
Abstract
Human neuronal cells differentiated from induced pluripotent cells have emerged as a new model system for the study of disease pathophysiology and evaluation of drug efficacy. Differentiated neuronal cells are more similar in genetics and biological content to human brain cells than other animal disease models. However, culture of neuronal cells in assay plates requires a labor-intensive procedure of plate precoating, hampering its applications in high-throughput screening (HTS). We developed a simplified method with one-step seeding of neural stem cells in assay plates by supplementing the medium with a recombinant human vitronectin (VTN), thus avoiding plate precoating. Robust results were obtained from cell viability, calcium response, and neurite outgrowth assays using this new method. Our data demonstrate that this approach greatly simplifies high-throughput assays using neuronal cells differentiated from human stem cells for translational research.
Keywords
Introduction
Disease modeling using induced pluripotent stem cells (iPSCs) has recently been applied to a variety of diseases in the study of disease phenotype and pathophysiology. 1 The iPSCs are self-renewing and can be differentiated into various types of human cells, such as neurons, cardiomyocytes, and hepatocytes. Disease models play a critical role in evaluation of drug efficacy during drug development. However, animal models, particularly rodent models, may not mimic certain human diseases appropriately. Insufficient disease models for neurodegenerative and neuropsychiatric diseases have hindered the development of new therapeutics for these maladies in the last two decades. 2 Recent advancement in iPSC technology has enabled large-scale production of neuronal cells differentiated from patient iPSCs that models neurological disorders, including Parkinson’s disease (PD), 3 Alzheimer’s disease (AD), 3 amyotrophic lateral sclerosis (ALS), 3 spinal muscular atrophy (SMA), 4 and familial dysautonomia. 5 Neuronal cells differentiated from patient iPSCs exhibited specific disease phenotypes, such as a decrease of mitochondrial function in PD dopaminergic neurons, 6 accumulation of amyloid-β and p-tau/total tau in AD neurons, 7 hyperexcitability in ALS motor neurons, 8 apoptosis in SMA motor neurons, 9 and cholesterol accumulation in Niemann–Pick disease type C (NPC). 10 Different types of neuronal cells have been generated from iPSCs, including neural stem cells (NSCs), astrocytes, oligodendrocytes, motor neurons, and dopaminergic neurons. These human neuronal cells, particularly patient-derived cells, can serve as cell-based disease models to evaluate compound efficacy and screen compound libraries for drug development, in addition to studying disease pathophysiology.
Practically, use of neuronal cells differentiated from iPSCs for various experiments involves labor-intensive laboratorial work. Culturing neuronal cells in assay plates requires plate precoating with extracellular matrix (ECM) proteins and/or positively charged polymers to support cell attachment and growth. The procedure of plate precoating involves multiple steps of reagent addition and plate washes, which not only reduces screening throughput but also yields large well-to-well and plate-to-plate variations. 11 Precoating of plates is also a bottleneck in high-throughput screening (HTS) using neuronal cells. 11
Here, we report development of a simple method of one-step seeding and culturing of neuronal cells in assay plates using medium containing a truncated recombinant human vitronectin (rhVTN-N), which has been used as a plate coating substrate for iPSC feeder-free culture. 12 Because plate precoating and plate washing are not needed in this method, it greatly simplifies experiments using neuronal cells differentiated from iPSCs ( Fig. 1A ). We have validated this method with several assays, including cell viability, calcium response, and neurite outgrowth. The results demonstrate that this method enables HTS using NSCs and neurons differentiated from stem cells. Therefore, this method of one-step seeding of NSCs in assay plates with the rhVTN-N-supplemented medium is useful for HTS employed to evaluate compound efficacy, to measure compound neural toxicity, and to identify new leads by screening of compound libraries.
