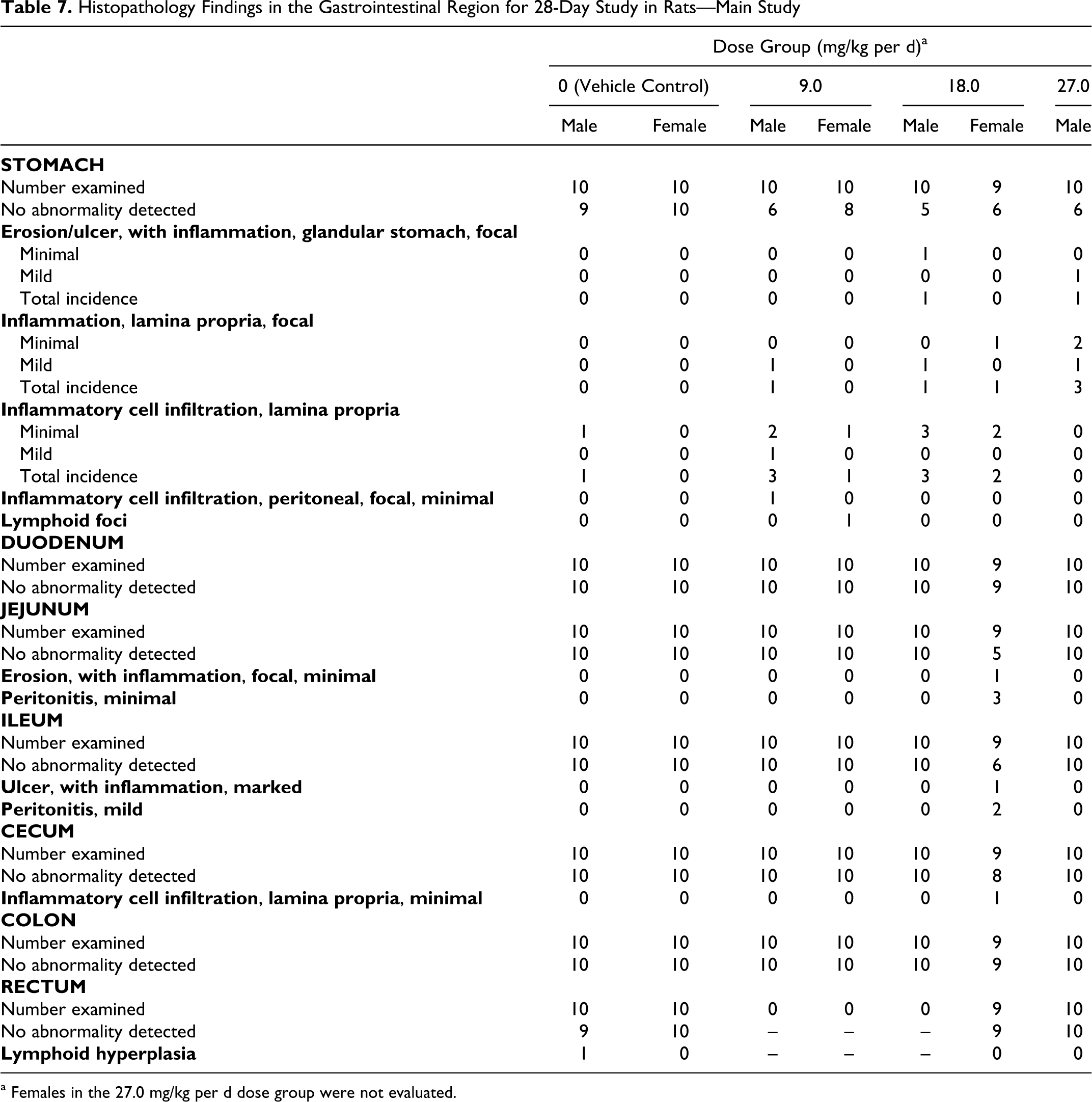
a Females in the 27.0 mg/kg per d dose group were not evaluated.
