
Abstract
Cyclosporine A is a well-known immunosuppressant drug that is currently used for prevention of allograft rejection. The current study was conducted to explore the therapeutic potential of cyclosporine A against 3-nitropropionic acid (3-NP)–induced neurotoxicity, an animal model of Huntington disease (HD). Systemic administration of 3-NP (10 mg/kg) for 14 days significantly impaired body weight, motor activity, biochemical parameters (raised lipid peroxidation, nitrite concentration, depletion of superoxide dismutase [SOD] and catalase), and mitochondrial enzymes. Cyclosporine A (2.5, 5, and 10 mg/kg) treatment significantly attenuated behavioral, biochemical, and cellular alterations. Furthermore,
Keywords
Huntington disease (HD) is one of the movement disorders, caused by preferential degeneration of medium spiny γ-aminobutyric acid (GABA)ergic neurons located in the striatum and associated with gait and cognitive deficits. 1 Huntington disease is an inherited autosomal dominant disorder with complete penetrance, resulting from a mutation in the huntingtin gene on chromosome 4. The nature of the mutation is an expansion of CAG repeats in exon 1 of the gene. 2,3 The expansion of the CAG repeats causes a region of polyglutamine on the “huntingtin” protein that enhances the function of caspase 3. Because this protease is known to play a role in apoptosis (a type of programmed cell death), it is thought that the huntingtin mutation leads to inappropriate apoptosis and destruction of cells. 4 Several mechanisms have been proposed to explain HD pathogenesis, including oxidative stress, mitochondrial defects, and excitotoxicity.
3-Nitropropionic acid (3-NP) is a mycotoxin that is isolated from fungus Arthinium and first described in 1970s when several children accidentally ingested contaminated sugarcane, resulting in motor dysfunction, with symptoms similar to those of HD. 3-Nitropropionic acid, which resembles the substrate succinate, irreversibly binds to succinate dehydrogenase (SDH) and inhibits its activity in the citric acid cycle, ultimately interfering with ATP synthesis and leading to selective degeneration of the striatal medium spiny GABAergic neurons. 5 –7 Because 3-NP induces several behavioral and cellular alterations similar to those seen in patients with HD, 3-NP is regarded as an important experimental tool to screen drug therapy for this complex disease. 8,9
Nitric oxide (NO) is a gaseous messenger synthesized from the oxidation of
Cyclosporine A extracts from Hypocladium inflatum gam are currently being used as an immunosuppressant drug against organ transplantation rejection cases. Cyclosporine A exerts its action by binding to small intracellular regulatory proteins, cyclophilins (CyPs). 16,17 In addition to its immunosuppressive property, cyclosporine A exerts a powerful neuroprotective and neurotrophic action. Leventhal et al previously reported that cyclosporine A protects the striatal neurons from 3-NP toxicity. 18 Immunosuppressants exert neuroprotective and neurotrophic actions against traumatic brain injury, sciatic nerve injury, focal, and global ischemia in animals. 19 However, the mechanisms of these neuroprotective actions are not understood so far. 19
Therefore, the current study has been designed to explore the possible functional relationship between cyclosporine A and NO pathways in 3-NP-induced neurotoxicity. A series of experiments have been carried out to evaluate the possible NO modulation on the protective effect of cyclosporine A.
Materials and Methods
Animals
Male Wistar rats (300-350 g) bred in Central Animal House facility of the Panjab University, Chandigarh, were used. Animals were acclimatized to laboratory conditions prior to experimentation. Animals were kept under standard conditions with food and water ad libitum. All the experiments were carried out between 09:00 and 15:00 hours. The protocol was approved by the Institutional Animal Ethics Committee and carried out in accordance with the Indian National Science Academy Guidelines for the use and care of animals.
