
Abstract
Glycine receptor 3 (GlyRα3) is a ligand-gated ion channel of the cys-loop family that plays a key role in mediating inhibitory neurotransmission and regulation of pain signaling in the dorsal horn. Potentiation of GlyRα3 function is therefore of interest as a putative analgesic mechanism with which to target new therapeutics. However, to date, positive allosteric modulators (PAMs) of this receptor with sufficient selectivity to enable target validation studies have not been described. To address this lack of pharmacological tools, we developed a suite of in vitro assays comprising a high-throughput fluorescent membrane potential screen and a medium-throughput electrophysiology assay using IonFlux HT together with conventional manual patch clamp. Using these assays, we conducted a primary screening campaign and report the structures of hit compounds identified as GlyR PAMs. Our functional characterization data reveal a hit compound with high efficacy relative to current known potentiators and selectivity over GABAAR, another major class of inhibitory neurotransmission receptors of importance to pain. These small-molecule GlyR PAMs have high potential both as early tool compounds to enable pharmacological studies of GlyR inhibitory neurotransmission and as a starting point for the development of potent, selective GlyRα3 PAMs as novel analgesics.
Introduction
Glycine receptors (GlyRs) are members of the cys-loop receptor ligand-gated ion channel family.1,2 In humans, there are three cloned α subunits (α1–3) and a single β subunit3,4 with the physiological receptor reported to be a heteropentamer of either 3α2β or 2α3β.5-7 However, in heterologous expression systems, GlyRs comprised of homomeric (α subunit only) and heteromeric (αβ subunits) are both functional.1,2 In addition to the orthosteric glycine binding site of these receptors, there are a number of allosteric sites that can potentiate or inhibit the action of glycine. 8 Under physiological conditions, activation of GlyRs causes an influx of chloride ions into the cell and results in membrane hyperpolarization. As such, GlyRs play a key role in mediating the effects of glycine as an inhibitory neurotransmitter.
Inhibitory neurotransmission in the spinal cord is fundamental to the processing of nociceptive signals and an important area for analgesic drug discovery. 9 GlyRα3 is highly expressed in the dorsal horn of the spinal cord and is a vital intermediate in inflammatory pain signaling pathways. 10 Prostaglandin E2 (PGE2), through its cognate receptor, triggers central sensitization, and this is achieved at least partly through inhibition of GlyRα3 activation, specifically by phosphokinase A (PKA)–mediated phosphorylation of a serine residue (S346) in the second intracellular loop.10,11 GlyRα3 receptor knockout mice have a reduction in pain sensitization caused by either PGE2 or peripheral inflammation.10,12 Additionally, the key role that glycinergic neurones play in nociceptive and itch pathways was recently revealed by targeted ablation and activation of these neurons in mice. 13
Evidence has therefore established GlyRα3 potentiation as a putative analgesic mechanism with which to target new therapeutics. However, to date, there is a lack of pharmacological tools with which to fully characterize the role of GlyRs in regulating nociceptive input at the dorsal horn. Current research relies on a combination of knockout mouse data and allosteric modulators with known polypharmacology, for example, 2,6-di-tertbutylphenol (DTBP) and dehydroxyl-cannabidiol (DH-CBD).12,14-16 Our aim was therefore to develop a suite of in vitro assays with which to identify novel small molecules able to potentiate GlyRs and serve as the starting point for the development of potent, selective tool compounds with which to characterize their role in pain and analgesia.
In this article, we have constructed homomeric and heteromeric glycine α3 receptor expressing cell lines (α3-GlyR and α3β-GlyR, respectively) and characterized them electrophysiologically. We have then conducted a high-throughput screen with a fluorescent membrane potential assay format, configured to discover positive allosteric modulators (PAMs). Through subsequent use of both medium-throughput automated electrophysiology and low-throughput manual patch clamp, we have demonstrated that activity of genuine hits from this screen translate through to electrophysiological platforms. The structures of hit PAMs are described, along with further characterization of one compound showing superior efficacy relative to current known potentiators and selectivity over GABAAR, another major class of inhibitory neurotransmission receptors of importance to pain.