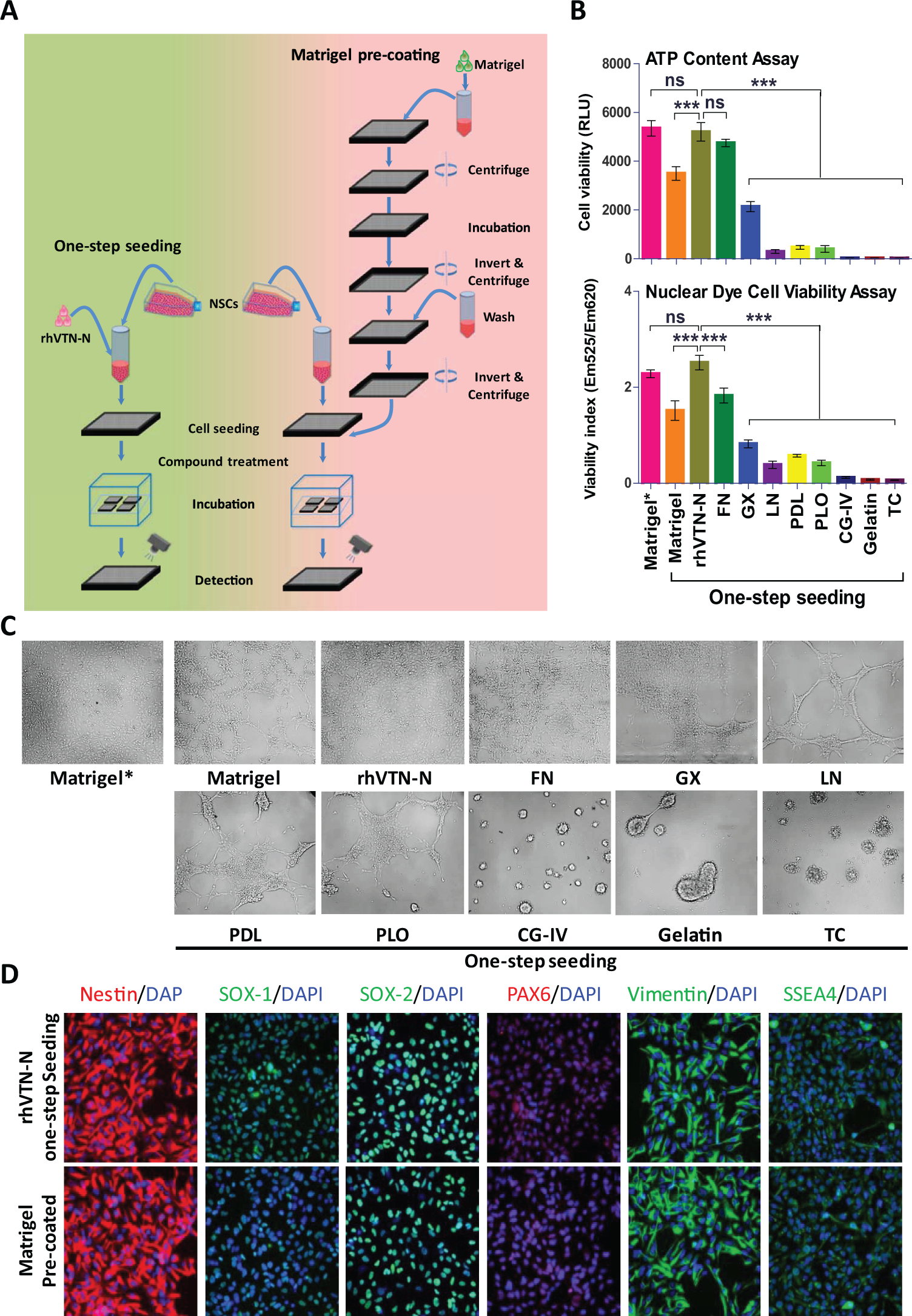
Development of the method for directly seeding NSCs without plate precoating in 1536-well plates. (
Materials and Methods
Cell Lines and Cell Culture
Wild-type (WT) iPSCs generated from normal fibroblasts (GM23450, Coriell Cell Repositories) were cultured in Matrigel (catalog no. 354277, Corning, Corning, NY)-precoated six-well plates with Essential 8 Medium (A1517001, Life Technologies, Waltham, MA). After being cultured at least 10 passages, NSC induction (Supplemental Materials and Methods) was initiated using the Neural Induction Medium kit (Life Technologies). The resulting NSCs were cultured in the complete StemPro NSC Serum Free Medium (A1050901, Life Technologies) containing knockout DMEM/F-12, StemPro neural supplement, 20 ng/mL of basic fibroblast growth factor (bFGF), 20 ng/mL of epidermal growth factor (EGF), and 1× GlutaMAX on the Matrigel-precoated flasks. 10 Immunohistochemistry staining with antibodies against nestin, SOX1, SOX2, PAX6, vimentin, and SSEA4 was performed for characterization of the NSCs induced from human iPSCs.
One-Step NSC Seeding Optimization in 1536-Well Plates
First, nine frequently used substrates (
We also found that the neural expansion medium composed of Neurobasal medium, Advanced DMEM/F-12 medium, and GIBCO Neural Induction Supplement (21103-049, 12634010, and A16477-01, Life Technologies) could not be used in this method because the NSCs were not attached to the assay plates and unable to grow when they were seeded in this medium supplemented with rhVTN-N (
One-Step NSC Seeding with rhVTN-N-Supplemented Medium for Neuronal Differentiation in 384-Well Plates
Neuronal differentiation was conducted as previously described.
10
The medium was composed of Neurobasal medium (21103-049, Life Technologies), B27, GlutaMAX, 10 ng/mL of brain-derived neurotrophic factor (BDNF), 10 ng/mL of glial cell–derived neurotrophic factor (GDNF), 1 µM of cAMP, and 200 ng/mL of
Neurite Outgrowth Assay
NSCs were seeded in 384-well plates under the same conditions as described above. After 4 weeks of differentiation, neuronal cells were stained with anti-β-III tubulin antibody and Hoechst 33342 nuclear dye as described in the immunofluorescence staining section. Fluorescence imaging was performed in the IN Cell 2200 imaging system with a 40× objective lens using standard FITC and DAPI filter sets. Images were analyzed using IN Cell Developer v1.9.2 analysis software (GE Healthcare, Little Chalfont, UK). Briefly, the DAPI channel was used to identify nuclei using an intensity segmentation algorithm (objects greater than 650 RFU ranging in size from 5 to 75 µm). β-III tubulin (FITC)–stained neurites were also identified by intensity segmentation (greater than 1150 RFU) with a postprocessing sieve to remove debris objects less than 2 µm in size. The mean neurite length, measured in microns, was averaged to the number of nuclei per image.
High-Throughput Cytotoxicity Assays in iPSC NSCs
ATP content assay, nuclear dye cell viability assay, and caspase-3/7 assay (Caspase-Glo, Promega) were conducted according to the manufacturers’ protocols using the one-step seeded NSCs in the rhVTN-N-supplemented medium in 1536-well plates. NSCs were plated at 3000 cells/well in 5 µL of StemPro NSC Serum Free Medium complete culture medium with 10 µg/mL of rhVTN-N in 1536-well plates (white, solid-bottom assay plates for ATP content assay and caspase-3/7 assay; black, clear-bottom plates for nuclear dye cell viability assay) and incubated for 4 h at 37 °C. Compounds were added to the assay plates at 23 nL/well using an NX-TR pintool station (WAKO Scientific Solutions, San Diego, CA). After 16 (caspase-3/7 assay) or 48h (ATP content assay and nuclear dye cell viability assay) of incubation at 37 °C with 5% CO2, the assay reagents at 3 µL/well were added to the assay plates. After incubation for the indicated times, the plates were detected by luminescence for the ATP content assay and caspase-3/7 assay or fluorescence for the nuclear dye cell viability assay in a plate reader.