Drugs and Treatment Schedule
The following drugs were used in the current study. 3-Nitropropionic acid,
Group 1 vehicle-treated group, group 2 received 3-NP (10 mg/kg, IP) for 14 days, group 3 received cyclosporine A (10 mg/kg by canula) alone, groups 4 to 6 received cyclosporine A (2.5, 5, and 10 mg/kg by canula) + 3-NP (10 mg/kg, IP) for 14 days, group 7 received
In the current study,
Measurement of Body Weight
Animals were weighed on the first and last day of the experimentation. Percentage change in body weight was calculated as
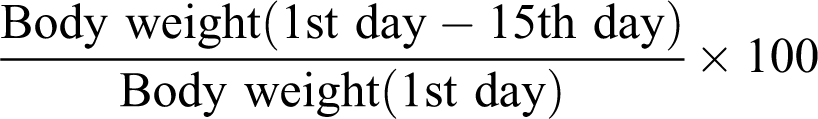
Behavioral Assessments
Assessment of gross behavioral activity (locomotor activity)
The locomotor activity was monitored using an actophotometer (IMCORP, Ambala, India). The motor activity was detected by infrared beams above the floor of the testing area. Animals were placed individually in the activity chamber for a 3-minute acclimation period before starting actual activity tasks. Each animal was observed over a period of 5 minutes and activity was expressed as counts per 5 minutes. 21
Rotarod Activity
The motor coordination and grip performance of the animals were evaluated using the rotarod apparatus (Techno, Ambala, India). The rats were exposed to a prior training session to acclimatize them to rotarod performance. Rats were placed on a rotating rod with a diameter of 7 cm (speed 25 rpm). The cutoff time was 180 seconds and each rat performed 3 separate trials after a 5-minute gap. The average time of the fall was recorded. 8
Dissection and Homogenization
On 15th day, animals were randomly divided into 2 groups, 1 for biochemical estimations and the other for mitochondrial enzyme complex estimations immediately after the behavioral assessments. The animals were sacrificed by decapitation and the brains were dissected out. The cortex, striatum, and hippocampus were separated by putting on ice. A 10% (w/v) tissue homogenate was prepared in 0.1 mol/L phosphate buffer (pH 7.4). The homogenate was centrifuged at 10 000g for 15 minutes. Aliquots of the supernatant were separated and used for biochemical estimations.
Measurement of Oxidative Stress Parameters
Measurement of lipid peroxidation
The quantitative measurement of lipid peroxidation in the brain was performed according to the method of Wills. 22 The amount of malondialdehyde (MDA), a measure of lipid peroxidation, was measured by reaction with thiobarbituric acid at 532 nm using a Perkin Elmer lambda 20 spectrophotometer (Norwalk, Connecticut). The values were calculated using the molar extinction coefficient of the chromophore (1.56 × 105 mol/L−1cm−1) and expressed as a percentage of the vehicle-treated group.
Estimation of nitrite
The accumulation of nitrite in the supernatant, an indicator of the production of NO, was determined by a colorimetric assay with Greiss reagent (0.1% N-(1-naphthyl) ethylenediame dihydrochloride, 1% sulfanilamide, and 2.5% phosphoric acid) as described by Green et al. 23 Equal volumes of supernatant and Greiss reagent were mixed, and this mixture was incubated for 10 minutes at room temperature in the dark. Absorbance at 540 nm was measured with a Perkin Elmer lambda 20 spectrophotometer. The concentration of nitrite in the supernatant was determined from a sodium nitrite standard curve and expressed as a percentage of the vehicle-treated group.
Catalase estimation
Catalase activity was assayed by the method of Luck, 24 in which the breakdown of hydrogen peroxide (H2O2) is measured at 240 nm. Briefly, the assay mixture consisted of 12.5 mmol/L H2O2 in phosphate buffer (50 mmol/L of pH 7.0) and 0.05 mL of supernatant from the tissue homogenate (10%), and the change in absorbance was recorded at 240 nm. The results were expressed as millimoles per liter of H2O2 decomposed per milligram of protein per minute.
Superoxide dismutase activity
Superoxide dismutase (SOD) activity was assayed according to the method of Kono 25 in which the inhibition of the reduction of nitrazobluetetrazolium (NBT) by SOD is measured at 560 nm using a spectrophotometer. Briefly, the reaction was initiated by the addition of 20 mmol/L of hydroxylamine hydrochloride to the mixture containing 96 mmol/L of NBT and 0.1 mL of the sample. The results were expressed as units per milligram protein.
Protein estimation
Protein concentration was measured by the Biuret method using bovine serum albumin (BSA) as a standard. 26
Mitochondrial Complex Estimation
Isolation of rat brain mitochondria
Rat brain mitochondria were isolated by the method of Berman and Hastings. 27 The brain regions were homogenized in isolation buffer with ethylene glycol tetraacetic acid (EGTA; 215 mmol/L mannitol, 75 mmol/L sucrose, 0.1% BSA, 20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1 mmol/L EGTA, pH 7.2). The homogenate was centrifuged at 13 000g for 5 minutes at 4°C. The pellet was resuspended in isolation buffer with EGTA and spun again at 13 000g for 5 minutes. The resulting supernatant was transferred to new tubes and topped off with isolation buffer containing EGTA and spun again at 13 000 g for 10 minutes. The pellet containing pure mitochondria was resuspended in isolation buffer without EGTA.
