
Abstract
We have recently demonstrated that the alkaloid colchicine (COL) inhibits glucocorticoid receptor (GR) transcriptional activity. In addition, we described proteasome-mediated degradation of GR in COL-treated HeLa cells. While these effects were previously attributed to cell cycle arrest in G2/M phase, this explanation is not applicable for nonproliferating cells such as human hepatocytes (HH). In the current study, we compared COL-mediated microtubule disruption and cell cycle arrest with selected GR functions in HeLa cells and HH as models of proliferating and quiescent cells, respectively. Microtubule disruption led to irreversible decrease in GR binding capacity and protein level in HeLa cells. None of the parameters was restored 24 hours after COL withdrawal. In contrast, dexamethasone (DEX) binding was increased in HH at the beginning of the treatment, with following transient activation of extracellular signal-regulated kinase (ERK). The findings of these investigations emphasize the GR-signaling differences between primary and transformed cells.
Introduction
Glucocorticoid receptor (GR) plays an important role in glucose homeostasis, drug metabolism, or anti-inflammatory response. We have recently demonstrated that colchicine (COL), a tropolone alkaloid, inhibits GR transcriptional activity, which resulted in downregulation of several biotransformation enzymes in primary human hepatocytes (HH). 1 In addition, we described proteasome-mediated degradation 2 of GR in HeLa cells treated with COL. The cellular effects of COL (including those on GR) were always attributed to the cell cycle arrest in G2/M phase. However, this explanation is not applicable for nonproliferating, that is, quiescent cells, such as HH.
Indeed, it is known that the function of GR depends on the particular phase of the cell cycle. 3,4 Initially, it was reported that the number of GR in HeLa cells, expressed as bound fraction of dexamethasone (DEX), oscillates throughout the cell cycle. 5 The number of cytosoplasmic GR increased in G1/S phase and was maintained throughout S and G2 phases, whereas nuclear binding of hormone was reduced in G2 and remained low until early G1. 6 Similarly, an increase in DEX binding between G1 and S phases and a decrease in G2/M phase of the cell cycle was observed in human lymphoid cell lines 7 or human peripheral lymphocytes. 8 In conclusion, a new GR protein synthesis near the S phase was suggested. However, direct proof was missing.
More light into the cell cycle regulation of GR function was introduced in early 90s. Inhibition of GR transcriptional activity and site-specific alterations in GR phosphorylation were observed in mouse fibroblasts synchronized in G2 phase of cell cycle. 9
Hsu and DeFranco demonstrated that DEX-dependent degradation of GR protein as well as the GR-mediated transcriptional repression were unaffected in G2 phase in contrast to transactivation on simple glucocorticoid-responsive element (GRE). 10 It implied that distinct activities involved in homologous receptor downregulation and transrepression operated regardless of cell cycle status. The levels of GR protein were comparable in asynchronous cells and in G2 synchronized ones.
Surprisingly, other authors observed that GR transcriptional activity is not altered in G2 phase but drops down in M phase of the cell cycle. 11 In addition, only the stably transfected MMTV promoter was repressed, whereas the same promoter transiently transfected was inducible. The authors concluded that GR itself is fully functional throughout the entire cell cycle, but GR responsiveness is repressed in mitosis due to the chromatin condensation rather than due to specific modification of GR.
Analyses in Chinese hamster ovary WCL2 cells overexpressing GR showed that basal phosphorylation of GR was significantly higher in G2/M phase as compared to S phase, whereas hormone-dependent hyperphosphorylation of GR was not altered in G2/M phase in contrast to S phase. 12 Hormone binding capacity of the cells was increased in S and G2/M phases as compared to G0/G1 phase and the increase was accompanied by a parallel increase in GR protein content. This is in contrast with the finding that G2/M synchronizing agents vincristine, nocodazole, and COL caused proteasome-mediated degradation of GR protein in human cervix carcinoma cells HeLa. 2,13
The weak spots of some studies performed to date are (1) use of overexpressing systems/exogenous GR that may differ from regulation of endogenous one; (2) absence of correlation between ligand binding capacity and GR protein content in the studies carried out before immune-detection discovery; and (3) the lack of data on nonproliferating cells.
In the current study, we compared COL-mediated effects both in proliferating and in nonproliferating cells. We used primary cultures of HH as a representative of quiescent, human healthy cells possessing endogenous functional GR. For comparison, the model of human cervix carcinoma cells HeLa, a representative of proliferating, transformed human cells that are equipped with endogenous functional GR, was chosen. We monitored the effects of COL on microtubules integrity, cell cycle distribution, GR binding capacity, and protein content. In addition, we analyzed reversibility of these effects after COL withdrawal.
Materials and Methods
Chemicals
Williams' medium E, Dulbecco’s modified Eagle medium (DMEM), fetal calf serum (FCS), penicillin, streptomycin,
Cell Cultures
Primary cultures of HH
Liver samples were obtained from surgery of hepatic tumors. Tissue acquisition protocol was in accordance with the requirements issued by ethical commission in France and Czech Republic. Hepatocytes were isolated according to a published protocol. 14 The cells were plated on collagen-coated culture dishes using cell density 14 × 104 cells/cm2. Culture medium, used as described before, 15 was enriched for plating with 5% FCS (vol/vol). The medium was exchanged for a serum-free medium 4 to 6 hours later and the culture was allowed to stabilize for additional 24 hours. After this period, the cells were starved in DEX-free medium for additional 24 hours and then throughout the whole experiment.
HeLa
Human Negroid cervix epitheloid carcinoma cells (ECACC No. 93021013) were cultured in DMEM supplemented with 10% FCS, 100 U/mL streptomycin, 100 µg/mL penicillin, 4 mmol/L
Experimental Setup
HeLa cells and/or HH were incubated for 16 hours with 1 µmol/L COL and/or dimethyl sulfoxide (DMSO) overnight. Thereafter, cultures were carefully washed 5 times with PBS and further cultured for 24 hours. Experimental parameters were measured in time intervals of 2, 4, 8, 12, and 24 hours. In this article, the time interval is referred as “Time from release.” All experiments in HeLa cells were performed in culture medium containing 10% FCS; for experiments in HH, DEX- and serum-free medium was applied.