Half maximal inhibitory (IC50) or activating (EC50) concentration of compound were calculated using Prism software (GraphPad Software, San Diego, CA). The percent cell viability in the ATP content and nuclear dye cell viability assays was normalized using the positive control compound thapsigargin (270 µM)-treated cells as 0% in cell viability, which has been shown to greatly reduce neuronal cell viability by depleting the intracellular calcium stores, 14 and DMSO solvent–treated wells for the 100% viable cells. The percent activities of the caspase-3/7 assay were normalized using the positive control compound staurosporine (75 µM)-treated cells as 100% in caspase activity, which has been shown to increase caspase-3/7 activities, and DMSO-treated wells for the 0% caspase activity. Signal-to-basal ratio (S/B), coefficient of variation (CV), and Z factor were calculated and used to evaluate the assay performance (Supplemental Materials and Methods). For cell viability evaluation of differentiated neuronal cells in 384-well plates, the cell viability measurement was conducted after the cells were cultured for 1, 2, and 4 weeks with ATP content assay and nuclear dye cell viability assay.
Homogenous LysoTracker Dye Staining Assay for Measurement of Enlarged Lysosomes in NPC Cells
Cells derived from patients with NPC exhibit the NPC disease phenotype of enlarged lysosome due to cholesterol accumulation in lysosomes that can be measured by the LysoTracker dye staining assay in a fluorescence imaging detection mode or in a fluorescence intensity mode by a plate reader.10,15,16 NSCs were plated at 1000 cells/well in 5 µL of complete StemPro NSC Serum Free Medium supplemented with 10 µg/mL of rhVTN-N in 1536-well, black, clear-bottom assay plates and incubated for 4 h at 37 °C and 5% CO2. Columns 1 and 2 were used as the negative control with the WT NSCs, and columns 3–48 were NPC NSCs. Methyl-β-cyclodextrin (MΒCD) and hydroxypropyl-β-cyclodextrin (HPBCD), which reduce cholesterol accumulation in NPC cells and thus decrease the enlarged lysosomes in NPC cells, were added to the assay plate at 23 nL/well as positive controls. After a 96 h incubation, 200 nM of LysoTracker Red dye (Life Technologies) was added to the assay plates to stain lysosomes. The fluorescence intensity (Ex = 570 nm, Em = 590 nm) was measured with a bottom read mode in the Tecan plate reader. The data from the homogenous LysoTracker staining assay were normalized using the WT NSCs as a negative control and DMSO-treated NPC NSCs as a positive control to calculate IC50 values, S/B ratio, CV, and Z factor.
High-Throughput Fluorescent Calcium Response Assay
ATP (a purinergic receptor agonist), carbachol (a muscarinic receptor agonist), and calcium ionophore ionomycin were used as stimuli for intracellular calcium release (referred to as agonists). The purinergic receptor antagonists suramin hexasodium and AR-C 118925XX, and muscarinic acetylcholine receptor antagonists tolterodine and telenzepine, as well as calcium chelators tetra(acetoxymethyl ester) (EGTA-AM) and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), were used as the antagonist compounds for the relevant stimulus (referred to as antagonist). The 25 µL NSCs were plated at 10,000 cells/well in the 384-well, black, clear-bottom plates in StemPro NSC Serum Free Medium complete culture medium supplemented with 10 µg/mL of rhVTN-N. The neurons were differentiated for 4 weeks as described above in the NSC one-step seeding with rhVTN-N-supplemented medium in commercial PDL-precoated 384-well plates. The fluorescence calcium dye (Cal-52-AM, AAT Bioquest) was prepared in DMSO at 2.5 mg/mL. The calcium dye loading solution was prepared by adding 10 µL to a mixture of 9 mL of Hanks’ balanced salt solution (HBSS) and 1 mL of 10× calcium assay buffer with quencher dye (Screen Quest 10× calcium assay buffer with Phenol Red Plus, AAT Bioquest). The loading solution was added at 25 µL/well and incubated for 3 h at 37° C with 5% CO2, followed by a 30-min incubation at room temperature. For calcium agonist response experiments, agonists were prepared at 3× of the final concentration in HBSS buffer for a 16-concentration titration (1:2 dilution) in a 384-well compound plate in triplicate. The assay plate and compound plate were then placed into the FDSS µCELL kinetic fluorescence plate reader (Hamamatsu, Japan) to measure fluorescence intensity kinetically for 5 min at 1 read/s (1 Hz). After the baseline fluorescent signals were measured for 15 s, 25 µL/well of 3× agonist solutions in a titration were added to the assay plate from the compound plate of each well at the speed of 50 µL/s, and the fluorescent intensity was continually recorded at 1 Hz, for 300 s in total. The ratio (F/F0) of fluorescent intensity at each time point (F) after adding agonists to the average fluorescence intensity of baseline (F0) was calculated. The maximum ratio (Fmax/F0) of each well was used for calculation of EC50 values.
For the antagonist experiments, the EC80 concentration of each agonist was added as the stimulus to induce the calcium response. Prior to the calcium response experiment, the cells were treated with the antagonists for 30 min at 25 µL/well with 16 concentrations of compound titration at a 1:2 ratio (see
Single-Cell Calcium Response Recording
The NSCs and differentiated neurons were loaded with a calcium indicator as described above. The fluorescence images before and after addition of calcium releasing agonists were recorded at 1 Hz in an IN Cell 2200 automated imaging station equipped with an integrated liquid handling system (GE Healthcare). All fluorescence images were acquired kinetically at 37 °C in the IN Cell 2200 automated imaging system using a 20× objective lens with standard DAPI and FITC excitation/emission filter sets. Cells were imaged at 1 Hz (1 frame per second) for a total of 180 s, with autofocus enabled at each time point. At time point 1, both DAPI (for nuclei) and FITC (for calcium response) images were acquired. But for the remaining 179 cycles, only FITC images were acquired. At time point 5, 25 µL/well of agonist compound solution or control solution was added via an onboard liquid handling system. Images were collected at standard 1 × 1 binning and analyzed with the IN Cell Workstation software v3.7.2 using the multitarget analysis algorithm. Briefly, Hoechst-stained nuclei (channel 1) were identified using top-hat segmentation looking for objects with a typical area of 75 µm2, and green Cal-520 dye–stained cells (channel 2) were identified using multiscale top-hat segmentation looking for objects with a typical size of 200 µm2. For the kinetic analysis, the nuclear images acquired at time point 1 were used as a unique cell ID for all subsequent time points and the Cal-520 images were analyzed at each time point for each cell. The average cellular fluorescence intensity ratio (F/F0) for individual cells was plotted over the time. F0 is the mean fluorescence intensity of 10 initial reads without addition of a stimulus, and F is the fluorescence intensity at a given time. The ratio of maximum fluorescence intensity (Fmax/F0) collected from each cell was used for determination of positive responding cells. To define the positive calcium in a cell, an average of the maximal intensity ratio (Fmax/F0) plus three times the standard deviation (mean + 3*SD) in the DMSO control was used as a threshold. The percent of cells with calcium response was calculated using the following equation: Calcium responding cells (%) = (Number of cells with their (Fmax/F0) ratios ≥ (Fmax (control)/F0) + 3 × SD)/Total cell number in the wells that were imaged.