Complex I (nicotinamide adenine dinucleotide dehydrogenase activity)
Complex I was measured spectrophotometrically by the method of King and Howard. 28 The method involves the catalytic oxidation of nicotinamide adenine dinucleotide (NADH) to NAD+ with subsequent reduction of cytochrome C. The reaction mixture contained 0.2 mol/L glycyl glycine buffer pH 8.5, 6 mmol/L NADH in 2 mmol/L glycyl glycine buffer, and 10.5 mmol/L cytochrome C. The reaction was initiated by the addition of 10 μL solubilized mitochondrial sample (0.00968 mg protein). The absorbance change at 550 nm was followed for 2 minutes.
Complex II (SDH activity)
Succinate dehydrogenase was measured spectrophotometrically according to the method of King. 29 The method involves the oxidation of succinate by an artificial electron acceptor, potassium ferricyanide. The reaction mixture contained 0.2 mol/L phosphate buffer pH 7.8, 1% BSA, 0.6 mol/L succinic acid, and 0.03 mol/L potassium ferricyanide. The reaction was initiated by addition of the 25 μL mitochondrial sample (0.025 mg protein) and the absorbance change at 420 nm was followed for 2 minutes.
Complex IV (cytochrome oxidase assay)
Cytochrome oxidase activity was assayed in brain mitochondria according to the method of Sottocasa et al. 30 The assay mixture contained 0.3 mmol/L reduced cytochrome C in 75 mmol/L phosphate buffer. The reaction was initiated by addition of the 10 μL solubilized mitochondrial sample (0.00968 mg protein) and absorbance change at 550 nm was followed for 2 minutes.
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-diphenyl-H-tetrazolium bromide assay
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-H-tetrazolium bromide (MTT) assay is based on the reduction of MTT by hydrogenase activity in functionally intact mitochondria. The MTT reduction rate was used to assess activity of the mitochondrial respiratory chain in isolated mitochondria by the method of