Materials and Methods
Materials
All chemicals (except in-house Pfizer compounds) were obtained from Sigma-Aldrich (Gillingham, UK). Cell culture reagents were purchased from Life Technologies (Gent, Belgium).
Cell Line Generation
Human glycine receptor α3 variant 2 cDNA (accession number NM_006529.1) was cloned into pLenti6.2 lentiviral expression vector (Life Technologies). Lentiviral stocks were generated and used to create stably expressing pools in a CHOK1 background with subsequent dilution cloning to generate a clonal cell line (CHO-α3-GlyR).
Human glycine receptor β subunit (accession number NM_000824) was cloned into pLenti4 expression vector (Life Technologies). Lentiviral stocks were generated and used to transduce CHO-α3-GlyR; further dilution cloning was performed to create a heteromeric stable cell line (CHO-α3β-GlyR).
Human glycine receptor α1 variant 2 cDNA (accession number NM_000171) was cloned into pLenti6.2 lentiviral expression vector (Life Technologies). Lentiviral stocks were generated and used to create stably expressing pools in a CHOK1 background with subsequent dilution cloning to generate a clonal cell line (CHO-α1-GlyR). GABA α1β3γ2 and α3β3γ2 cell lines were purchased from Eurofins (St. Charles, MO).
CHO-α3-GlyR was cultured in DMEM containing 10% fetal bovine serum (FBS), 2 mM GlutaMAX, 12.5 mM HEPES, 1× nonessential amino acids, and 500 µg/mL blasticidin at 37 °C and 5% CO2 in a humidified incubator. CHO-α3β-GlyR was cultured in the same conditions with the addition of 100 µg/mL zeocin.
Fluorescent Membrane Potential Assay
Cells were resuspended in culture medium and plated into 384-well µClear poly-
For single point (SP) screening, compounds were diluted in DMSO to 4 mM (final assay concentration [FAC] of 5.9 µM) in 150 nL using an Echo550 (Labcyte, Sunnyvale, CA) to which 20 µL of HBSS +/+ containing 100 µM glycine (FAC of 20 µM corresponding to EC20) was added. Compound was added to the cell plates on an FDSS6000 (Hamamatsu, Welwyn Garden City, UK) following a baseline read and increase in fluorescence measured. The response was quantified as maximum peak height ratio (excitation at 472 nm/emission at 540 nm) over a 120 s postcompound addition. Data analysis was carried out with either GraphPad Prism (v6.03; La Jolla, CA) using a nonlinear-fit four-parameter equation weighted by least squares to account for increases in variability or ActivityBase (v8.0.5.4; IDBS, Guildford, UK). Compound effect was calculated as percent activity relative to control wells, with the maximum signal (100% activity) being defined by a saturating concentration of glycine (10 mM) and the minimum signal (0% activity) defined using EC20 glycine (20 µM).
IonFlux HT Electrophysiology Assay
The IonFlux HT (Fluxion, San Francisco, CA), an automated electrophysiology platform based on a microfluidic system described in detail by Spencer et al., 17 was used to perform medium-throughput assays. Extracellular solution (ECS) comprised 148 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM glucose, adjusted to pH 7.4 using NaOH and 310 mOsm. The intracellular solution (ICS) comprised 140 mM CsCl, 5 mM NaCl, 5 mM EGTA, and 10 mM HEPES, adjusted to pH 7.3 with CsOH and 305 mOsm.
Test compounds were prepared from 10 mM 100% DMSO stocks on an Echo550. For SP assays, 300 nL was dispensed into a 96-well V-bottom polypropylene plate in duplicate and diluted to 300 µL in ECS (for agonist testing) or 50 µM glycine (for PAM testing), resulting in a FAC of 10 µM in 0.1% DMSO. For concentration–response curve (CRC) assays, 5-point full-log dilutions were prepared using Echo550 and backfilled with DMSO where necessary, to give 300 nL per well, which was diluted to 300 µL in 50 µM glycine to yield compound FACs of 10 µM to 1 nM in 0.1% DMSO.