Data Analysis and Statistics
IC50 or EC50 concentration values of compound confirmation data were calculated using Prism software (GraphPad Software). Unless otherwise noted, all values are expressed as the mean ± SD (n = 3). For statistical analysis, results were analyzed using one-way or two-way analysis of variance (ANOVA) and differences were considered statistically significant if p < 0.05.
Results
rhVTN-N-Supplemented Medium for One-Step Seeding of Neural Stem Cells
Plate precoating with positively charged synthetic polymers and/or biological reagents containing ECM proteins such as Matrigel, fibronectin (FN), and LN is required for culture and growth of neuronal cells in assay plates. The plate precoating process involves multiple steps of reagent addition, incubation, and plate washing, which are labor-intensive and time-consuming, as well as a possibility of microbe contamination. Although synthetic polymers such as PDL and PLO can be coated on plates in advance, the ECM proteins must be freshly coated in a thin layer to assay plates prior to the experiments because the coated proteins cannot be dried in the plates. To simplify the experiments using neuronal cells, we designed a one-step seeding of neuronal cells by adding ECM proteins to cell culture medium without a need for plate precoating (
Fig. 1A
). We first tested the addition of one of nine commonly used coating reagents in cell culture medium for one-step seeding of NSCs in tissue culture–treated plates without plate precoating (
One-Step Seeding of Neural Stem Cells for Neuron Differentiation
In order to determine if a one-step seeding method could be applied to neuron differentiation in assay plates, we plated NSCs in 384-well assay plates. We initiated the neuron differentiation during one-step seeding by adding the rhVTN-N-supplemented neuronal differentiation medium to commercial PDL-coated plates or tissue culture–treated plates. Neurons differentiated in the LN and PLO doubly precoated plates were used as a control. 10 For neurons cultured in the rhVTN-N-supplemented medium in commercial PDL-coated plates, the cell growth rate and cell viability were similar to those cultured in the control plates precoated with LN and PLO. However, most neuronal cells died after 2 weeks in culture in the rhVTN-N-supplemented medium in plates without PDL coatings ( Fig. 2A ). After 4 weeks of differentiation in the assay plates, the cells showed positive staining of β-III tubulin, microtubule-associated protein (MAP2), neurofilament-L (NF-L) with