Histopathology Findings in the Gastrointestinal Region for 28-Day Study in Rats—Recovery
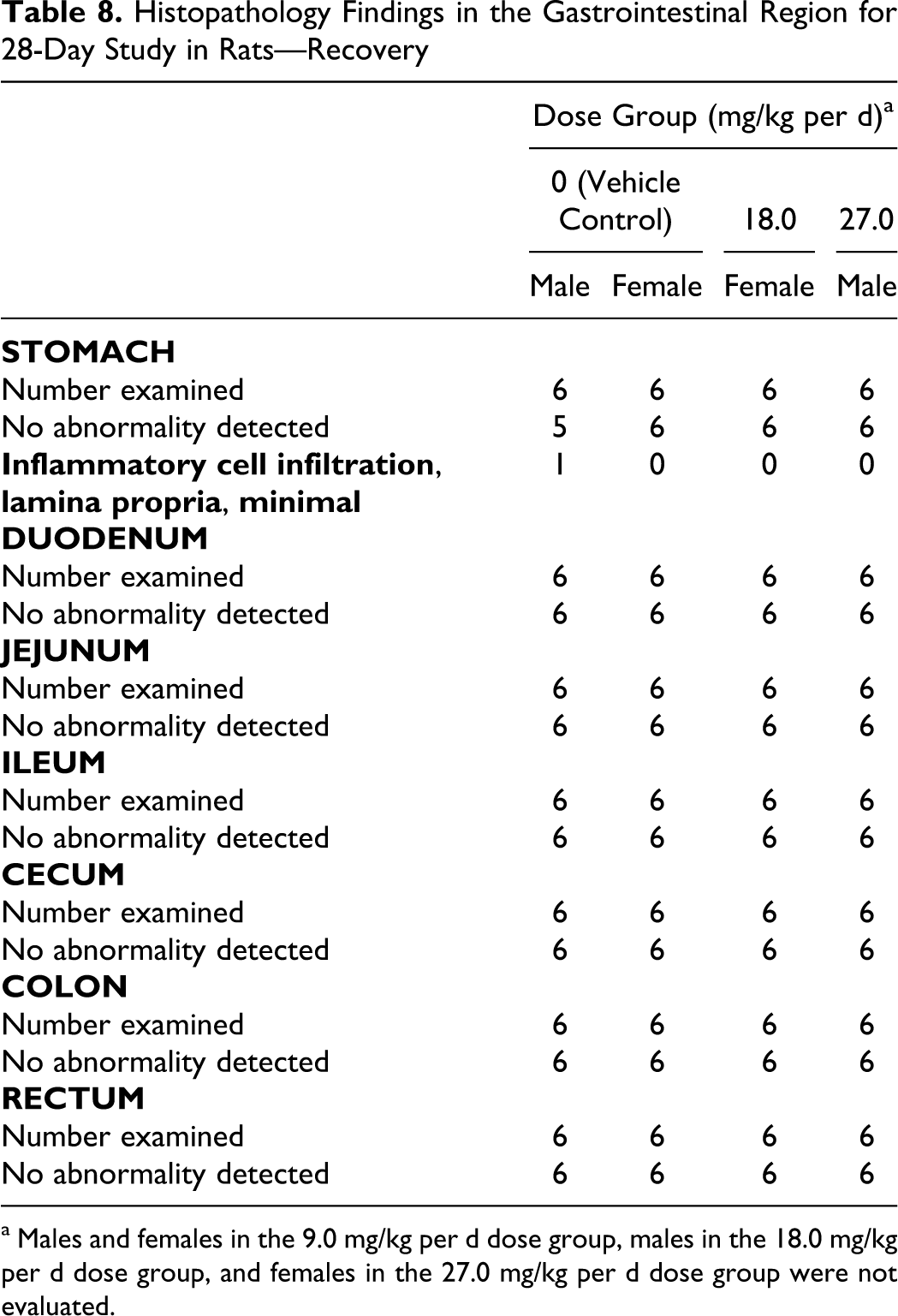
a Males and females in the 9.0 mg/kg per d dose group, males in the 18.0 mg/kg per d dose group, and females in the 27.0 mg/kg per d dose group were not evaluated.
The following histopathological findings were observed in the gastrointestinal tract of 1 female in the 18.0 mg/kg per d dose that was killed prematurely due to clinical signs of toxicity: mild peritonitis in the cecum, ileum, and jejunum, minimal peritonitis in the colon, and duodenum, a moderate focal ulcer in the glandular stomach, and mild peritoneal inflammation in the stomach. Females in the 27.0 mg/kg per d dose group were either found dead or killed prematurely due to clinical signs of toxicity. Histopathological analyses were not performed on these animals.