CHO-α3-GlyR were harvested and resuspended to 1.5 × 106 cells/mL in ECS immediately prior to addition to the IonFlux plate. For SP format, reagents were added to the wells of an IonFlux plate as follows: 90 µL of ICS to trap wells, 75 µL to all compound wells (C1–C8); 1 mM glycine to C1, test compound + 50 µM glycine to C2–C5 (PAM format), test compound only to C6 (agonist format), 50 µM glycine to C7, ECS + 0.1% DMSO to C8. For the CRC format, wells C2–C6 were replaced with five concentrations of test compound + 50 µM glycine.
Electrophysiological recordings were carried out at room temperature with GlyR currents recorded in a whole cell configuration patch clamp. IonFlux software v2 was used for cell capture, seal formation, and obtaining whole cell and data acquisition. Once whole cell formation was obtained, cells were maintained at a holding potential of −60 mV and data acquisition started. The protocol consisted of 8 × 135 s sweeps; each sweep contained two additions of EC20 glycine followed by control or test compound addition in both agonist and PAM format. The control addition in each of the first three sweeps was 1 mM glycine (maximum control), ECS (minimum control), and 50 µM glycine (EC20 control). The remaining sweeps tested five compound additions with washout in between. The same protocol was used for both SP and CRC format assays. A pulse from −60 to −70 mV was used at the beginning of each sweep for seal resistance measurements. Each sweep consisted of 30 s baseline, 10 s EC20 addition, 30 s washout, 10 s EC20 addition, 30 s washout, 10 s compound addition, and 15 s washout using a sampling frequency of 500 Hz. Chloride current was measured following the addition of compound in the presence of EC20 glycine compared with EC20 glycine alone. A visual representation of the protocol through an exemplar current trace is shown in
Data were exported into GeneData Screener (v13.0.0; Genedata, Basel, Switzerland) for analysis. The whole trace was visualized and cursors set up for 5 s at the end of each period of the sweep/protocol. A baseline average value was measured within the (5 s) cursor at the end of each baseline/washout period. A minimum value of the current amplitude generated upon the second EC20 glycine addition and the compound addition in both agonist and PAM formats was measured within the (5 s) cursor at the end of each addition. The baseline measurement was subtracted from both the EC20 glycine addition and the compound addition current amplitude values to give an absolute current amplitude value (for both). The effects of compounds were quantified by calculating the percentage enhancement of the chloride current generated upon compound and glycine addition above the glycine current alone using the following formula:
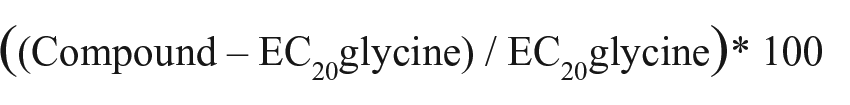
Percentage enhancement values were used to plot the data using GraphPad Prism and perform a nonlinear regression curve fit.
Manual Patch Clamp Assay
Electrophysiology
Voltage clamp recordings were made from CHO-α3-GlyR. Cells were plated onto poly-L-lysine (PLL) coated coverslips the day before experimentation. Patch pipettes were pulled from thick-walled borosilicate glass using a PC-10 puller (Narashige, London, UK). Tip resistances ranged from 2 to 4 MΩ when filled with ICS. ICS and ECS used for manual patch clamp studies were as described above for the IonFlux HT assay. Cells were clamped at −60 mV using a Multiclamp 700B amplifier (Molecular Devices), and series resistance was compensated for by up to 80%. Cells were discarded if series resistance exceeded 15 MΩ or if it significantly changed during acquisition. Data were digitized using a Digidata 1550 (Molecular Devices), and online recording/offline analysis was performed using PClamp 10 software (Molecular Devices).
Experimental design and data analysis
Compound applications were made using a pinch valve gravity-based perfusion system (Intracel, Royston, UK). The EC value of 50 µM glycine was approximated for each cell whereby the current induced by a saturating glycine concentration (1 mM) was compared with the current induced by 50 µM glycine. Typically, 50 µM glycine corresponded to EC5–10. In the rare event that the EC value of 50 µM glycine exceeded EC20, recordings were discarded. The PAM activity of test compounds was assessed as follows: control glycine responses were evoked by applying 50 µM glycine every 20 or 30 s until stable current amplitudes were observed in at least three consecutive glycine applications. Once a reproducible 50 µM glycine response was established, 50 µM glycine was coapplied with 10 µM test compound (1 µM in the case of compound 1). In some experiments, test compounds were applied alone (i.e., in the absence of glycine) to test for possible agonist effects. No more than one test compound was tested per coverslip. Offline, the current amplitude relative to baseline of two stable glycine applications was averaged to give a control glycine response. Current responses to test compounds plus glycine were assessed by measuring the steady-state current amplitude, or alternatively, if the test compound response introduced desensitization, the peak current amplitude was measured. PAM activity of test compounds was determined by calculating percent enhancement of control.