negative nestin, sex-determining region Y-box 2 (SOX-2), and glial fibrillary acidic protein (GFAP) ( Fig. 2B ), indicating relatively mature neurons. The neurite outgrowth rate of these neurons was also similar to that of the control ( Fig. 2C ), as measured by a mean length of neurite projections stained with β-III tubulin. The results indicate that neuronal cell differentiation in plates requires both rhVTN-N-supplemented medium and the use of commercial PDL-coated plates.
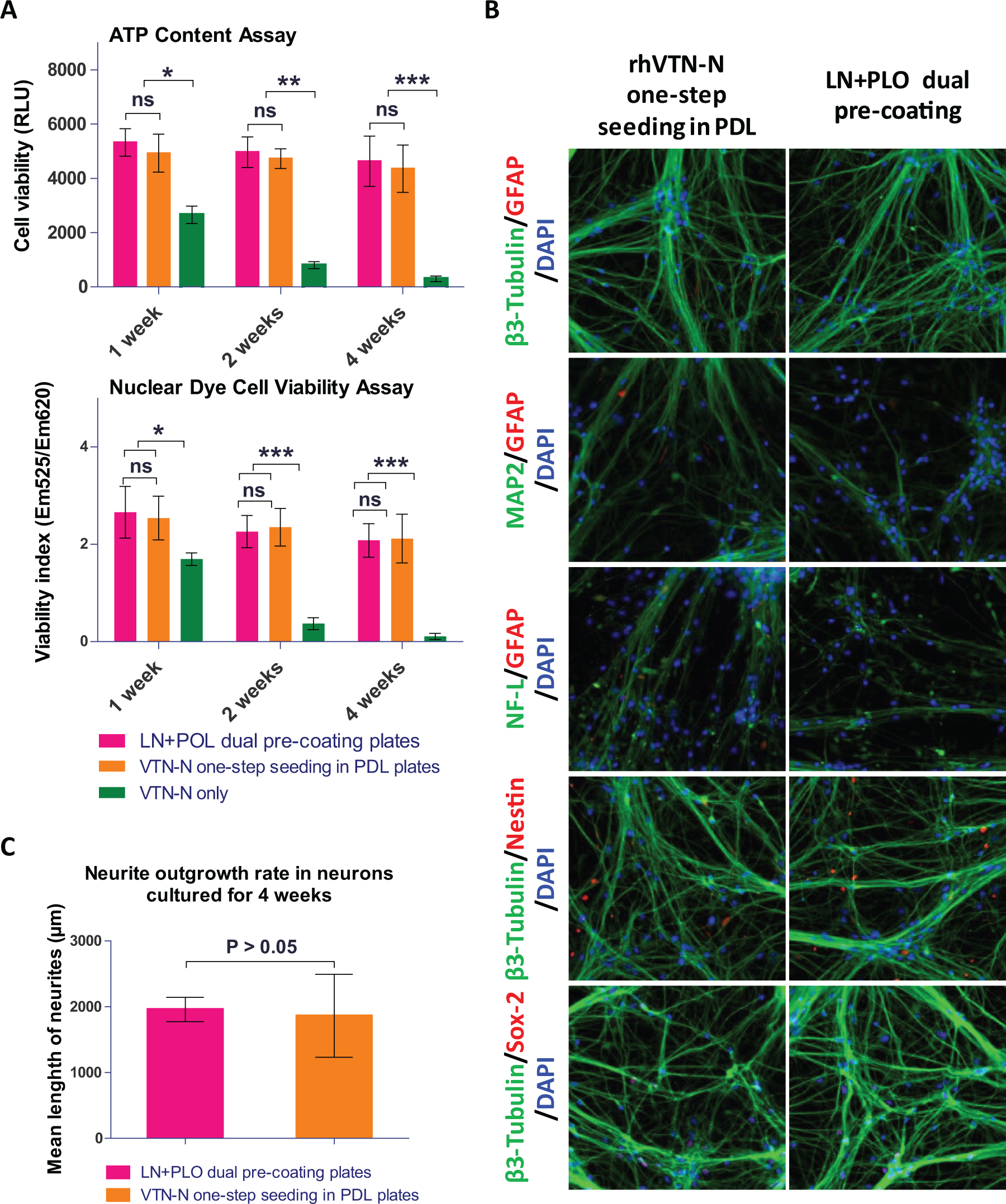
Results of direct seeding of NSCs for neuron differentiation. (
One-Step Seeding of Neural Stem Cells in rhVTN-N-Supplemented Medium for High-Throughput Cell Viability Assays
To validate this method, we first determined the viability of NSCs directly seeded in 1536-well plates with the rhVTN-N-supplemented medium. A set of parameters for HTS assays were first determined with cell viability experiments, including the ATP content assay, nuclear dye cell viability assay, and cell apoptosis assay for caspase-3/7 activity. The scatter plots of these three assays exhibited robust assay profiles ( Fig. 3A ). The S/B ratios of ATP content, nuclear dye, and caspase-3/7 assays were 123.8-, 34.3-, and 1.6-fold; CV values were 8.6%, 7.2%, and 4.4%; and Z factors were 0.74, 0.73, and 0.57, respectively. The results indicated that these assays are robust and suitable for HTS.
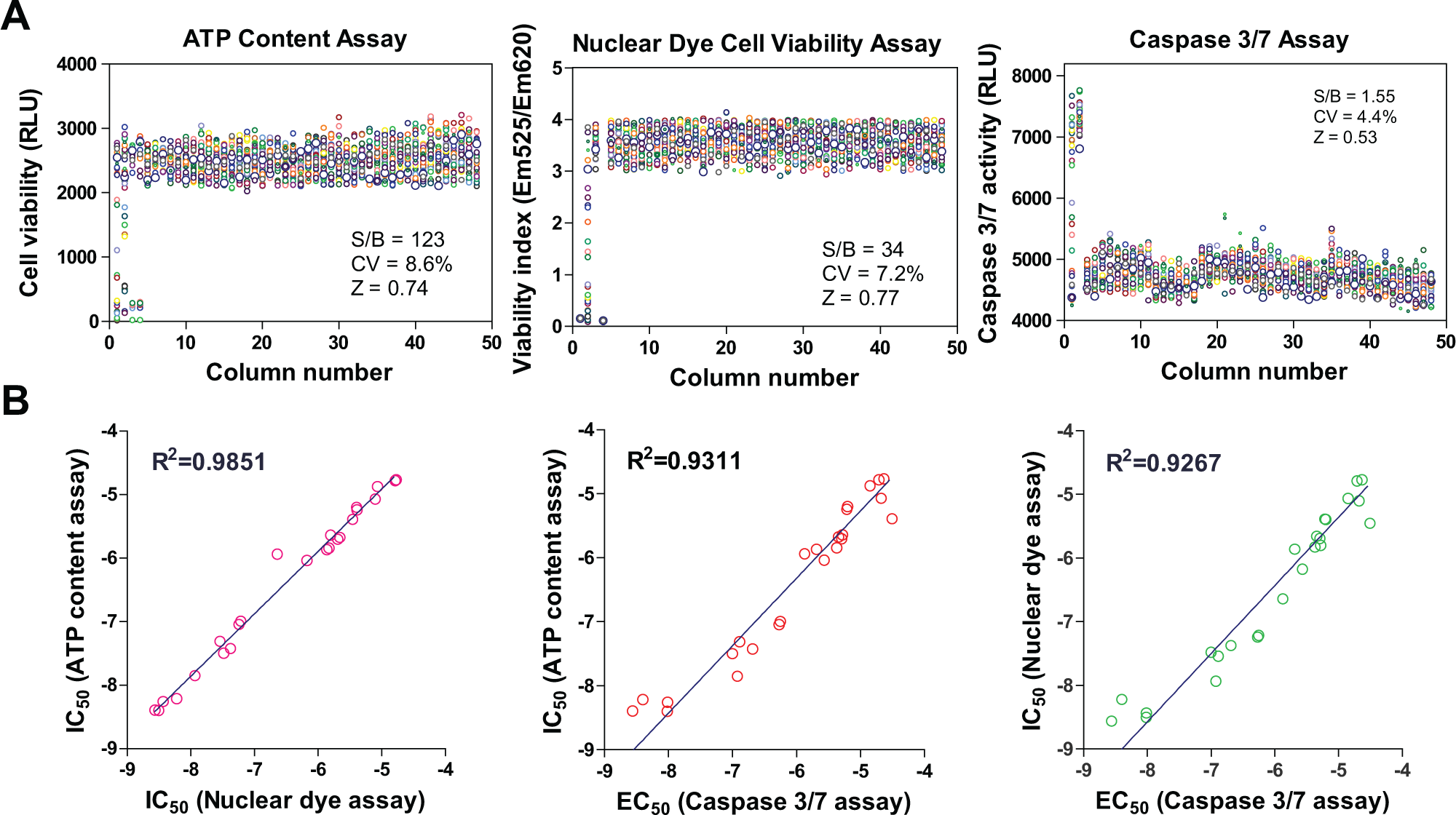
Assay performances of one-step seeding of NSCs in 1536-well plates. (
We also selected 24 neuronal toxic compounds from the literature (
Homogenous LysoTracker Staining Assay for Measurement of Enlarged Lysosomes in NPC Cells
We also applied this method to a phenotypic compound screening assay using NSCs differentiated from iPSCs of an NPC patient. NPC NSCs exhibit an NPC disease phenotype of enlarged lysosome due to cholesterol accumulation; this can be detected by the LysoTracker dye staining assay.10,15 The fluorescence signal of LysoTracker dye staining in NPC cells can be measured by fluorescence microscopy and also by a fluorescence plate reader. 15 We found that the S/B ratio was 3.05-fold and the Z factor was 0.57, as determined by a LysoTracker dye staining assay in 1536-well plates when measured using a plate reader ( Fig. 4A ). The IC50 values of two control compounds, MΒCD and HPBCD, were 2.4 and 33.7 µM, respectively ( Fig. 4B ), similar to a previous report. 10 The results demonstrated that the one-step seeding method can also be used with patient-derived NSCs in a HTS context.