Other findings included evidence of chronic progressive nephropathy and mineralization in the kidney, the incidence and/or severity of which increased with increasing dose between the 9.0 and 18.0 mg/kg per d dose groups. These findings persisted throughout the recovery period.
Histopathological findings in the nasal cavity were suggestive of possible mechanical irritation associated with direct contact with the dosing apparatus. Minimal hyperplasia and/or inflammatory cell infiltration of the transitional epithelium in the foremost section of the nasal cavity was observed in several high-dose males at termination, in some vehicle control females, and in 1 control male during the recovery phase. These differences in appearance of the transitional epithelium were considered related to technical procedures (ie, the plane of the section) and not to treatment (Table 9). Similarly, the inflammatory cell infiltrate observed in a single high-dose male after 28 days of recovery was considered a spontaneous finding and not related to treatment (Table 10). There were no findings indicative of a safety concern in the nasal passages of the rats.
Histopathology Findings in the Nasal Region for 28-Day Study in Rats—Main Study
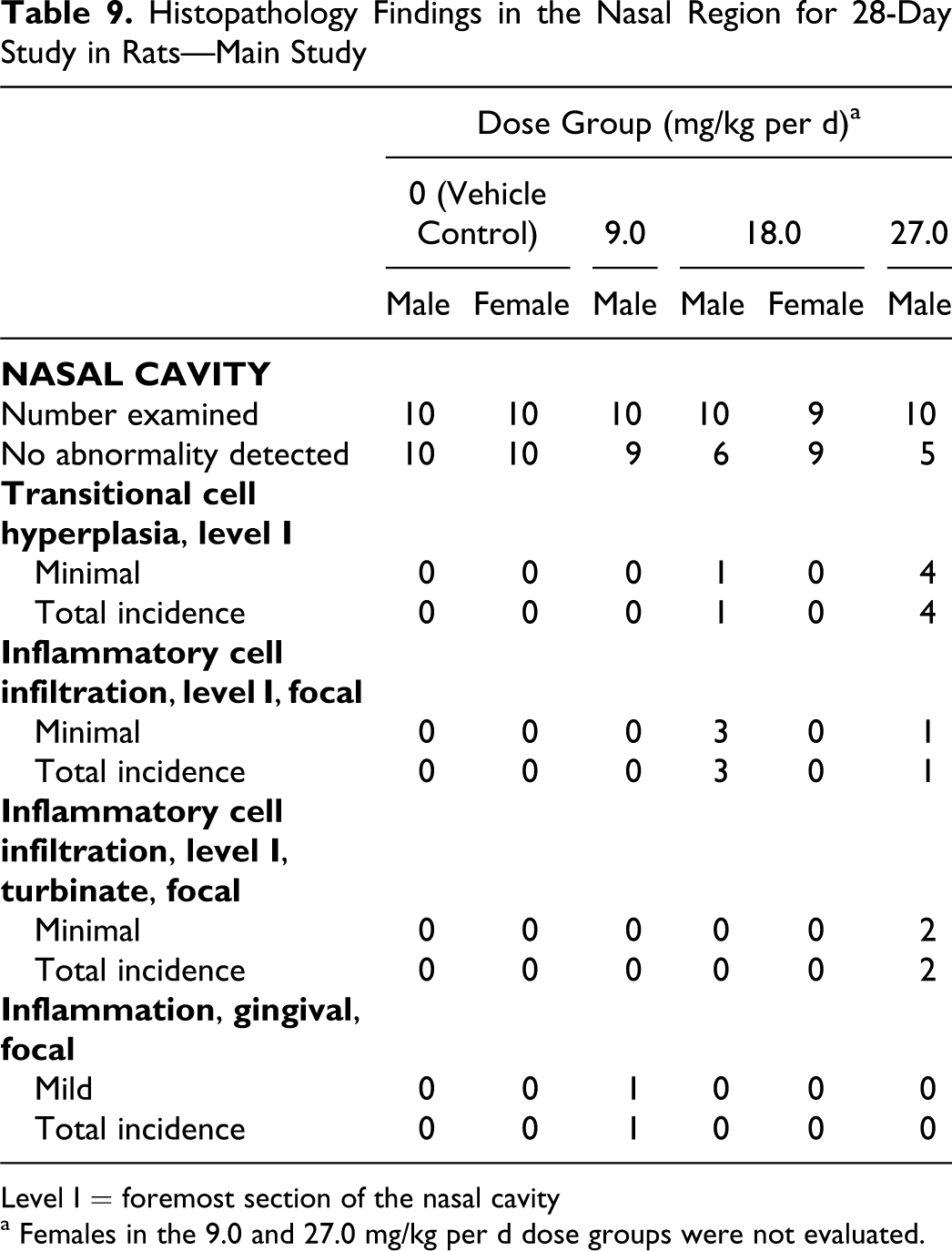
Level I = foremost section of the nasal cavity
a Females in the 9.0 and 27.0 mg/kg per d dose groups were not evaluated.