Results
α3-GlyR Cell Line Development and Validation
A clonal stable cell line expressing homomeric GlyRα3 was constructed using lentiviral transduction of CHO cells. Prior to commencing high-throughput assay development, the CHO-α3-GlyR clonal line was characterized electrophysiologically to validate the integrity of heterologous-expressed GlyRα3 channel function. Figure 1A shows the concentration-dependent channel activation by glycine (EC50 = 88 µM, 95% CI: 77–101 µM) measured using IonFlux HT; the EC50 is consistent with previous published values, and we detected no response in parental CHO up to the highest concentration tested (results not shown). Figure 1B shows the distribution of current across 62 individual cells measured using manual patch clamp. The mean maximal current evoked by 1 mM glycine was 10,590 pA (95% CI: 9417–11,762 pA). Chloride current evoked by 50 µM glycine (approximately EC5–10 of activation) was inhibited as expected by the known nonselective GlyR blocker, picrotoxin ( Fig. 1C , E ), and potentiated by the nonselective GlyR PAMs, DTBP and DH-CBD ( Fig. 1D , F ). Maximal potentiation by DTBP (median value of 277% from n = 5) and DH-CBD (median value of 699% from n = 6) was time dependent, requiring approximately 1 min (DTBP) and 10 min (DH-CBD) to reach steady-state potentiation (data not shown).
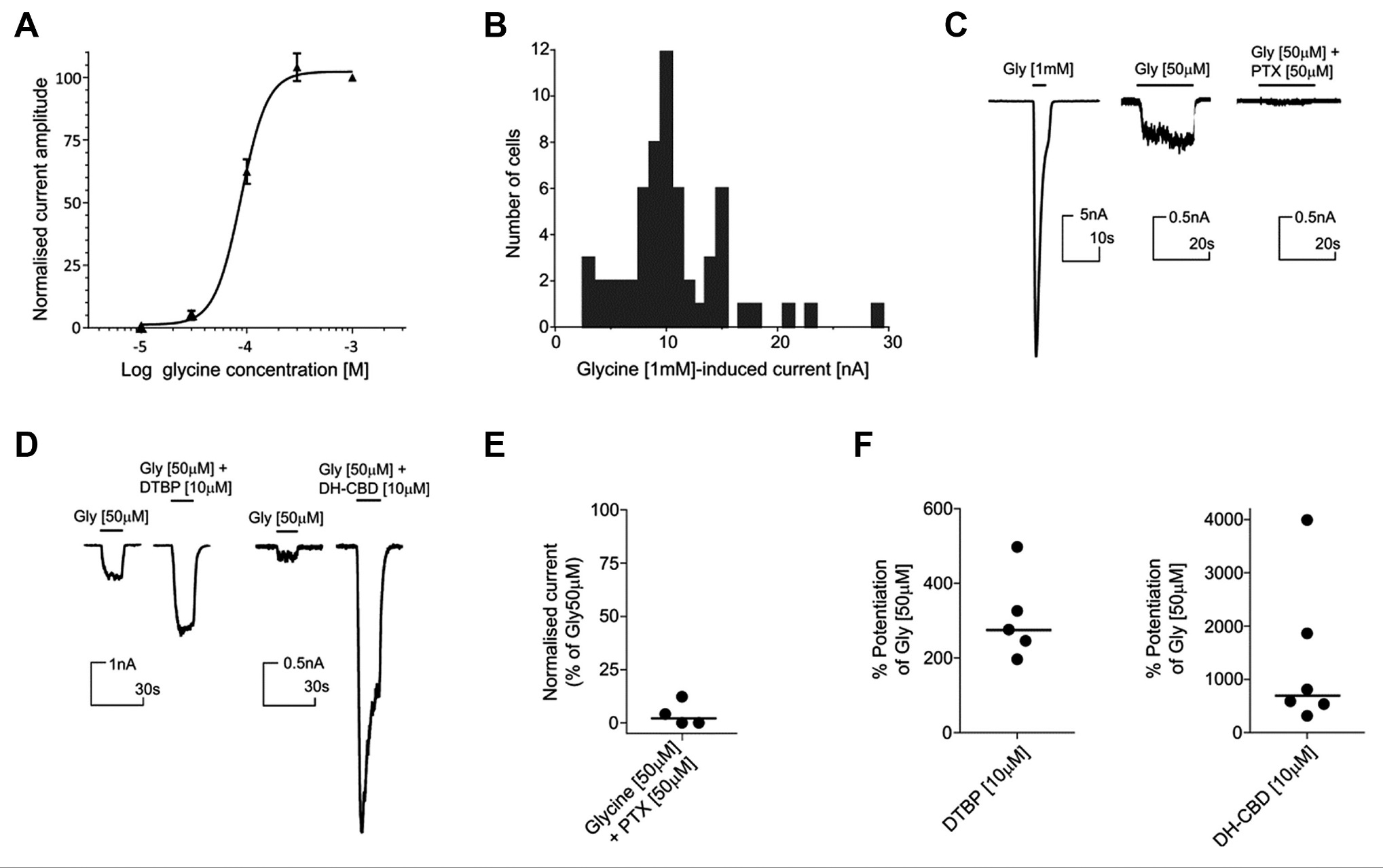
Functional characterization of CHO-α3-GlyR. (
Development of a Plate-Based Membrane Potential Assay