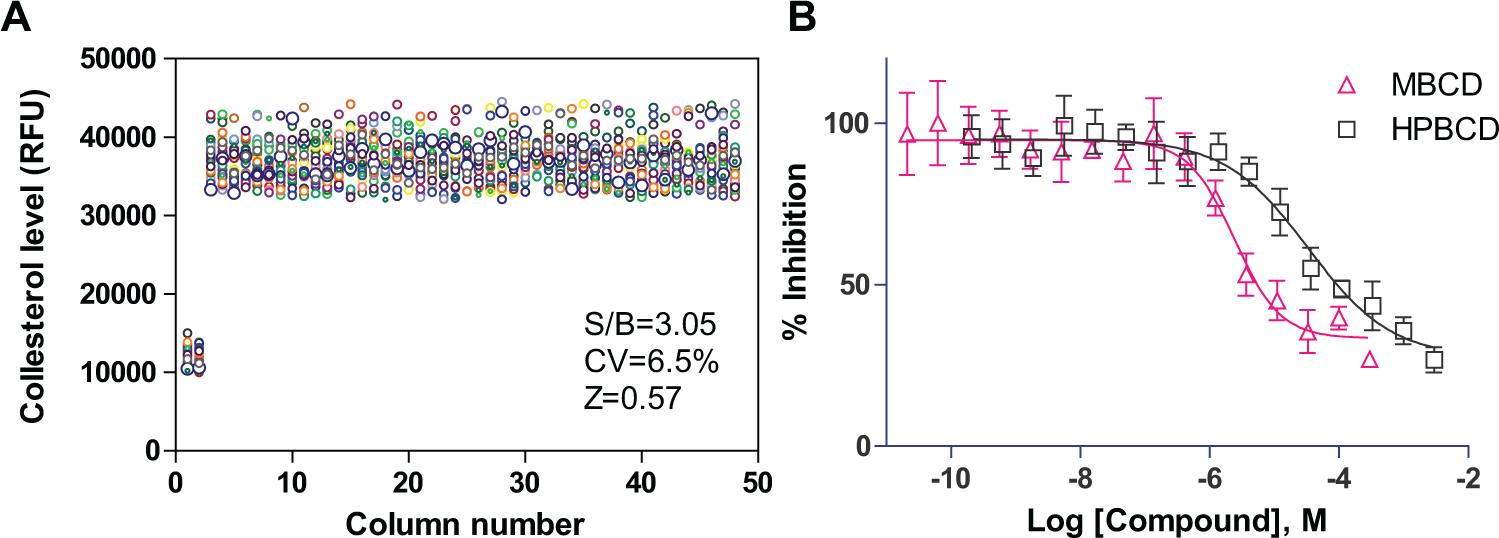
Results of the homogenous LysoTracker dye staining assay using NPC patient NSCs directly seeded in 1536-well plates with rhVTN-N-supplemented medium. (
High-Throughput Fluorescent Calcium Response Assays Using Neural Stem Cells and Neurons
To further validate this method, we tested it with a high-throughput calcium response assay in a 384-well assay plate (
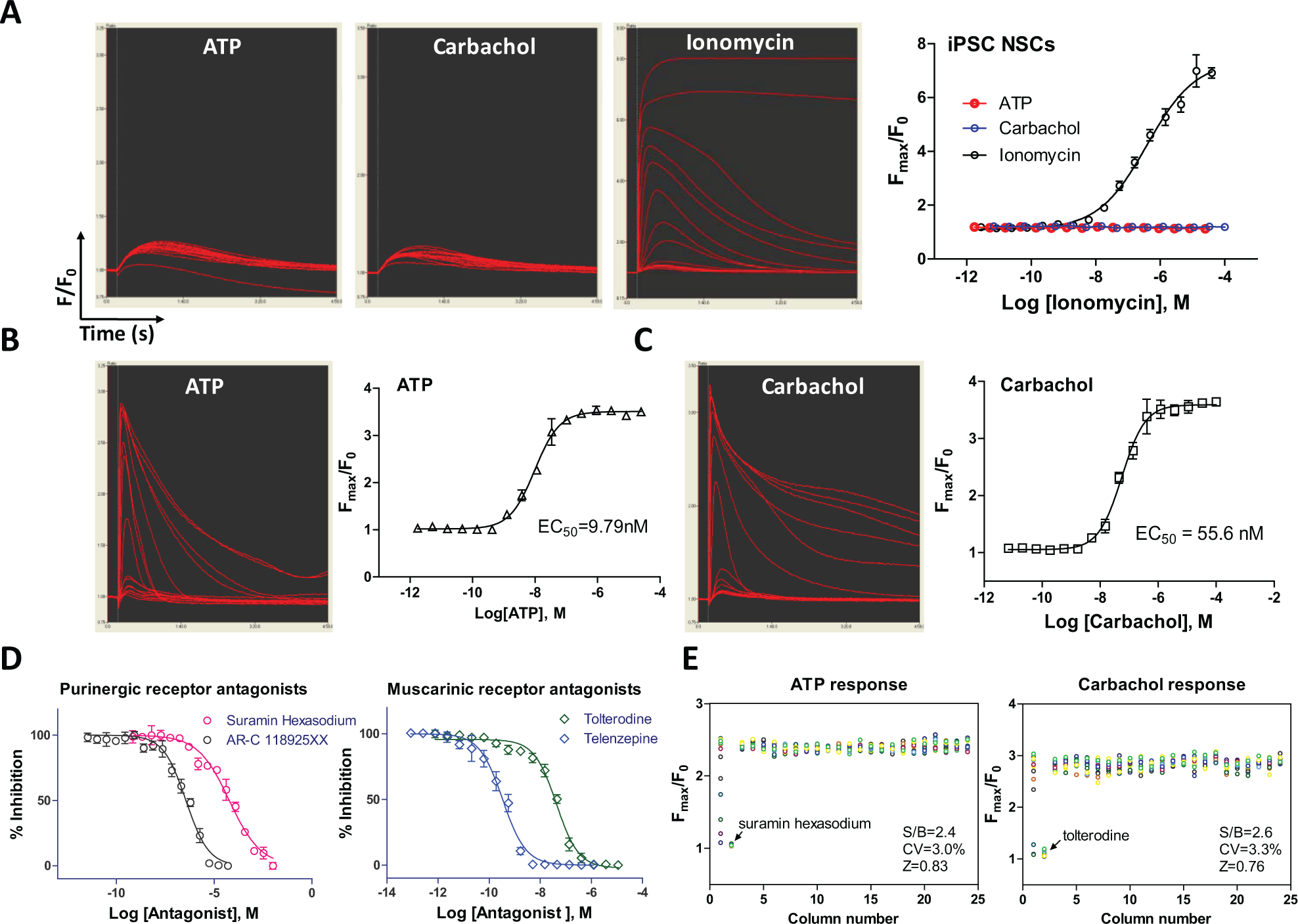
Results of calcium response assay using NSCs directly seeded in rhVTN-N-supplemented medium in 384 plates. (
While NSCs only express low levels of receptors and ion channels,
18
the differentiated neurons express much higher levels of ion channels and receptors, including calcium channels, purinergic receptors, glutamate receptors, and ryanodine receptors.
19
We then measured the intracellular calcium responses to ATP and carbachol in differentiated neurons plated in commercial PDL-coated 384-well plates with the rhVTN-N-supplemented medium. The EC50 values of the ATP and carbachol were 9.8 and 55.7nM, respectively (
Fig. 5B
,
C
). The calcium responses stimulated by ATP and carbachol were concentration dependently inhibited by the purinergic receptor antagonists suramin hexasodium (IC50 = 59.5 µM) and AR-C 118925XX (IC50 = 359.8 nM) and the muscarinic acetylcholine receptor antagonists tolterodine (IC50 = 42.8 nM) and telenzepine (IC50 = 0.3 nM), respectively (
Fig. 5D
). These calcium responses in neurons were robust with the favorable HTS assay parameters (
Fig. 5E
,
Single-Cell Calcium Response in Neural Stem Cells and Neurons
Because not all of the cells in a population respond equally to a stimulus, a single-cell recording of calcium response offers a much more sensitive detection system that distinguishes the responding cells from nonresponding cells in a cell population. Using a kinetic fluorescence imaging system, we measured the single-cell calcium response in NSCs and neurons, in addition to the above whole-well calcium response. In NSCs, only a small portion of cells responded to the ATP or carbachol stimulation (
Fig. 6A
), while the majority of cells responded to ionomycin (
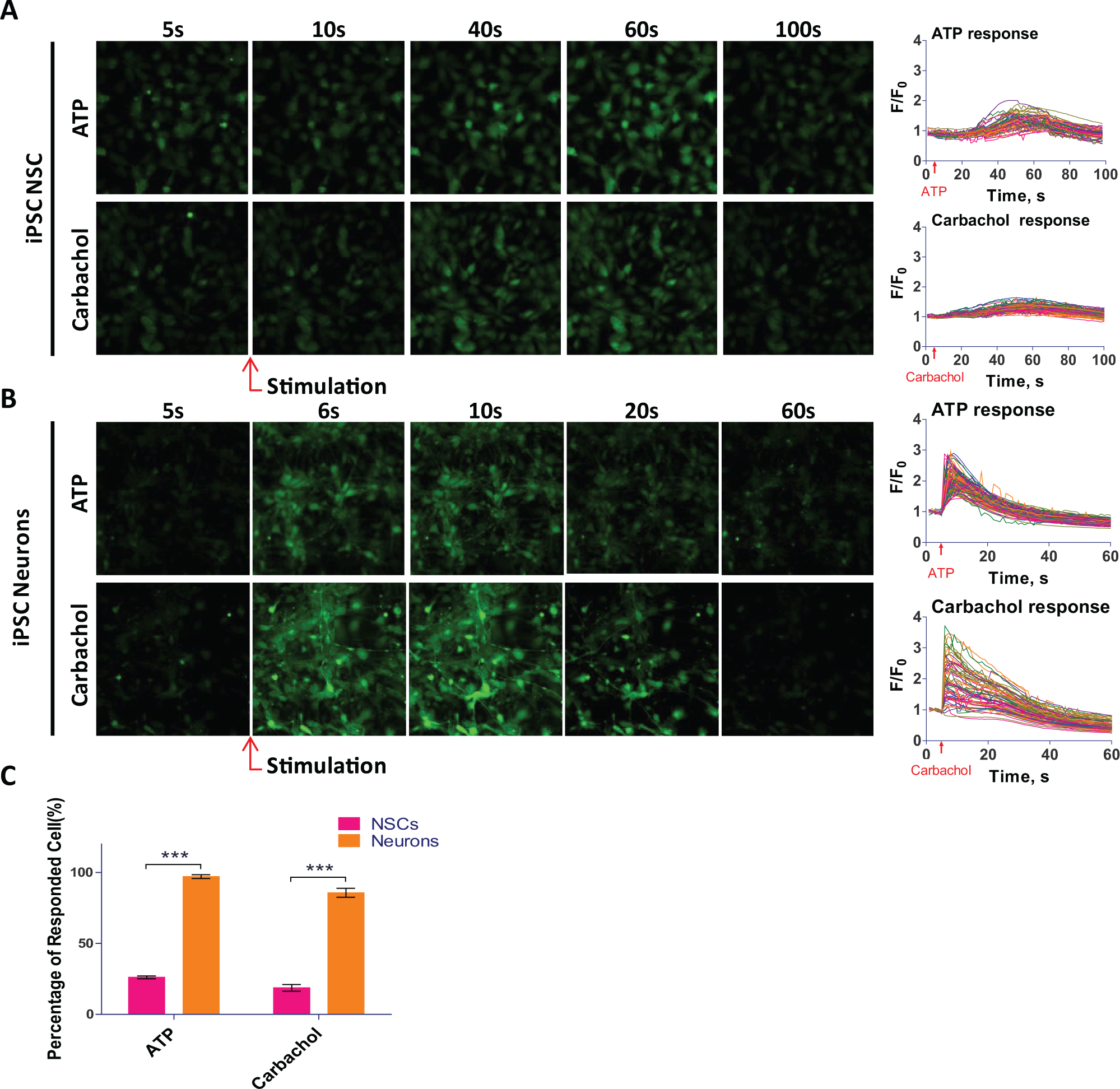
Results of single-cell calcium responses in NSCs and neurons with NSCs directly seeded in rhVTN-N-supplemented medium in 384-well plates. (