Histopathology Findings in the Nasal Region for 28-Day Study in Rats—Recovery
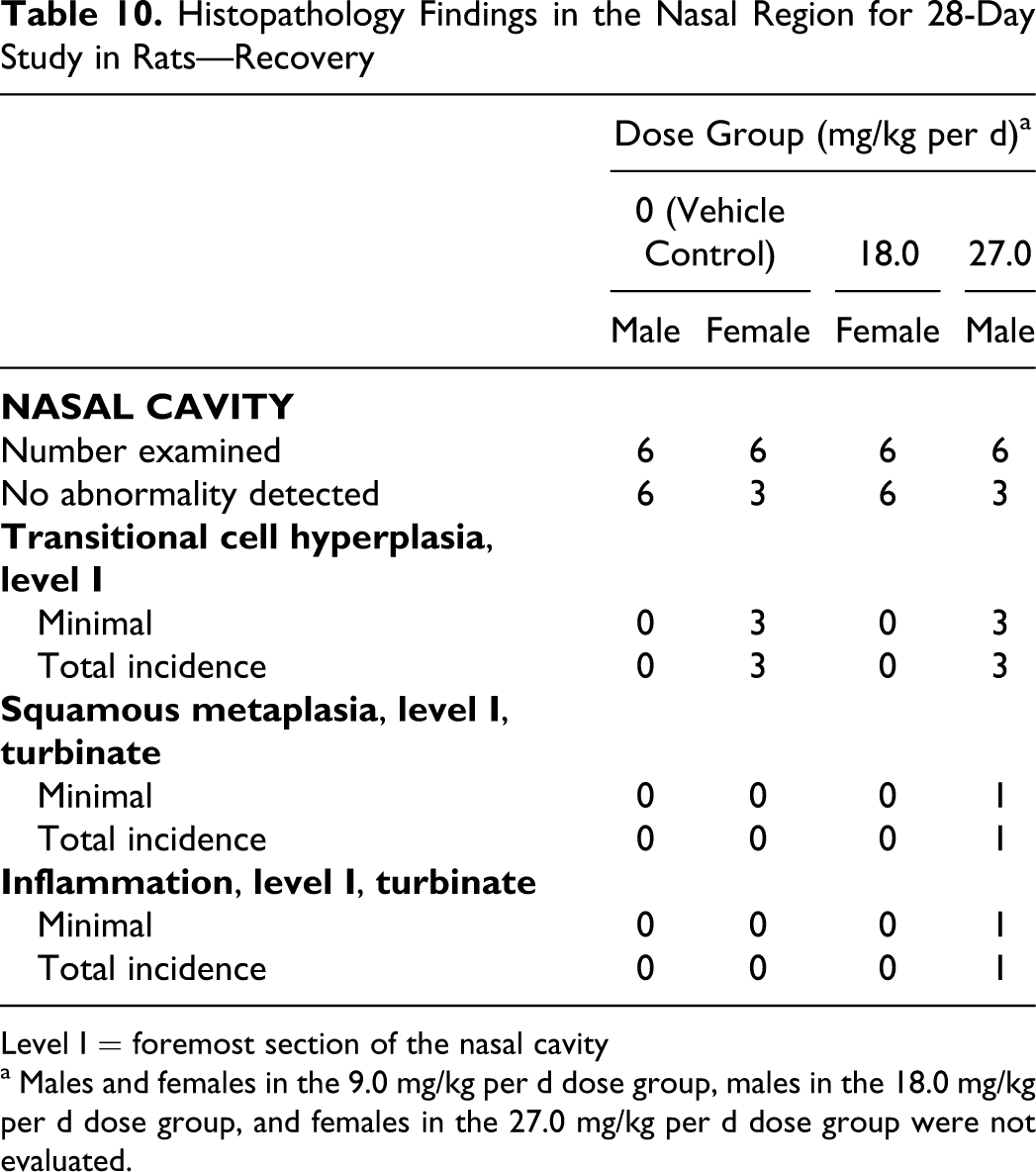
Level I = foremost section of the nasal cavity
a Males and females in the 9.0 mg/kg per d dose group, males in the 18.0 mg/kg per d dose group, and females in the 27.0 mg/kg per d dose group were not evaluated.