Immunofluorescence
After the treatement, the cells were fixed (60 mmol/L PIPES, 25 mmol/L HEPES, 10 mmol/L EDTA, 10 mmol/L MgCl2, 3 vol%/vol% formaldehyde, 0.05 vol%/vol% glutaraldehyde, pH 6.9), permeabilized (150 mmol/L NaCl, 50 mmol/L Tris-HCl, 0.2 vol%/vol% Triton X-100, pH 7.5), and their nonspecific sites were saturated with bovine serum albumin (BSA; 3 vol%/vol% in PBS). As the first antibody, mouse monoclonal anti-α-tubulin (CLON DM1A; T9026; Sigma) was used in a dilution of 1:1000 supplemented with 1% BSA for 1 hour; Alexa Fluor 546 served as the second antibody (Invitrogen; A11018) in a dilution of 1:1000 again incubated for 1 hour in the presence of 1% BSA. Staining of nuclei with Hoechst 33258 was performed in parallel (1 µg/mL; 5 minutes). Microtubules status was then evaluated using fluorescent microscopy with a Leica DMRA wide-field microscope using a 63× oil immersion objective with A4 filter (Leica, Ex. BP 360/40 nm, dichroic 400 nm, Em. BP 470/40 nm) for Hoechst and Y3 filter (Leica, Ex. BP545/30 nm, dichroic 565 nm, Em. BP 610/75 nm) for Alexa Fluor 546. For imaging, the MetaMorph Imaging System v.4.5 (Universal Imaging Corp, Marlow, UK) was used.
Flow Cytometry Analysis
Following treatments, cells were washed with PBS, detached from culture surface using trypsin, resuspended in PBS containing EDTA, and centrifuged at 2500 g for 10 minutes at 4°C. Cells were again washed in ice-cold PBS and fixed with −20°C chilled ethanol (70%; vol/vol) by low-speed vortexing. For detection of DNA content analysis, we used propidium iodide staining (propidium iodide, 50 µg/mL; glycerol, 5%; Triton X-100, 0.1%). The fluorescence was measured on flow cytometer and analyzed by CellQuest software (BD Biosciences, San Jose, California).
Ligand Binding Assay
Cytosolic extracts were prepared as follows. Cells were washed twice with PBS and a volume of buffer 1 (25 mmol/L HEPES; 1 mmol/L EDTA; 0.1 mmol/L sodium orthovanadate; 1 tablet of protease inhibitors COMPLETE [Roche]) equal to the volume of cell sediment was applied. Thereafter, the cell suspension was homogenized by syringe with needle and left on ice for 15 minutes. Subsequently, the volume of buffer 2 (buffer 1 with 20% of glycerol) equal to actual volume of suspension was added and the suspension was centrifuged (35 000 rpm/4°C per hour; Ti 70.1 rotor). The final supernatant, the cytosolic fraction, was used. Then, 143.5 µL of the cytosol was mixed with 5 µL of 300 nmol/L [3H]-DEX alone or in combination with 1.5 µL DMSO or unlabelled DEX (1 mmol/L) and incubated overnight at 4°C. With 100 µL of the incubation reaction, bound and free steroids were separated by gel filtration using Sephadex LH-20 (Pharmacia, Guyancourt, France), and radioactivity was measured with 600 µL of the total eluate of 1000 µL in a liquid scintillation counter Packard Tri-Carb 2100TR (Canberra Packard, Savigny-le-Temple, France). The presence of GR in the cytosolic fraction was verified using Western blot (WB).
Protein Analyses
Total extracts were prepared as described elsewhere. 2,16 The protein content in the extracts was determined by the biscinchoninic acid method. The extracts were analyzed on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels (8%) and protein transfer was carried out onto polyvinylidene fluoride (PVDF) membrane. The membrane was stained with Ponceau S red dye for control of transfer and then saturated with 8% nonfat dried milk for 2 hours. Blots were probed with primary antibodies against human GR ([E-20] rabbit polyclonal; dilution 1/500) and actin ([I-19] goat polyclonal; dilution 1/1000), both purchased from Santa Cruz Biotechnology, Inc, or against phospho-p44/42 mitogen-activated protein kinase (MAPK; ERK1/2; [Thr202/Tyr204], rabbit polyclonal antibody, 9101S); p44/42 MAPK (ERK1/2; rabbit polyclonal, 9102); phospho-c-Jun-N-terminal kinase (JNK; Thr183/Tyr185; rabbit polyclonal antibody, 9251S); JNK (rabbit polyclonal, 9252); phospho-p38 MAPK ([Thr180/Tyr182], rabbit polyclonal antibody, 9211S); p38 (rabbit polyclonal, 9212); and caspase-3 (rabbit polyclonal, 9662), all purchased from Cell Signaling Technology Inc (Danvers, Massachussets). Chemiluminescence detection using horseradish peroxidase–conjugated secondary antibodies and Santa Cruz Western Blotting Luminol Reagent (sc-2048) was performed.
Results
Microtubules Integrity Is Restored After COL Withdrawal
The first step in this study was to evaluate the microtubules integrity status. Incubation of HeLa cells and HH with COL (16 hours; 1 µmol/L) caused the collapse of microtubular network in both types of the cells (Figure 1). We observed partial restoration of microtubules network (Figure 1) 24 hours after COL withdrawal regardless of proliferating or nonproliferating cells. Thus, the reintegration of microtubules was cell cycle–independent process for we observed the same in proliferating as well as in nonproliferating cells.