Discussion
We described here a simple method (
Additional surface treatment is often needed for culturing primary cells and neurons, although tissue culture–treated plasticware prepared by gas plasma is generally used. 20 The additional surface treatment provides an environment similar to that of the ECM that the cells experience in vivo. ECM consists of secreted proteins that mediate proper attachment, spreading, growth, and differentiation of neuronal cells. 21 In many types of cell culture, fetal bovine serum (FBS) supplementation provides ECM proteins and various growth factors to cells. But FBS is usually not added to culture medium for stem cells and neurons differentiated from iPSCs, because only a few specific growth factors can be added in the medium to prevent uncontrolled differentiation. Therefore, precoating of dishes and plates with ECM proteins and polymers is a common practice for culturing stem cells and neurons, although the process is labor-intensive with a high reagent cost. Neuronal cell culture requires freshly precoated plates, covered with thin layers of both ECM proteins and positively charged polymers. Because an ECM protein like LN is very easily absorbed by plastic and tends to form aggregates in plates at room temperature, precautions such as keeping the coating material on ice are usually needed for the plate precoating. 22
Positively charged synthetic polymers such as PDL, polyethyleneimine, and PLO are commercially available and commonly used to coat dishes and plates. They provide electrostatic interaction with the negatively charged cell membrane, supporting cell attachment. 23 These materials are usually stable and can be precoated in assay plates in advance or can be obtained from commercial venders. On the other hand, extracellular proteins such as Matrigel, FN, LN, and VTN have to be freshly coated onto assay plates. A dual precoating with LN and PLO in assay plates is commonly used for neuronal cell culture and neuronal cell differentiation. The process involves dispensing of a coating reagent to plates, incubation, and multiple plate wash steps.24,25 A recent protocol for using the rhVTN-N to precoat plates describes that it is unnecessary to wash plates after the plate coating, but it still requires removal of VTN (http://tools.thermofisher.com/content/sfs/manuals/vitronectin_man.pdf).