Ketorolac Tromethamine Exposure
On day 1, the Cmax values for males and females revealed good dose-to-Cmax linearity. Females were exposed at much higher levels than males as evidence by Cmax values that were approximately 2- to 3-fold higher for females compared to values for males at each dose level on day 1. Normalized Cmax values on day 28 were approximately 60% to 76% higher than the respective day 1 values for males and there was good linearity for the data for males on day 28. On day 28, the results for females departed from linearity. On day 28, the dose adjusted Cmax values for females in the 18.0 mg/kg per d dose group were comparable to the dose adjusted Cmax values for males in the 27.0 mg/kg per d dose group. The systemic exposure data (AUC) generally followed this pattern. The Tmax was achieved within 0.5 (the first sampling time) to 1 hour (Table 11).
Ketorolac Tromethamine Exposure for 28-Day Study in Rats
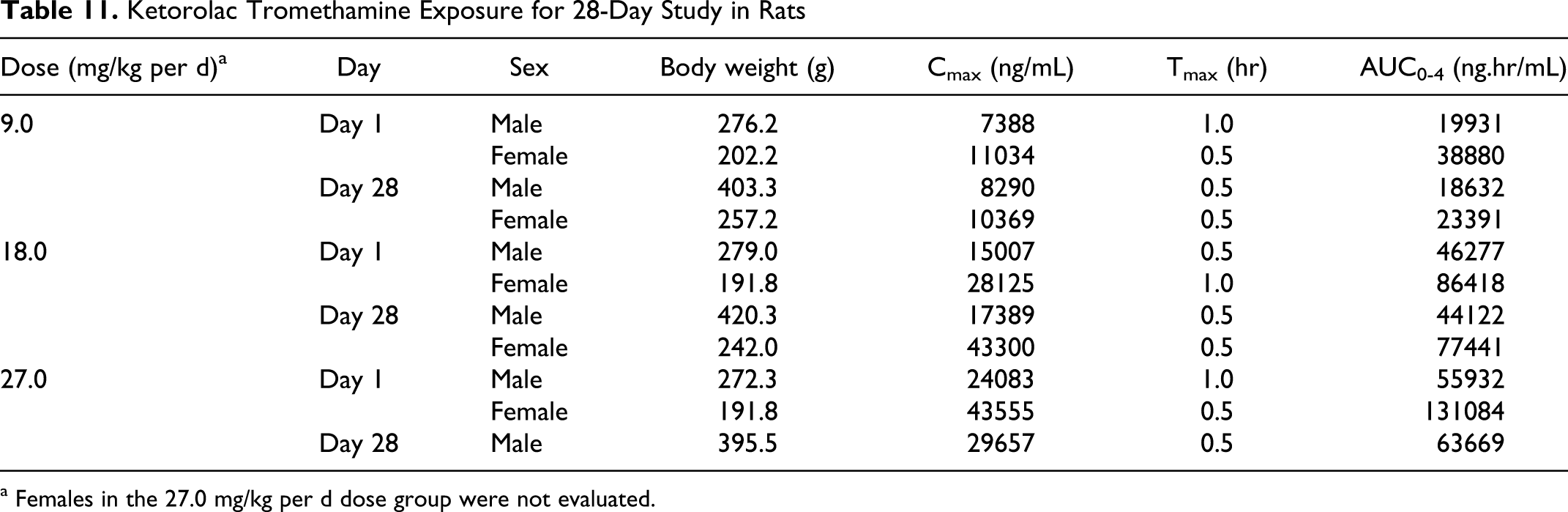
a Females in the 27.0 mg/kg per d dose group were not evaluated.