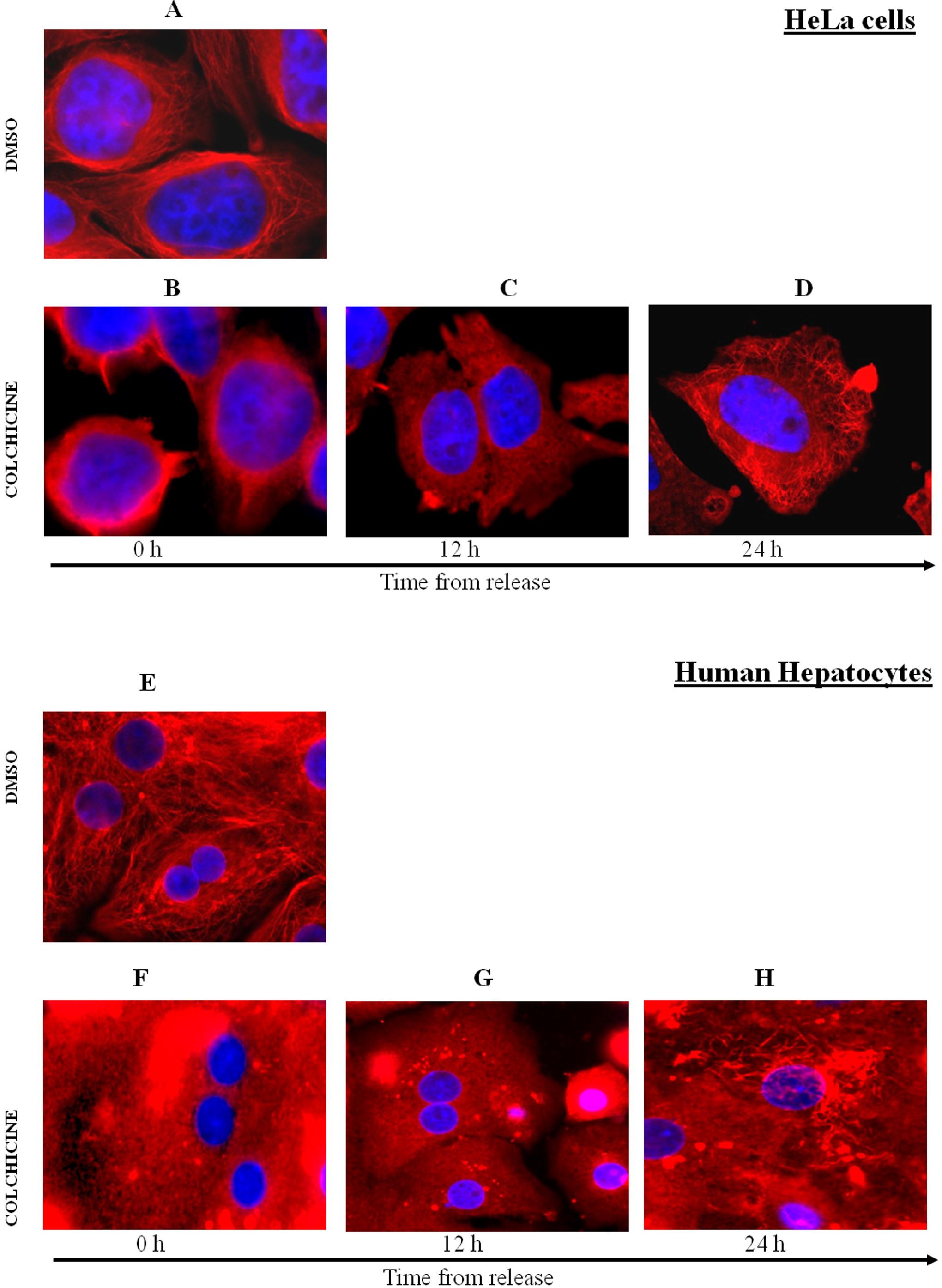
Immunofluorescence staining of microtubules in human hepatocytes (HH) and HeLa cells. In the dimethyl sulfoxide (DMSO)-treated cells, there is apparent detailed microtubules structure with blue nucleus in the middle (A and E). In the case of HH, even 2 nuclei can be seen (E). In the cells treated with colchicine (COL; 1 µmol/L), the microtubules are disintegrated (B and F). There is no apparent change 12 hours after COL withdrawal (C and G), but some fibers of microtubules can be seen in both types of cells another 12 hours later (D and H).
Colchicine Arrests HeLa Cells but Not Primary HH at G2/M Boundary of the Cell Cycle
Asynchronous HeLa cells were synchronized in G2/M phase of the cell cycle by COL (16 hours; 1 µmol/L), as revealed by flow cytometry analyses (Figure 2A, DMSO vs. COL at 0 hours). Throughout the following 24-hour period, a certain portion of HeLa cell population merged into the G2/M corresponding peak. Thus, the G2/M synchronization by COL was irreversible process. In contrast, HH showed characteristic distribution of cells. This pattern was not affected by COL treatment and remained unaltered even 24 hours after COL withdrawal (Figure 2B). We have previously observed similar phenomenon in primary rat hepatocytes. 17 Multiple peaks in HH can be attributed to the presence of 1 or 2 nuclei (Figure 1B) in single cell in the culture.