To eliminate the need for plate precoating, we first tested the addition of ECM proteins and polymers into cell culture medium for directly seeding cells onto plates without plate precoating. Nine reagents were examined, including rhVTN-N, Matrigel, FN, Geltrex, LN, collagen type IV, gelatin, and two polymers, PDL and PLO. (
NSCs,
27
neural progenitor cells,
28
and neurons differentiated from stem cells
13
have recently been employed for compound screens, but the screening throughput of these reported experiments was limited due to the requirement of plate precoating to support cell attachment and growth. Matrigel, FN, LN, VTN, PLO, and PDL have been used as substrates for plate precoating in neuronal cell culture (
Although the plate precoating can be handled in these experiments for testing a few compounds, it is a burden for HTS with a large amount of compounds. Reductions of robotic steps in a screening assay can significantly increase compound screening throughput, reduce the chances of interruptions in robotic operation due to errors and malfunctions of liquid handlers, and increase data quality by decreasing well-to-well and plate-to-plate variations. This method of one-step seeding of NSCs significantly reduces assay steps, which is specifically useful for robotic screening of large compound collections using neuronal cells.
In conclusion, we have developed a simple method for one-step seeding of NSCs in assay plates with an rhVTN-N-supplemented medium without the need for plate precoating. This method greatly increases the throughput and reproducibility of experiments using neuronal cells derived from human iPSCs for translational research. Additionally, the principle of this method has the potential to be extended to seed iPSCs and iPSC-derived progenitor cells, which in turn can be differentiated into other cell types for various experiments.
Footnotes
Acknowledgements
The authors thank Paul Shinn and the compound management team at NCATS for their assistance. The authors also thank DeeAnn Visk for editing and critiquing the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was funded by the Intramural Research Program of the National Center for Advancing Translational Sciences, National Institutes of Health.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.