Fourteen-Day Local Tolerance Study in Rabbits
All of the animals survived until their scheduled necropsy, and there were no observed test article-related effects.
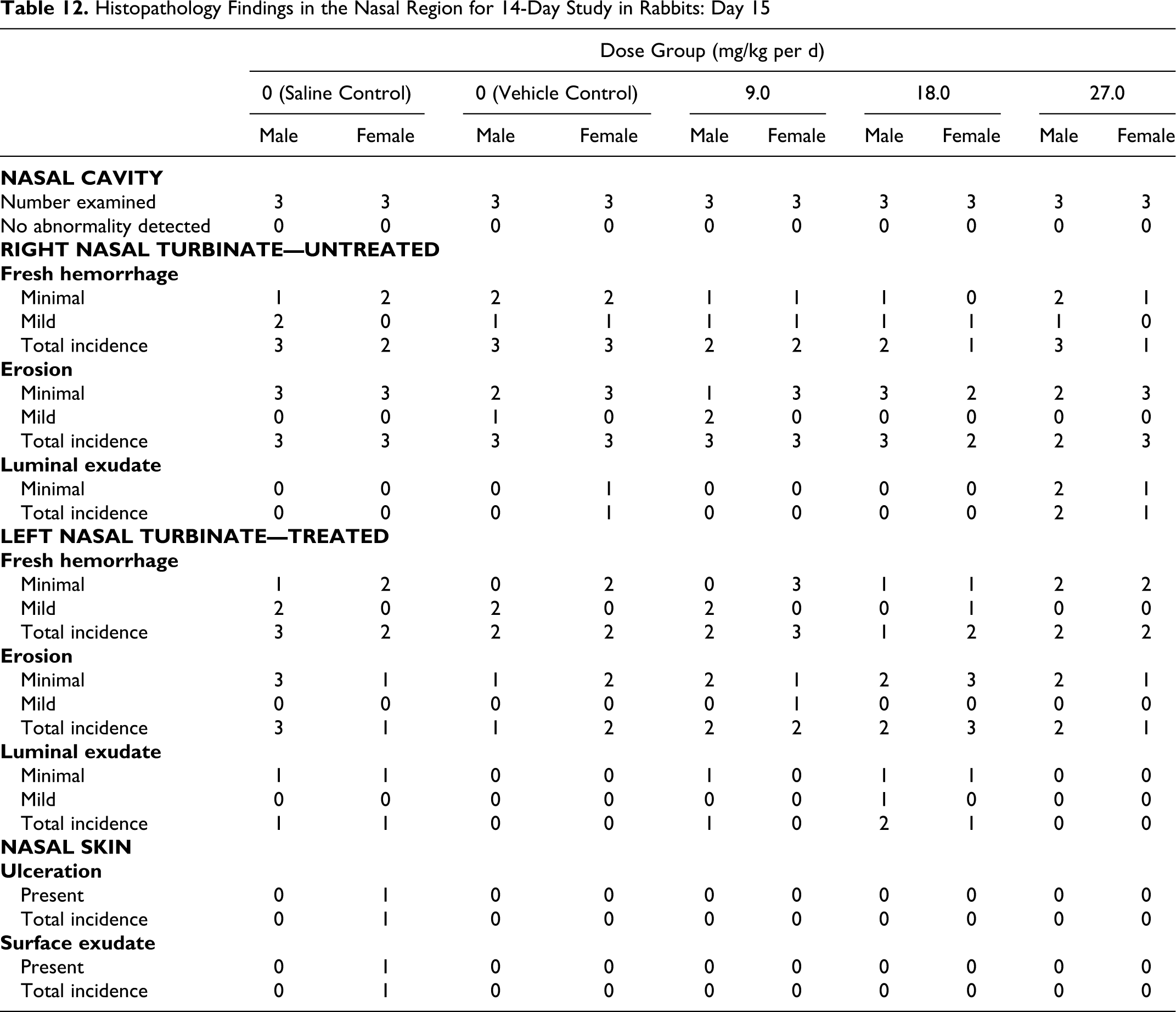
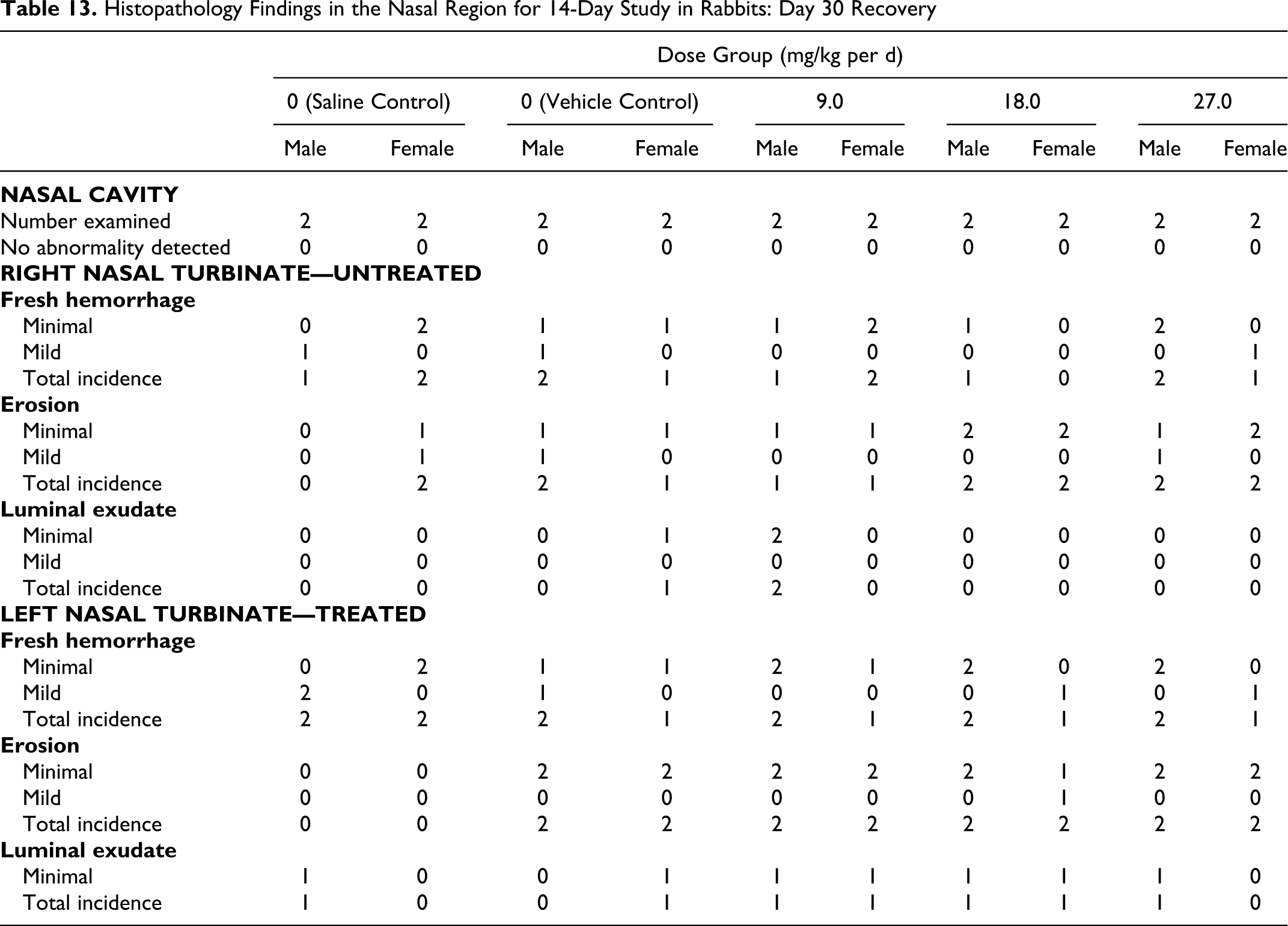
Histopathological findings in the nasal turbinates included fresh hemorrhage, erosion, and luminal exudate at both the day 15 terminal sacrifice and the day 30 recovery sacrifice. These findings were observed in similar incidence and severity in the saline control, vehicle control, and treated groups. In addition, ulceration and surface exudate were noted in a single female animal in the saline control group on day 15. All findings were considered related to the terminal necropsy procedures of harvesting the tissue or handling during the histology process and not related to the intranasal administration of either control or test articles (Tables 12 and 13).