Prior to high-throughput assay development, CHO-α3-GlyR was further stably transduced with β subunit encoding lentivirus to generate a CHO-α3β-GlyR cell line and thereby ensure maximum likelihood of identifying compounds active against the physiological heteropentameric receptor.
To enable screening and identify novel small-molecule modulators of the channel, CHO-α3β-GlyR was used to develop a high-throughput-compatible fluorescent membrane potential assay. Using this format, glycine activation in both the homomeric (α3-GlyR) and heteromeric (α3β-GlyR) cell lines evoked a robust change in membrane potential with EC50 values of 66 µM (95% CI: 42.5–102.3 µM) and 61 µM (95% CI: 49.5–77.0 µM), respectively ( Fig. 2A ). The homomeric and heteromeric populations were both susceptible to picrotoxin block, but consistent with previous reports, 18 the heteromeric receptor required greater concentrations of picrotoxin to achieve equivalent block ( Fig. 2B ). We demonstrated that the assay was sensitive to known potentiators, with the glycine CRC being left-shifted in the presence of maximal concentrations of DTBP (3.1-fold glycine EC50 shift to 23.2 µM; 95% CI: 17.7–30.5 µM) and DH-CBD (2.6-fold glycine EC50 shift to 27.0 µM; 95% CI: 19.8–38.1 µM) ( Fig. 2C ). Also consistent with previous reports, ivermectin was found to be an agonist at α3β-GlyR, rather than a PAM, 19 and both DTBP and DH-CBD demonstrated a small level of agonist activity in the absence of glycine. 16 Based on these validation data, an SP assay suitable for high-throughput screening (HTS) was configured with an EC20 glycine stimulus of 20 µM.