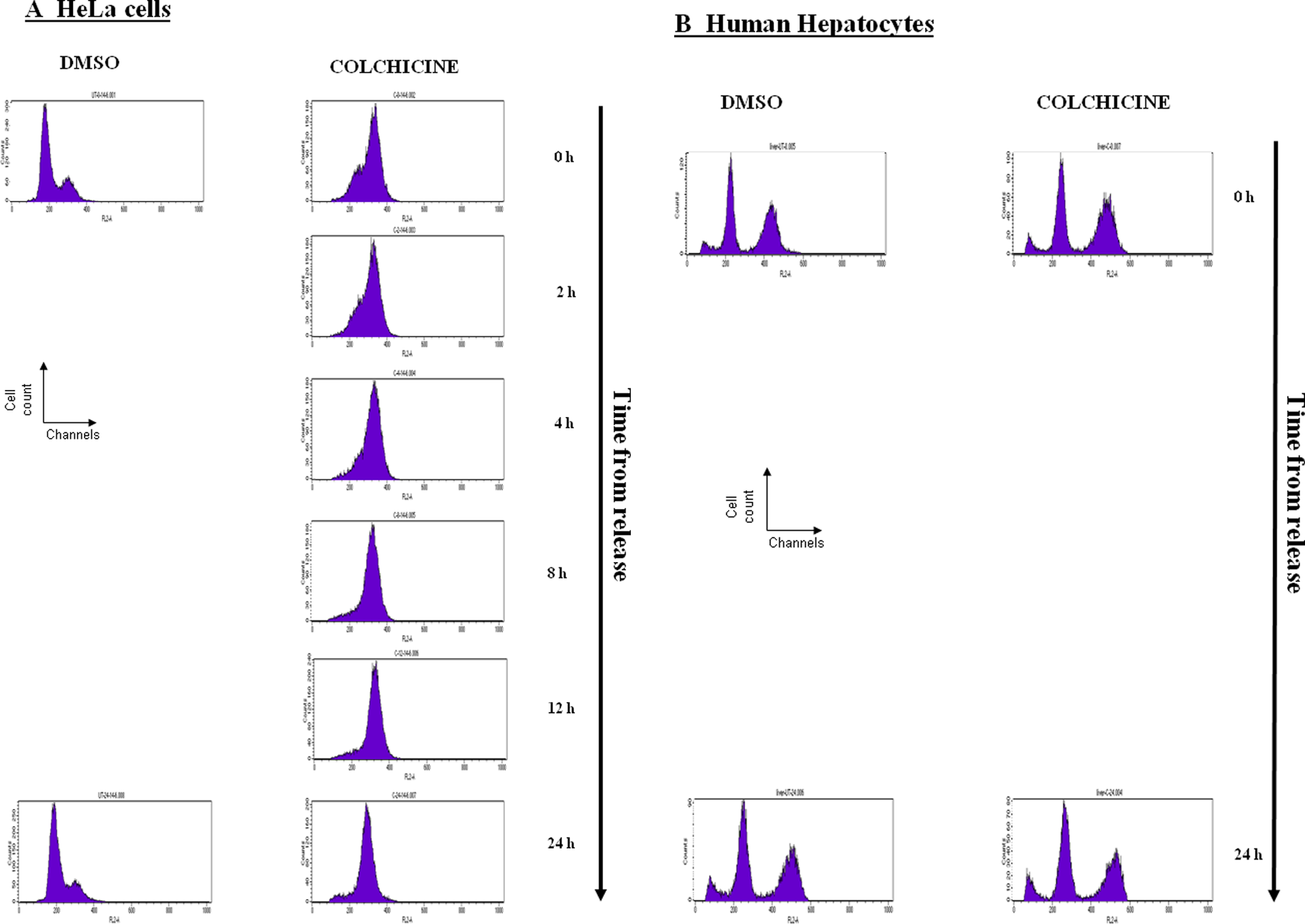
Flow cytometry of HeLa cells and primary human hepatocytes (HH). HeLa cells (A) treated with dimethyl sulfoxide (DMSO) as negative control show 2 peaks corresponding with G0/G1 and G2/M phases of cell cycle. Human hepatocytes (B) treated with DMSO show 3 peaks corresponding to cells in apoptosis phase (peak 1), G0 phase (peak 2) with 1 nucleus, and G0 (peak 3) with 2 nuclei. After colchicine withdrawal, all cells are stopped in G2/M phase peak in HeLa cells (A) only, while there is no apparent effect in HH (B). The calibration bars indicate the fluorescence intensity (horizontal bar) and the cell number. Similar results were obtained from 2 independent experiments.
Colchicine Restricts GR Hormone Binding in Primary HH but Not in HeLa Cells
To validate the GR binding ability in “time from release” period, we performed ligand binding assay (LBA). At this point, it is important to realize that LBA comprises 2 information: (1) qualitative—the affinity and occupancy of GR by DEX and (2) quantitative—the amount of GR protein.
Incubation of HeLa cells with COL (16 hours; 1 µmol/L) resulted in the decrease in DEX-binding to cytosolic GR as compared to incubation with DMSO (Figure 3A, upper panel, compare DMSO and COL at time 0 hours, black bars). Dexamethasone binding to cytosolic GR was increasing initially but at the end it dropped down to its minimum at 24 hours after COL withdrawal, whereas it remained unaltered in extracts from DMSO-preincubated cells (Figure 3A, upper panel, compare DMSO and COL at time 24 hours, black bars). In parallel, we have investigated the ability of cold DEX to compete with radio-labeled DEX in binding to cytosolic GR. The extent of competition between labeled and unlabeled DEX, expressed as the percentages of 3H-DEX displacement by unlabeled DEX, was almost constant over the time period tested, regardless of preincubation with COL (Figure 3A, lower panel).