Liu et al. 31 Briefly, 100 μL mitochondrial samples containing 0.0656 mg of protein were incubated with 10 μL MTT for 3 hours at 37°C. The blue formazan crystals were solubilized with dimethylsulfoxide and measured by an enzyme-linked immunosorbent assay (ELISA) reader with a 580 nm filter (Model 680 Microplate Reader, Bio-Rad, Japan).
Statistical Analysis
The data were analyzed using 1-way analysis of variance (ANOVA) followed by Tukey test. All the values are expressed as mean ± SEM. In all tests, the criterion for statistical significance was P < .05.
Results
Effect of Cyclosporine A on Body Weight, Locomotor, and Rotarod Performance and Its Modification by NO Modulators in 3-NP-Treated Rats
3-Nitropropionic acid (10 mg/kg) treatment caused significant decrease in body weight, locomotor, and rotarod endurance on 15th day as compared to vehicle-treated group. Furthermore, cyclosporine A (2.5, 5, and 10 mg/kg by canula) treatment significantly improved body weight, locomotor, and rotarod endurance as compared to 3-NP-treated group (P < .05). Pretreatment of
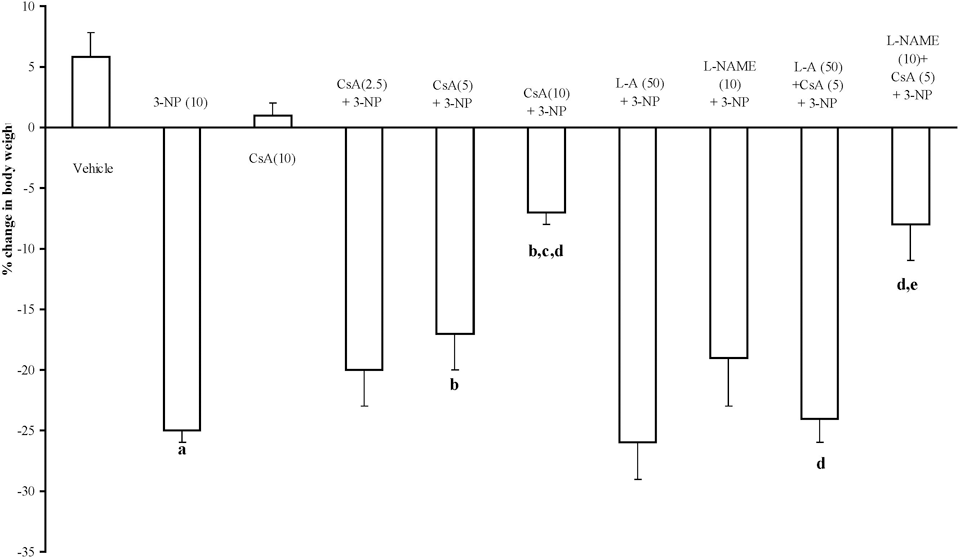
Effect of cyclosporine A,
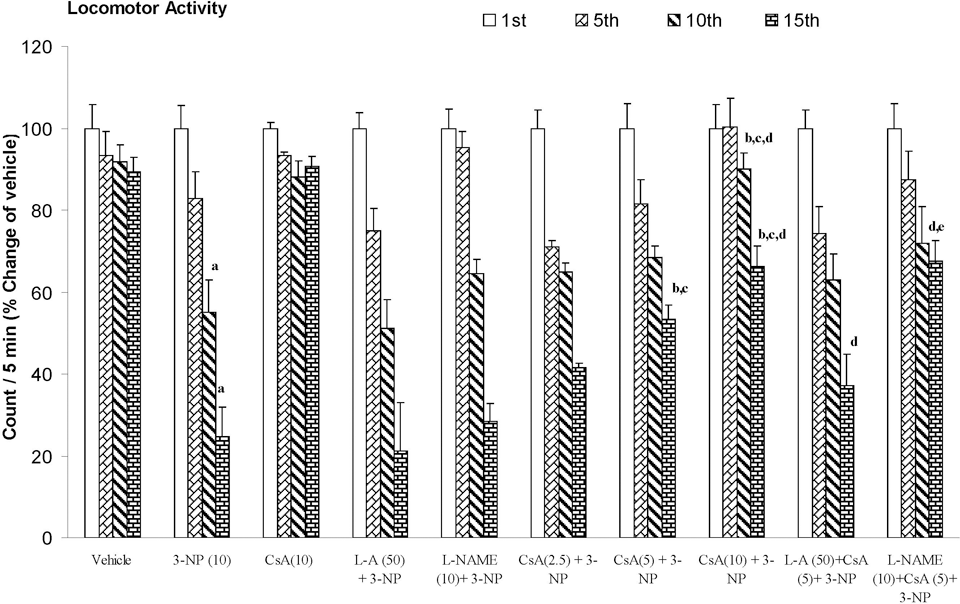
Effect of cyclosporine A,
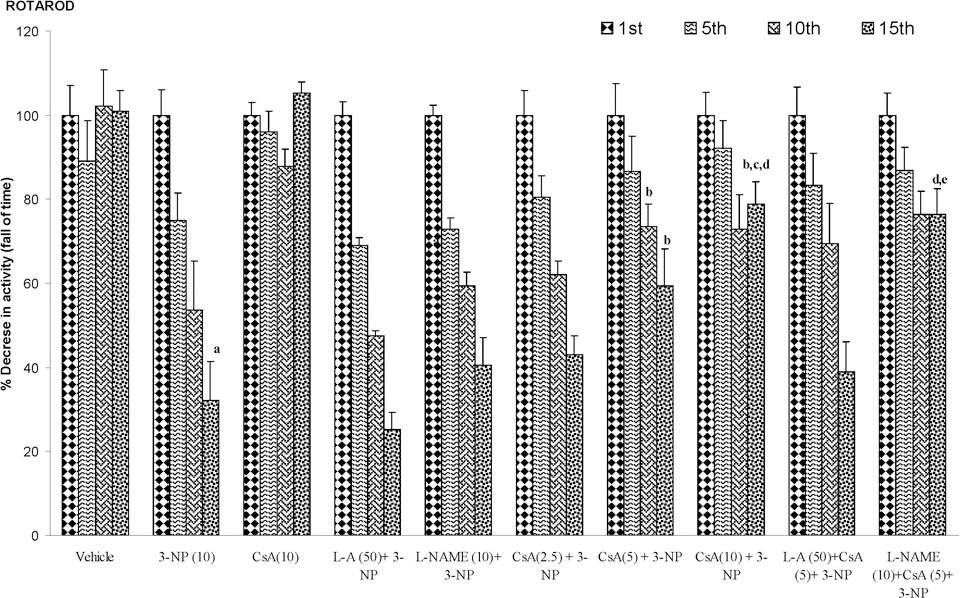
Effect of cyclosporine A,
Effect of Cyclosporine A on Brain Oxidative Damage (Lipid Peroxidation, Nitrite, SOD, and Catalase) and Its Modification by NO Modulators in 3-NP-Treated Rats
Systemic administration of 3-NP significantly increased lipid peroxidation (MDA levels) and nitrite concentration, while also reducing SOD and catalase enzyme activities in striatum, cortex, and hippocampus regions of the brain as compared to vehicle-treated group. However, cyclosporine A (2.5, 5, and 10 mg/kg by canula) treatment attenuated the oxidative damage (decreased MDA and nitrite levels and restored SOD and catalase enzyme activities) as compared to 3-NP-treated animals (P < .05; Table 1
).