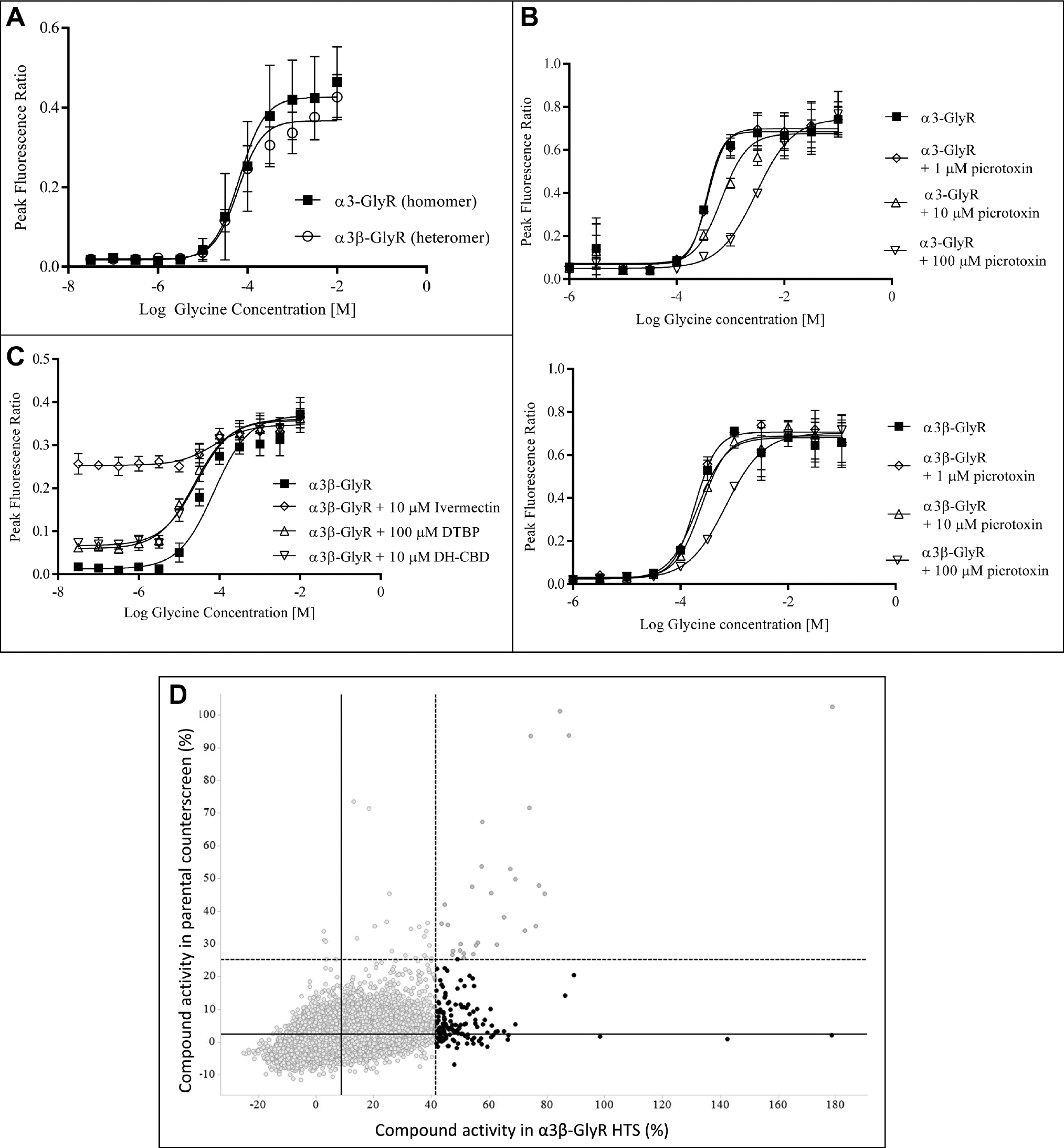
Development of an HTS-compatible fluorescent membrane potential assay. (
HTS to Identify α3β-GlyR PAMs
The fluorescent membrane potential assay was used to screen a small-molecule compound library (targeted ion channel subset of 56,558 compounds) to identify putative new α3β-GlyR potentiators. Since we anticipated a high false-positive rate arising from the screening of such compounds, together with a membrane potential assay format, a counterscreen with the parental cell line was performed in parallel. In this HTS environment, the α3β-GlyR assay performed robustly (screening QC data are shown in
Electrophysiological Characterization of HTS Hits
To facilitate the rapid characterization of HTS hits and ensure an early understanding of how membrane potential activity translated into an electrophysiological readout, the medium-throughput IonFlux HT assay was employed early in the screening strategy. The nature of this assay enabled profiling of all primary screening hits at a single concentration with a multiple addition protocol that quantified activity in the presence and absence of EC20 glycine (PAM and agonist mode, respectively).
Figure 3A
shows the activity of 4/147 compounds, which were confirmed as α3-GlyR potentiators in the IonFlux HT format from the set of membrane potential screening actives (denoted as compounds 1–4 in
Fig. 3A
), with only compound 1 demonstrating significant opener activity in the absence of glycine (data not shown).