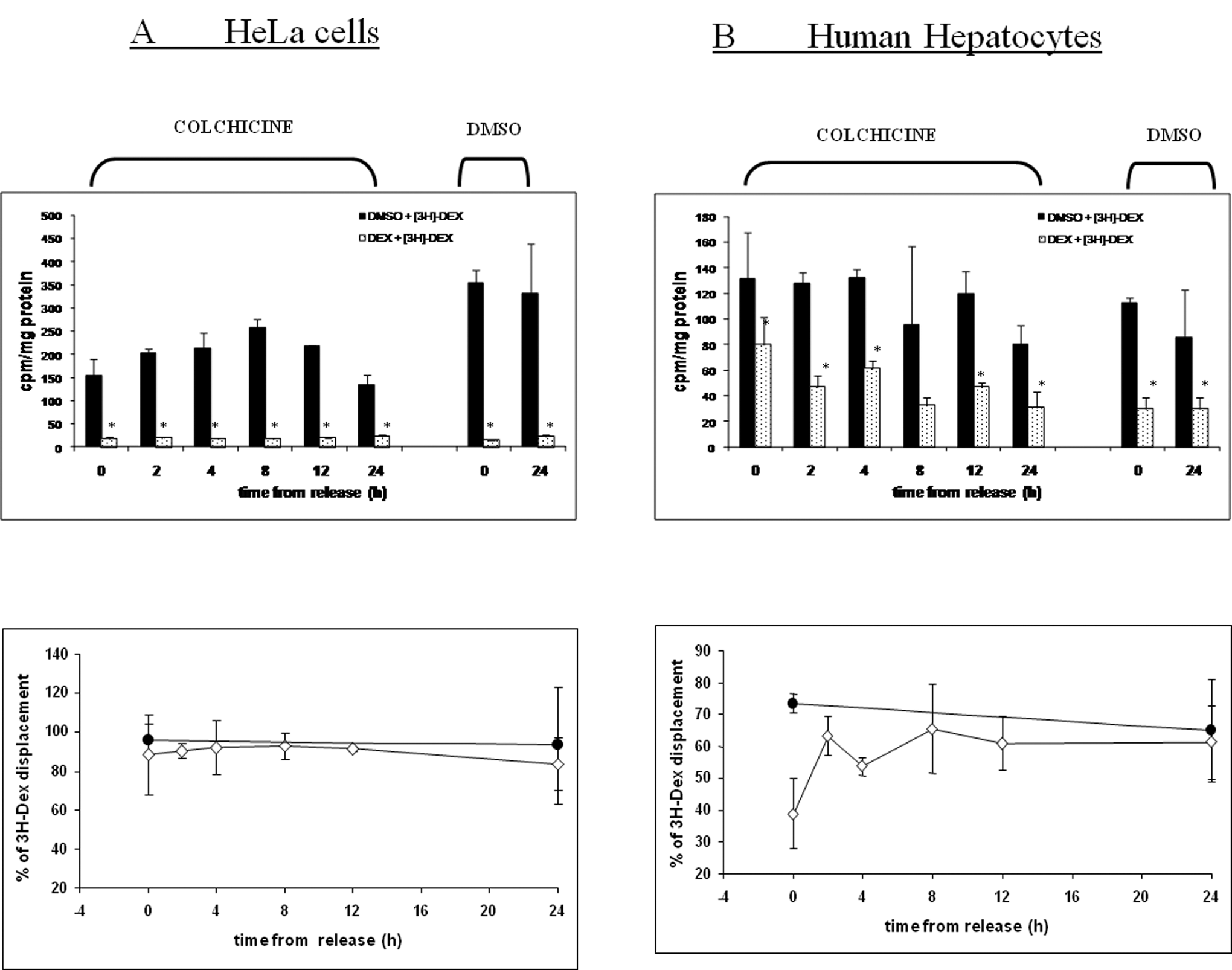
Ligand-binding analysis of colchicine (COL) effects on dexamethasone (DEX)-binding to glucocorticoid receptor (GR). Upper panels: The data in bar graph are expressed as counts per minute normalized on total protein content in sample (cpm/mg protein). Full black bars and spotted bars stand for [3H]-DEX combined with dimethyl sulfoxide (DMSO) and cold DEX as competitors, respectively. Bars are mean ± SD from duplicate measurements for 2 independent cytosolic fraction preparations. *Significantly different (P < .05) from black bars ([3H]-DEX + DMSO). Lower panels: The plots show the extent of displacement of [3H]-DEX by cold DEX, calculated as percentage of displacement = 100 × (1 − DEX/[3H]-DEX), where DEX refers to samples with unlabeled competitor and [3H]-DEX refers to samples with [3H]-DEX only. • = DMSO-preincubated cells; • = COL-preincubated cells. In the upper panels, it is seen that the GR-binding capacity (black bars = DMSO + [3H]-DEX) is diminished after COL treatment in HeLa cells (A) but not in HH (B) where it is slightly increased. Nevertheless, the competitiveness (spotted bars = DEX + [3H]-DEX) is fully preserved in Hela cells (A) but not in HH (B). The lower panels display the extent of this competition in untreated (•) versus COL-treated cells (•). Treatment with COL suppressed the competitiveness in HH but this was restored during “time from release” period (B, lower panel). There was no such effect in HeLa cells (A, lower panel).
In contrast to HeLa cells, incubation of HH with COL (16 hours; 1 µmol/L) resulted in modest increase in DEX-binding to cytosolic GR as compared to DMSO-preincubated cells (Figure 3B, upper panel, compare DMSO and COL at time 0 hours, black bars). This increased binding remained almost constant during the “time from release” period with an exception at 24 hours, where it dropped down. The extent of competition between cold and labeled DEX drastically decreased in HH preincubated with COL (35%-40% inhibition) as compared to DMSO-preincubated cells (75% inhibition; Figure 3B, lower panel). After 24 hours, the extent of competition was fully restored.
Colchicine Withdrawal Leads to GR Protein Content Decline
Because we had observed different radio-labeled hormone binding between extracts from COL- and DMSO-pretreated cells, we further investigated the GR protein content. Incubation of HeLa cells with COL (16 hours; 1 µmol/L) resulted in the decrease in cytosolic GR protein content as compared to incubation with DMSO (Figure 4A, compare DMSO and COL at time 0 hours). This result is in accordance with what we observed previously. 2 In addition, GR level continued declining after the COL withdrawal. Thus, low binding capacity (Figure 3A, upper panel) is most likely due to the decline in GR protein content (Figure 4A). The content of cytosolic GR in DMSO-preincubated cells was constant over the time period tested (Figure 4A). In contrast, the level of cytosolic GR protein was not drastically decreased in COL-preincubated HH (16 hours; 1 µmol/L), but it was rather modestly increased (Figure 4B, compare DMSO and COL at time 0 hours). However, during the “time from release” period, it slightly dropped down and at 24 hours it disappeared completely. This observation is in good correlation with binding capacity of GR in HH (Figure 3B, upper panel) represented by 3H-DEX samples without a competitor (black bars). The evidence that upper band corresponds to the full-length receptor was made by comparison of cytosolic extracts of HeLa and HH cells via WB analysis (data not shown).