Effect of Cyclosporine A, l-Arginine, and l-NAME on 3-Nitropropionic Acid–Induced Biochemical Changes in the Striatum, Cortex, and Hippocampus a
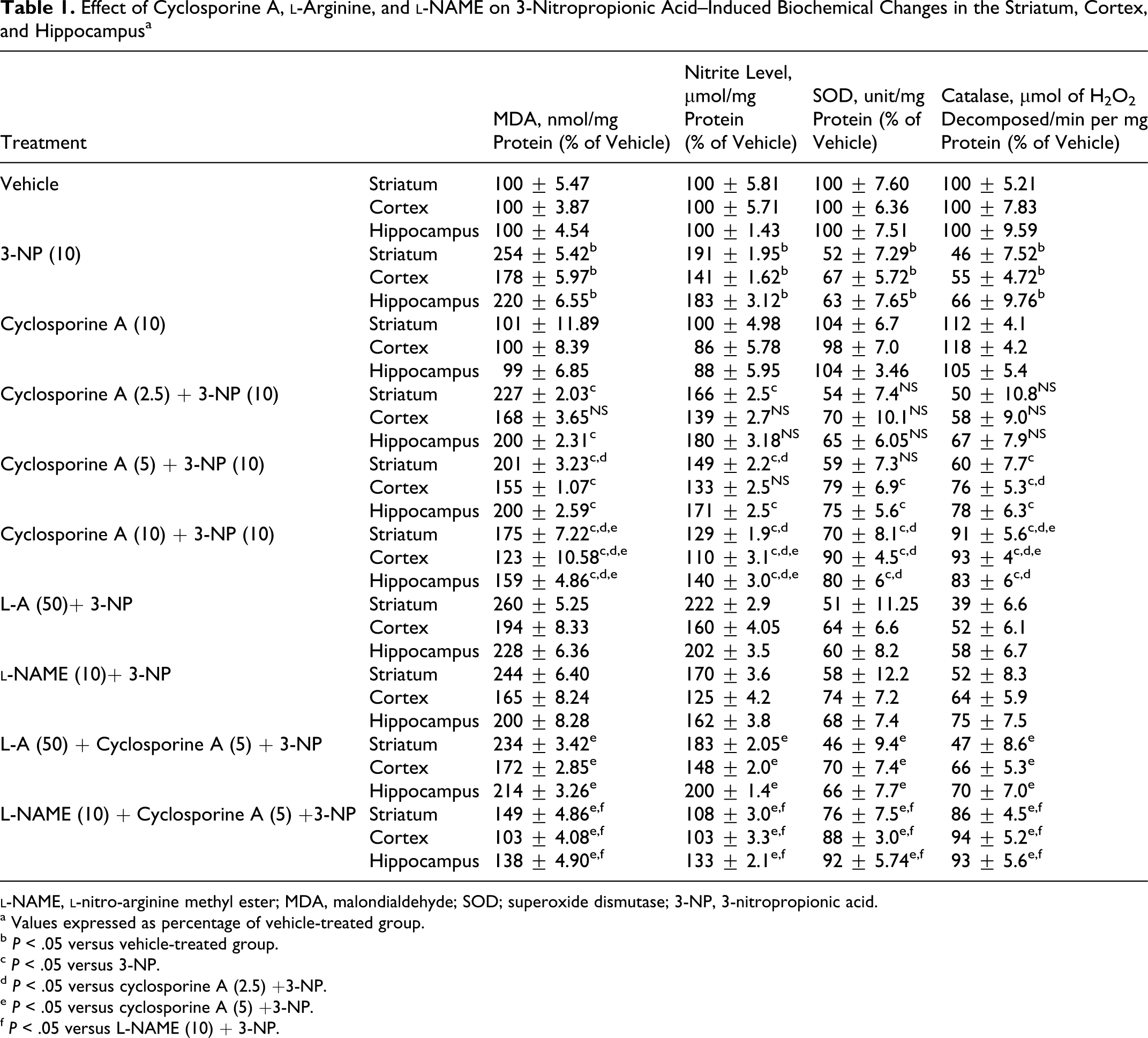
a Values expressed as percentage of vehicle-treated group.
b P < .05 versus vehicle-treated group.
c P < .05 versus 3-NP.
d P < .05 versus cyclosporine A (2.5) +3-NP.
e P < .05 versus cyclosporine A (5) +3-NP.
f P < .05 versus L-NAME (10) + 3-NP.