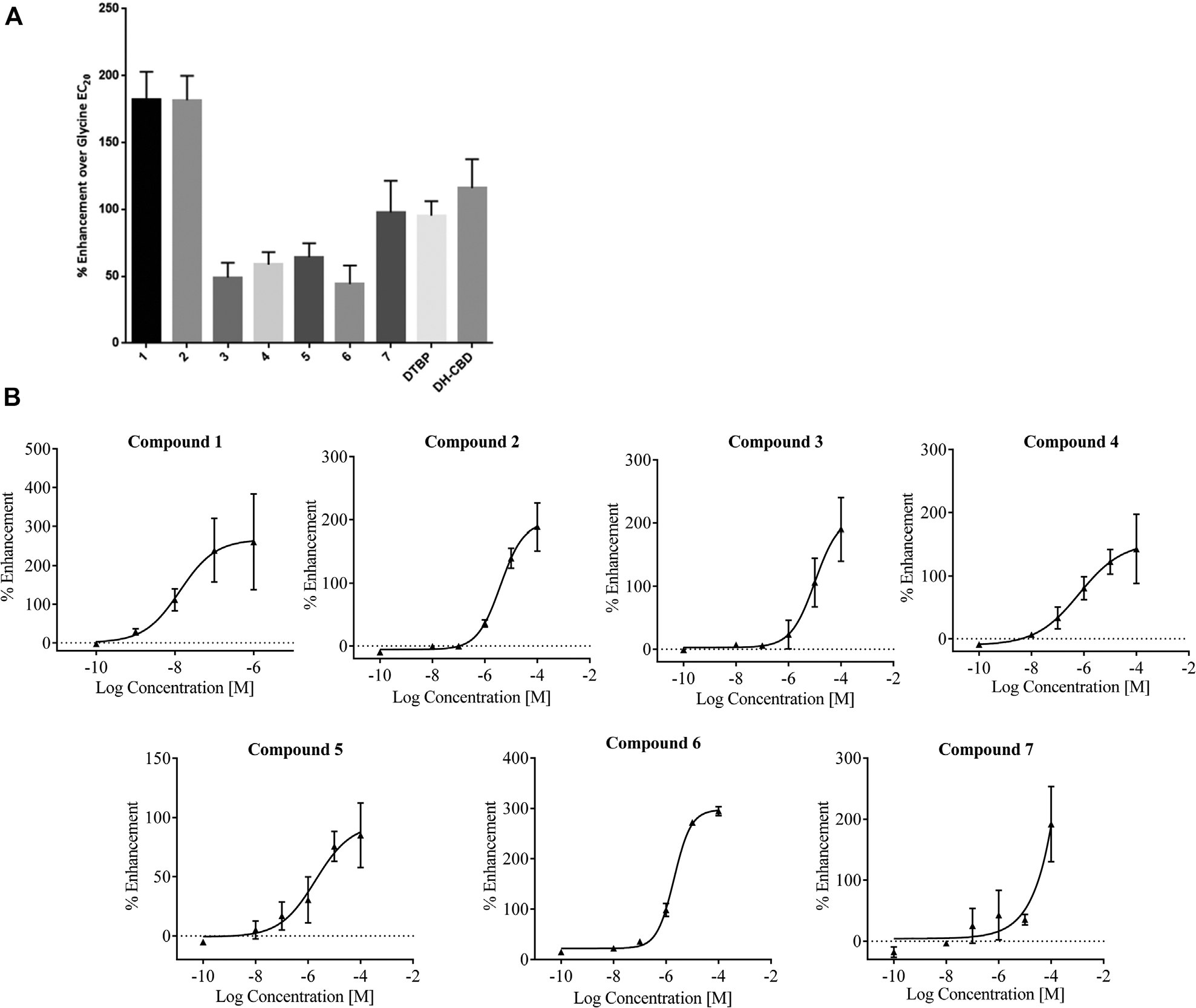
IonFlux HT characterization of α3-GlyR membrane potential screening hits. (
Activity Data for Confirmed HTS Hits.