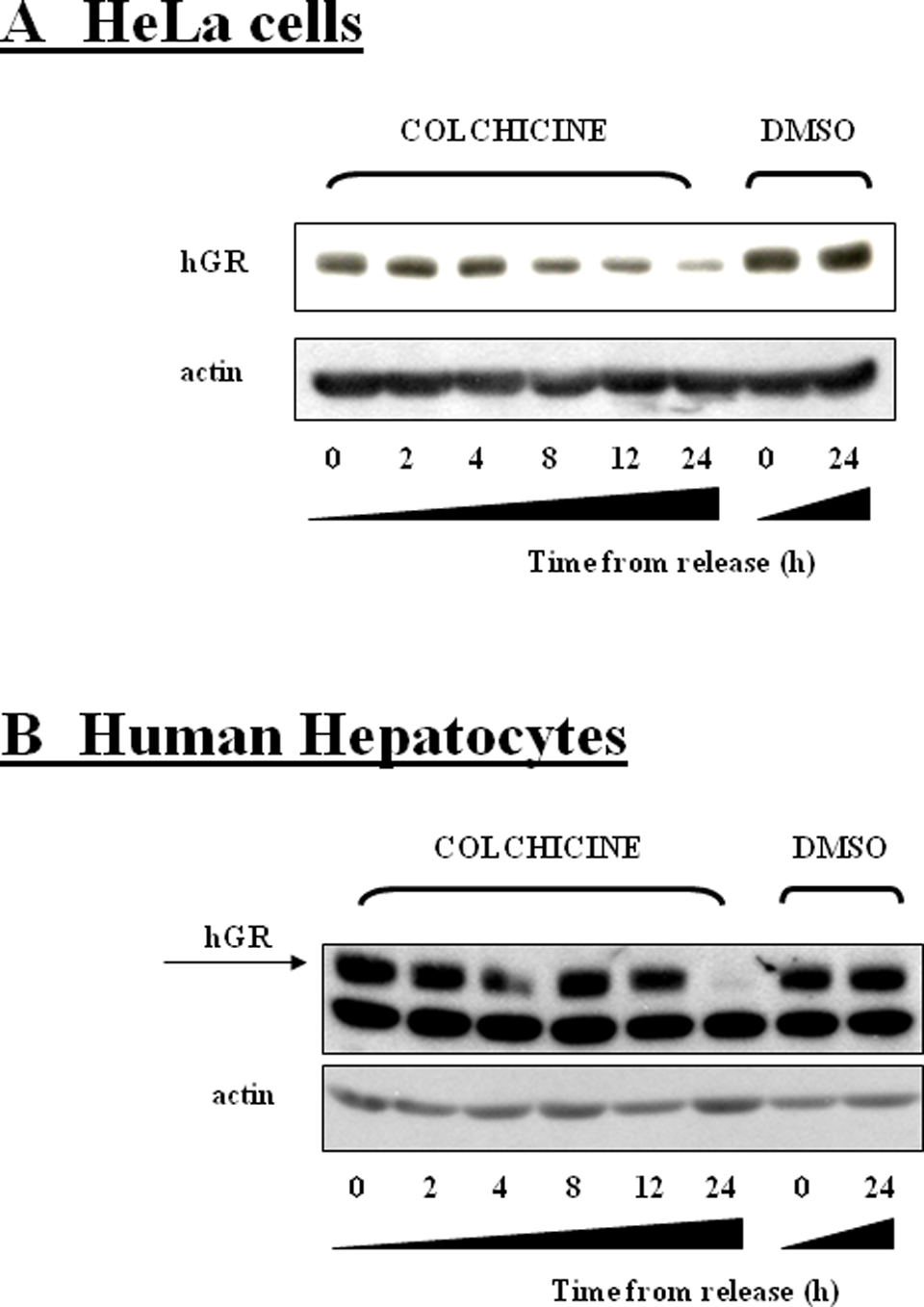
Western blot analysis of colchicine (COL) effects on cytosolic glucocorticoid receptor (GR) protein content. After the treatment with colchicine (1 µmol/L), the GR protein was diminishing during the “time from release” period, but it remained constant in dimethyl sulfoxide (DMSO)-treated Hela cell (A). In contrast, GR protein in human hepatocytes (HH) was slightly increased after COL treatment, but it rapidly diminished 24 hours after COL withdrawal while it remained constant in DMSO-treated hepatocytes (B). Similar behavior was observed in 3 independent experiments.
Apparent radio-labeled hormone binding at 24 hours after COL withdrawal may be due to the nonspecific binding of DEX. Because GR protein disappeared in HeLa as well as in HH regardless of the cell cycle status, we claim that this effect is cell cycle–independent but microtubules-dependent.
Release From COL-Induced Block Activates Transiently ERK in Primary HH but Not in HeLa Cells
Because we observed some similarities in the results from both types of cells (microtubules disruption, protein decline at 24 hours after COL withdrawal) as well as some differences (G2/M phase synchronization for HeLa vs. no effect for HH, increased GR binding affinity in HH vs. no effect in HeLa cells), we wanted to find a link between COL and its effects on hormone binding. For microtubule disruptors are known to activate MAP kinases, 18 –20 we decided to monitor their activation in our experimental setup. We chose the 3 most prominent members of this class of kinases, particularly JNK, p38, and extracellular signal-regulated kinase (ERK).
In the case of p38 and JNK activation, we did not detect either any activation (case of HeLa cells, Figure 5A) or a slight activation was observed for HH on the level of phosphorylated JNK when compared with untreated cells (Figure 5B). This activation did not change during the “time from release” period. Surprisingly, this cannot be said for extracelullar signal-regulated kinase (ERK).