Effect of Cyclosporine A on Mitochondrial Enzymes Levels and Its Modification by NO Modulators in 3-NP-Treated Rats
Systemic 3-NP (10 mg/kg) administration significantly reduced mitochondrial enzyme (I, II, IV, and MTT assay) activity as compared to vehicle-treated rats. Chronic administration of cyclosporine A (10 mg/kg by canula) attenuated mitochondrial enzyme activity as compared to 3-NP-treated group (P < .05; Figures 4-7).
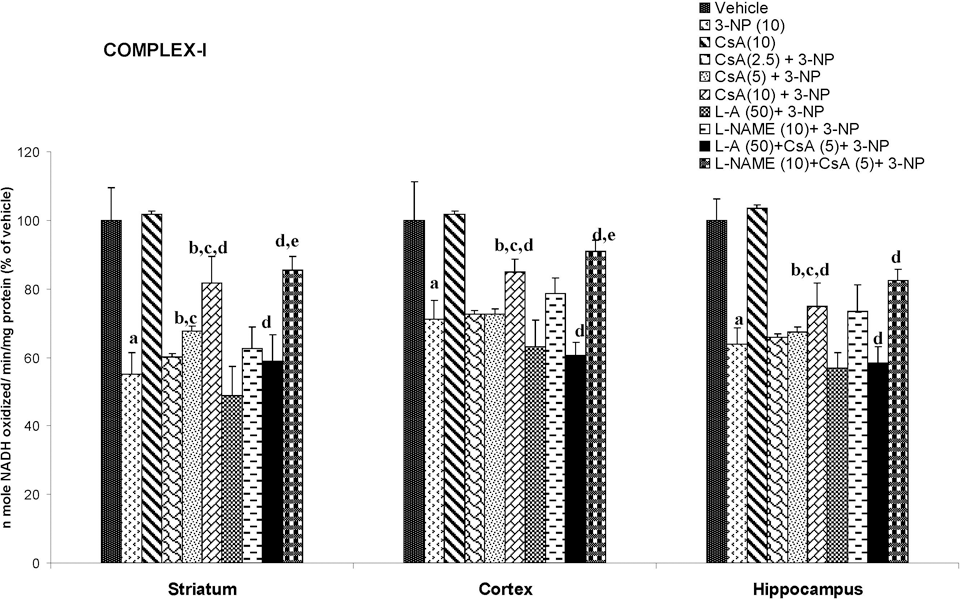
Effect of cyclosporine A,
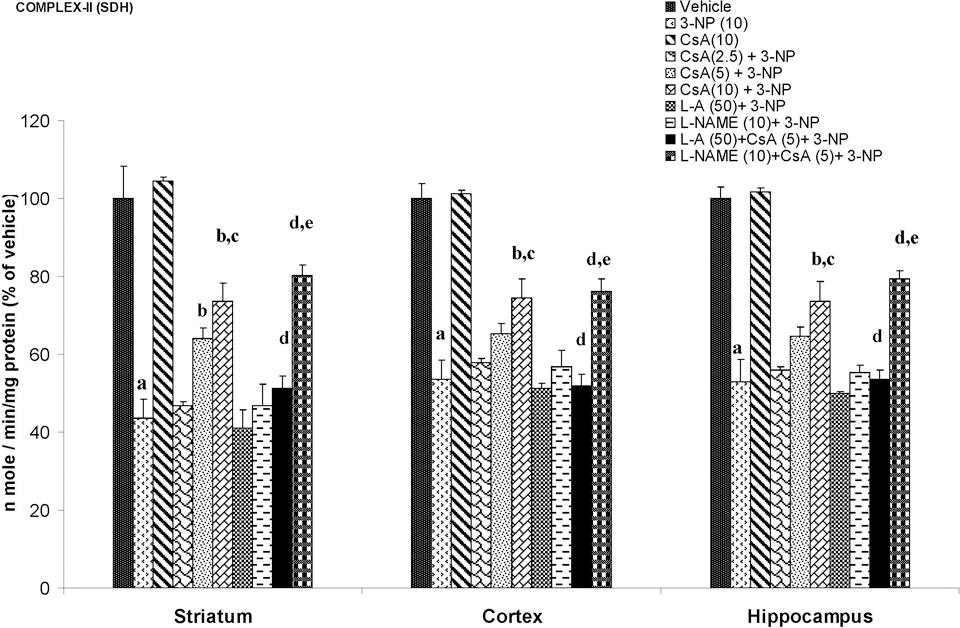
Effect of cyclosporine A,
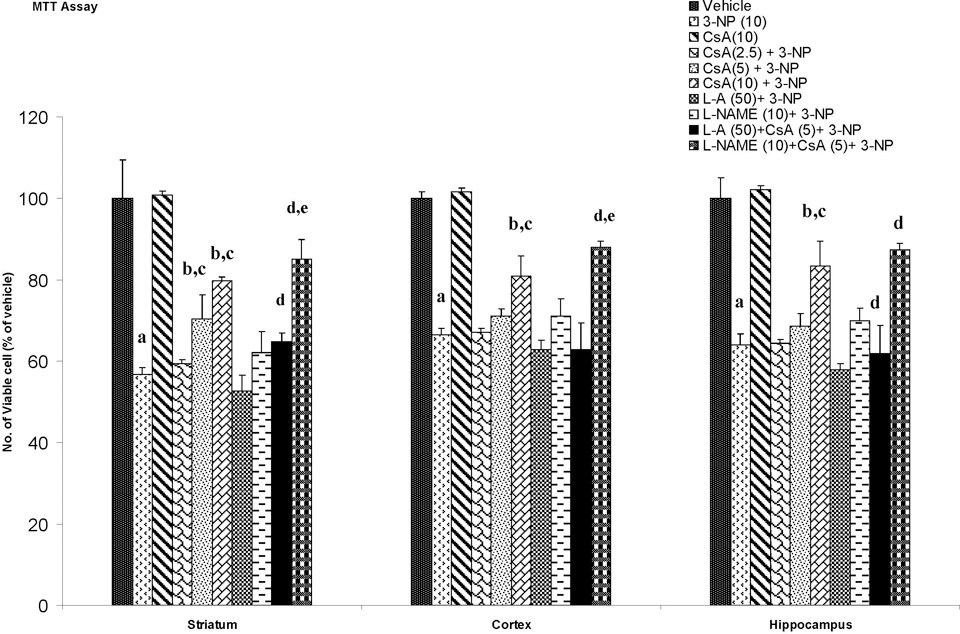
Effect of cyclosporine A,
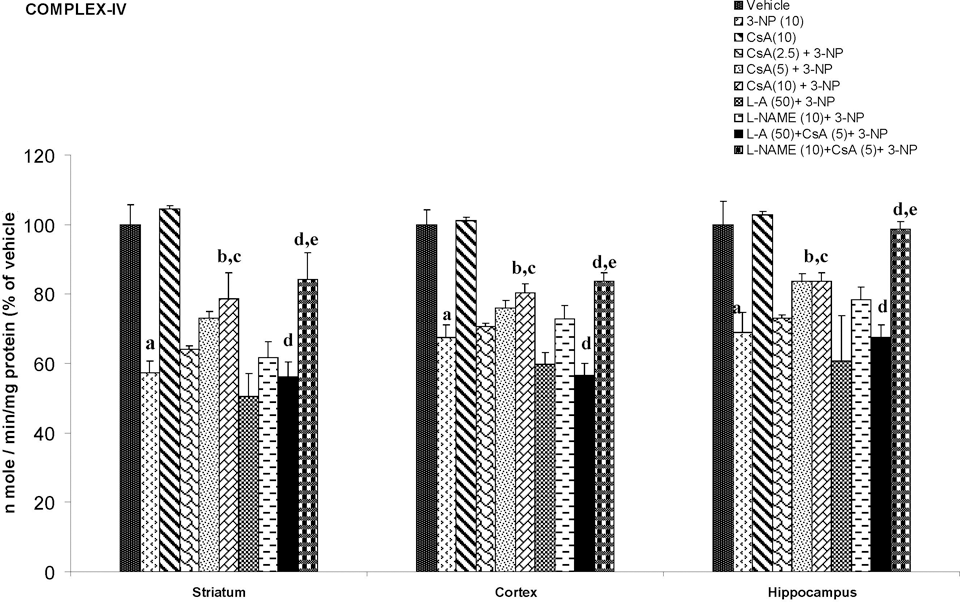
Effect of cyclosporine A,
Discussion
The current study investigates the effect of systemic administration of 3-NP on body weight, motor activities, and biochemical and mitochondrial parameters in rats. The locomotor activity and motor functions in 3-NP-treated animals were significantly decreased as compared to vehicle-treated group as evidenced by actophotometer and rotarod apparatus, respectively. The results showed that animals developed motor impairment following 3-NP administration for 14 days. This progression of motor abnormality could be due to degeneration of the neurons in the striatum. Another hallmark of the 3-NP neurotoxicity is decrease in the body weight, as observed in the present set of experiments, which might be result of depressed energy metabolism, which ultimately causes the decrease in ATP generation in the body. Cyclosporine A treatment significantly attenuated 3-NP-induced behavioral alterations. It has also been reported that cyclosporine A treatment significantly attenuated behavioral alterations in rats with hemi Parkinsonism 32 and transgenic mice for HD. 33 Previous study from our laboratory also report that cyclosporine A attenuates the 3-NP-induced cognitive impairment and glutathione redox dysfunction. 9
Pretreatment with
Oxidative stress is considered one of the major determinants of 3-NP neurotoxicity
21,36,37
as evidenced by increased oxidative damage and depletion of endogenous antioxidant levels.