Histopathology Findings in the Nasal Region for 14-Day Study in Rabbits: Day 15
Histopathology Findings in the Nasal Region for 14-Day Study in Rabbits: Day 30 Recovery
Discussion
The intranasal toxicity studies in rats and rabbits demonstrated that intranasal administration of SPRIX produces no local irritation and that the target organ toxicity by this route of administration is similar to that observed with other routes, namely, GI tract- and kidney-related effects. 12,13 Studies in rats and rabbits demonstrated that an intranasally administered SPRIX solution is well tolerated and produced no unexpected toxicities. Target organ toxicity associated with the drug class was observed in rats. No indications of local toxicity in the nasal cavity were observed.
The 5-day study in rats showed that SPRIX administered intranasally at doses of 54 mg/kg per d or greater in a 1-day regimen and 36 mg/kg per d or greater in a repeat dose 5-day regimen are associated with dose-limiting effects on body weight and the GI tract. Additionally, deaths were observed in phase 2 (5-day repeat dose phase) at doses of 36 mg/kg per d and greater. No signs of nasal irritation were observed on clinical or gross examination. As no nasal symptoms were observed in other animals, it is likely that the nasal discharge observed in the animal in the 36 mg/kg per d dose group that was euthanized on day 5 was not directly due to treatment and may have been associated with an excess of porphyrins in a very sick animal. On the basis of the phase 2 repeat dose study, rats tolerated a dose of 24 mg/kg per d, equating to an estimated nasal dose of approximately 0.58 mg/cm2 per d, for 5 days.
Intranasal administration of SPRIX for 28 days in rats was associated with GI toxicity, a recognized side effect of this class of drug, 10 in female and male rats treated with 18.0 and 27.0 mg/kg per d, and renal toxicity in females treated with 9.0 and 18.0 mg/kg per d. The GI toxicity was associated with mortality in a significant number of females in the 27.0 mg/kg per d dose group. Females were more sensitive to the systemic effects of SPRIX when compared to males, probably due to increased exposure in females. Females were exposed at much higher levels than males as evidenced by Cmax values that were approximately 2- to 3-fold higher for females compared to values for males at each dose level on day 1.
In the rat, the No Observed Effect Level (NOEL) for local effects was at least the target high dose of 27.0 mg/kg per d, associated with a nasal dose of approximately 0.65 mg/cm2 per d. The No Observed Adverse Effect Level (NOEL) for systemic effects was the target low dose of 9.0 mg/kg per d for males and was not identified (<9.0 mg/kg per d) for females.
In the rabbit, based on the parameters evaluated, the NOAEL was considered at least 27.0 mg/kg per d for 3 times daily nasal administration by spray pump for 14 consecutive days. The NOEL dose was at least the approximate high dose of 27 mg/kg per d or a nasal dose of approximately 2.25 mg/cm2 per d nasal surface area per day.
The nonclinical program for SPRIX, in addition to the information already available in regard to ketorolac tromethamine, provided adequate assurance that the proposed clinical use of this product does not present a significant safety risk to the intended clinical population. The toxicology studies demonstrated that administration of SPRIX by the intranasal route does not affect the overall toxicity profile of the drug. In placebo-controlled clinical trials, intranasal ketorolac administered to patients with moderate-to-severe pain 3 to 4 times a day for up to 5 days was well tolerated. 4 –6 The most common adverse event reported by patients more frequently in the intranasal ketorolac group compared to placebo was mild, transient nasal irritation. However, physician evaluation of nasal mucosa at the end of treatment did not reveal any clinically relevant changes or concerns in patients treated with intranasal ketorolac compared to placebo. 4 The toxicology studies presented in the current article confirm that intranasal administration of ketorolac does not result in upper respiratory system pathology.
Footnotes
Acknowledgment
The authors gratefully acknowledge the work of SRI International for the conduct of the rabbit study.
The authors declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: funded by ROXRO PHARMA, Inc. Two of the authors (TZ, KCB) are employed by SciLucent, LLC, which provides consulting services to ROXRO PHARMA, Inc. One of the authors (PM) is employed by Charles River Laboratories, where studies were conducted on behalf of ROXRO PHARMA, Inc.