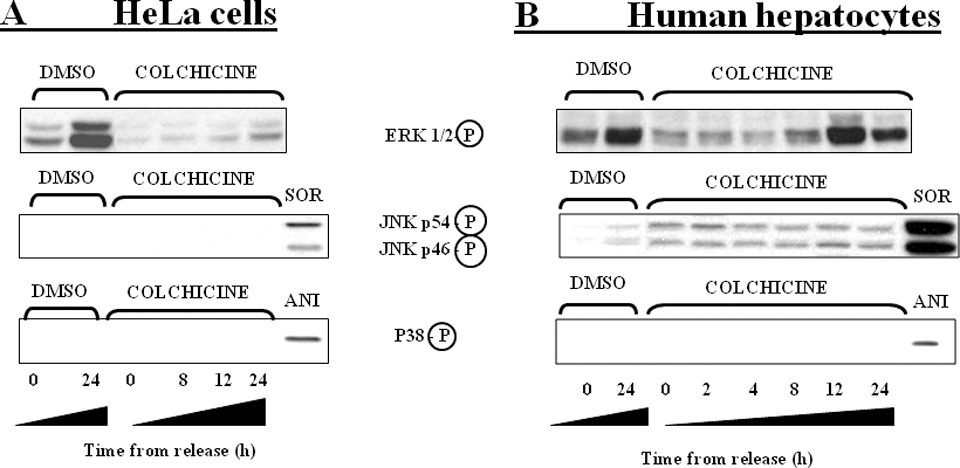
Western blot analysis of colchicine (COL) effects on mitogen-activated protein kinases (MAPKs) activation. After treatment with dimethyl sulfoxide (DMSO), extracellular signal-regulated kinase (ERK) activation was suppressed in HeLa cells (A, 0 hours) and in human hepatocytes (HH; B, 0 hours) while it was fully restored 24 hours later. Presence of COL suppressed ERK activation in both cellular models but there was a robust transient ERK activation in HH (B) 12 hours after COL withdrawal but none in HeLa cells (A). P38 was not detectably activated in any cell model tested (A and B). c-Jun-N-terminal kinase (JNK) was activated only slightly in HH (B). Positive controls sorbitol (SOR) and anisomycin (ANI) were used for exact identification of activated JNK and p38, respectively. Similar behavior was observed in 3 independent experiments.
In HeLa cells pretreated with COL (16 hours; 1 µmol/L), the level of phospho-ERK1/2 was strongly suppressed (Figure 5A). However, we observed the same for DMSO-pretreated cells. The level of activation for DMSO-pretreated cells was at the nominal values after 24 hours, while the COL pretreatment inhibited the restoration of activation even in the presence of serum-containing medium. Similar observation was seen in HH (Figure 5B). The major difference was the unexpected activation of ERK1/2 12 hours after the COL withdrawal and it preceded the GR protein diminution (Figure 4B). Total level of ERK1/2 was not affected in any cell model tested.
Colchicine Withdrawal Leads to Induction of Apoptosis in Primary HH and in HeLa Cells
It is known that cytoskeleton disruption leads to apoptosis in several cells in vitro. 21 –23 To confirm the role of apoptosis in our study, we monitored the level of caspase-3, a critical executioner of apoptosis, and its cleaved fragments. After the COL withdrawal, we detected a decreasing amount of full-length caspase-3 during the “time from release” period in Hela cells (Figure 6A) and in HH (Figure 6B). As a matter of fact, we detected an increasing amount of cleaved fragments of caspase-3 in both cells. Thus, COL withdrawal triggered an apoptotic process.
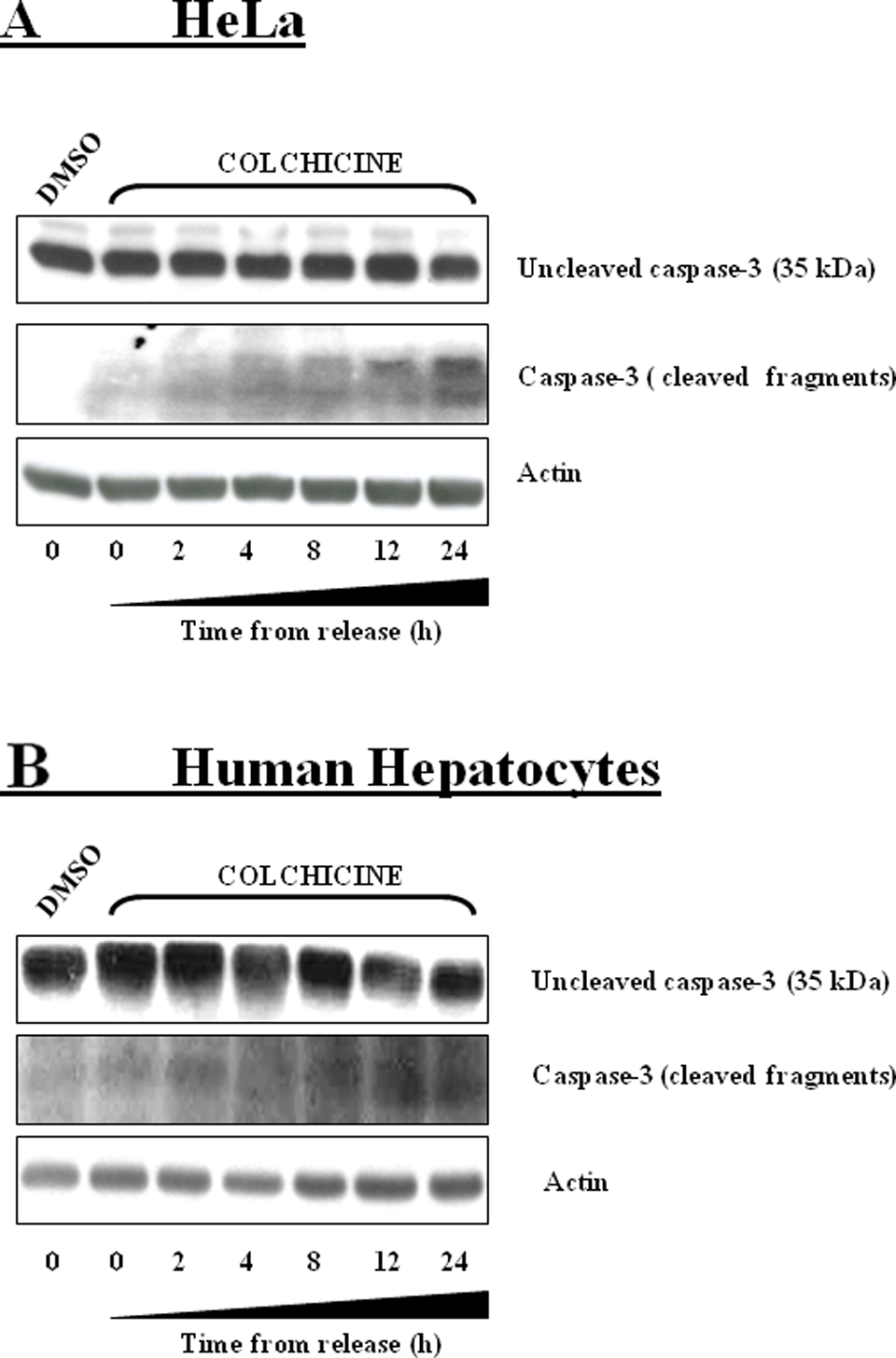
Western blot detection of caspase-3. After treatment with colchicine (COL), decreasing content of full-length caspase-3 (35 kDa) and increasing amount of its 2 fragments during the “time from release” period in HeLa cells was detected (A). Concerning the full-length caspase-3, we observed the same in human hepatocytes (HH; B) even though the cleaved fragments were hard to detect. Similar behavior was observed in 2 independent experiments.