8,21,38
In the current study, 3-NP significantly increased MDA, nitrite levels, and decreased the antioxidants enzyme (SOD and catalase) in the striatum, cortex, and hippocampal. The oxidative stress may further cause cell death in different regions of brain, so there may be some correlation between oxidative damage and cell death in 3-NP neurotoxicity. Cyclosporine A treatment significantly reversed the oxidative stress parameters, suggesting the antioxidant-like action of cyclosporine A. However, the primary role of cyclosporine A on the oxidative stress process or antioxidative defense mechanism is still not clear. Furthermore, the current study tried to explore the involvement of NO mechanism (if any) in the protective action of cyclosporine A.
Mitochondria are important organelles for energy production, Ca2+ homeostasis, and cell death. In apoptotic and necrotic death, an increase of mitochondrial membrane permeability transition (MPT) is considered one of the key events. 39 3-Nitropropionic acid induced a MPT, which plays an important role in the mechanism of apoptotic cell death. 3-Nitropropionic acid stimulated Ca2+ release from mitochondria, decreased membrane potential, induced mitochondrial swelling, and stimulated cytochrome c release from mitochondria. 40 It has been reported that 3-NP treatment caused a greater loss of mitochondrial membrane potential in striatal cells. 41 In the current study, 3-NP significantly blocked mitochondrial enzymes in mitochondria isolated from the striatum, cortex, and hippocampal regions. Cyclosporine A is a well-known blocker of mitochondrial transition pore (MTP) and NO has been shown to inhibit cytochrome c oxidase (complex IV), leading to an increase in ROS generation in mitochondria. 42 However, cyclosporine A treatment restored the mitochondrial enzyme activity in 3-NP-treated animals. The MTT assay is commonly used to assess any impairment in the mitochondrial function. The reduced MTT metabolism, as observed in our study, in different regions of treated rat brain indicates that chronic 3-NP exposure may affect the mitochondrial functions, whereas cyclosporine A treatment significantly reversed the effect of 3-NP impaired mitochondrial redox activity. Furthermore, NO modulators (both positive and negative) significantly reversed the effect of cyclosporine A, suggesting that NO mechanism is involved in the mitochondrial enzyme functions. Therefore, it is possible that cyclosporine A directly affects mitochondria enzymes activity by a mechanism involving NO. It is known that cyclosporine A preserves mitochondrial function by inhibiting the opening of MPT pores through binding to cyclophilin D, which specifically expressed in the mitochondrial matrix. 43 It seems that cyclosporine A might prevent neuronal cell death by increasing mitochondrial function, in addition to its action on suppression of calcineurin activity. 44 Cyclosporine A has limited passage into the brain parenchyma, 45,46 but it accumulates in pharmacological concentrations in the brain at high blood levels. 47
The current study highlights that cyclosporine A attenuates 3-NP-induced behavioral, biochemical, and mitochondrial dysfunction by involving antioxidant and NO modulation in rat brain. However, further cellular studies are required to understand the effect of cyclosporine A on oxidative stress and NO pathways and the interaction in different experimental systems.
Footnotes
Acknowledgments
The Senior Research Fellowship (Puneet Kumar) of the Indian Council of Medical Research (ICMR), New Delhi, is gratefully acknowledged.
The author(s) declared no conflicts of interest with respect to the authorship and/or publication of this article.
The authors are thankful for the financial support from the Indian Council of Medical Research (ICMR), New Delhi.