Discussion
In our current study, we demonstrate that COL selectively affects GR functions, which is supported in particular by the following findings. (1) Colchicine caused a decrease in GR protein content in HeLa cells but not in HH. However, there was apparent posttreatment decline in GR protein level in accordance with the microtubules network restoration in both cellular models independently of cell cycle status. (2) Colchicine affected competitiveness of labeled and nonlabeled hormone in cytosolic extracts of HH but not of HeLa cells. This effect was dependent on microtubules disruption/reintegration processes but independent of cell cycle status. Effects in hormone binding capacity correlated well with the changes in the GR protein content.
Partial microtubule network restoration process seems to be the only common thing for both tested cell models. In other measured parameters, we found opposite effects. The protein content was significantly reduced at the starting point of the experiment in Hela cells but not in HH (Figure 4). This is in accordance with previously described proteasome-ubiquitine-dependent degradation of GR protein in HeLa cells by microtubules disruptors. 2 However, the process of protein decline kept continuing for following incubation period even after COL withdrawal. In opposite to the observation in Hela cells, other authors reported increased amount of GR protein in WCL2 cells synchronized in G2/M phase. 12 This could be due to differences between endogenous human GR used in our study and overexpressed exogenous hamster GR investigated by Hu et al.
An interesting thing that arose from this study is the COL-mediated increase in GR affinity toward DEX in HH but not in HeLa cells (Figure 3, lower panels). This was confirmed by the inability of nonlabeled hormone to displace the radiolabeled one. It brings another piece of puzzle in understanding of GR action within hepatocytes. It can be clearly stated that while the transcriptional activity is suppressed by inhibited translocation, 1 the hormone binding affinity of GR increases. In addition, we observed decline in hormone binding capacity in HeLa cells, which is not in accordance with observation made earlier 6 where the authors observed circa 50% increase in DEX-binding capacity throughout S-G2-M phases as compared with G1. However, the protein quantification was not done and we cannot compare whether the effect was due to higher afinity or higher GR protein content. Consistently, a study performed a decade later proved that both hormone binding capacity and GR protein level were increased in G2/M phases of cell cycle. 12
Another common denominator in this study is the inhibition of ERK activation in both cellular models by DMSO (Figure 5). This effect is intensified by COL. However, while there is no apparent restoration of activation in HeLa cells, there is a transient one in HH. These data suggest an ERK participation in GR disappearance at 24 hours after COL removal. But what stimulus triggered ERK activation in HH remains enigmatic.
Similarly to what we observed previously, 24 COL activated JNK after 16 hours in HH (Figure 5B). However, the level of JNK activation remained unaltered and for this reason we do not anticipate a connection between this activation and sudden GR disappearance in HH. In addition, there was slight JNK activation in untreated cells 24 hours after DMSO withdrawal as well, probably reflecting the well-known phenomenon of “primary cultures mortality” in contrast to “immortal” cancer-derived cell lines.
In contrast to many cell cycle–related studies, we used a tropole alkaloid COL instead of the generally used double thymidine block to synchronize the cells. Thus, the different actions of these compounds on cells might reflect the above-mentioned discrepancies. This statement can be obviously confirmed by caspase-3 detection, which we made in both cellular models (Figure 6). Because COL is known to trigger apoptotic process in some cells, 25,26 the overall decrease in GR protein content may be attributed to this process.
There are not many studies that would explain GR signaling pathway modification after microtubule disruption in detail. Moreover, some claim that microtubules are not required for GR-mediated gene induction. 27 But at least for physiologically relevant cell model, HH, this is not the truth. 1 Because COL is a drug of choice for gout treatment and similarly acting microtubules destabilizers (vincristine, vinblastine) are often used anticancer drugs, these patients may suffer from large drug–drug interactions. Patients with cancer taking some of these drugs have disturbed the GR signaling pathway. Glucocorticoid receptor is known regulator of some P450s and also regulates “the regulators” like pregnane X receptor (PXR) and constitutive androstane receptor (CAR). 28 Thus, patients being on these drugs might have decreased metabolic capacity. Consequently, the level of supporting drugs (eg, antimycotics) must be dynamically modified to prevent a toxic effect of these drugs. In addition, another microtubules-interfering agent used in breast cancer therapy, taxol, which in contrast to above-mentioned drugs stabilizes the microtubules, interferes with cell signaling by PXR 29 ; hence, the final in vivo situation is more complex in this view. Moreover, the drug metabolism is not the only one system being altered by microtubules destabilizers. In addition to xenobiotic metabolism, the effect of GR on glucose homeostasis is well known.
Still, an essential question arises: What is it that distinguishes COL action in HH and in HeLa cells? Receptors? Transporters? Metabolism? Signaling events that lack the usage in nonproliferating cells? The true answer remains to be discovered.
Overall, the role of microtubules in GR function is controversial. However, the evidences that perturbation of microtubules network may alter stability, subcellular localization, and transcriptional activity of GR are undisputable. 30 –32 The major intention of this article was to draw the attention to the differences of GR functionality between proliferating (HeLa) and nonproliferating (HH) cells (both possessing functional GR) and to demonstrate that some GR functions are perturbed in microtubules-dependent way in quiescent cells (competitiveness of nonlabeled and labeled hormone, hormone binding capacity) but not in proliferating cells when using a microtubule-disrupting agent like COL.
Footnotes
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: grants GACR 304/10/0149 and 303/07/0128 and 305/08/P089 from the Grant Agency of the Czech Republic.