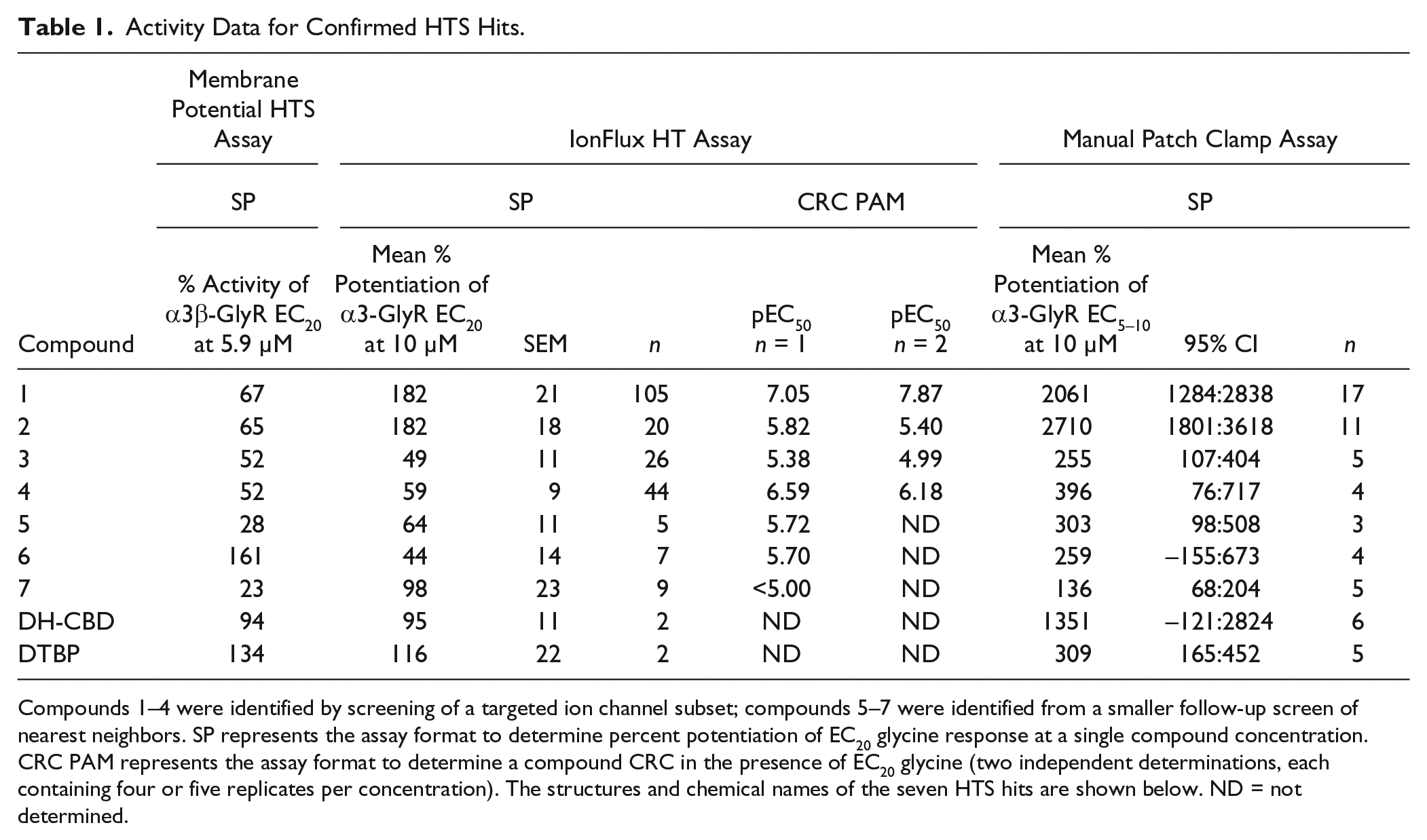
Compounds 1–4 were identified by screening of a targeted ion channel subset; compounds 5–7 were identified from a smaller follow-up screen of nearest neighbors. SP represents the assay format to determine percent potentiation of EC20 glycine response at a single compound concentration. CRC PAM represents the assay format to determine a compound CRC in the presence of EC20 glycine (two independent determinations, each containing four or five replicates per concentration). The structures and chemical names of the seven HTS hits are shown below. ND = not determined.
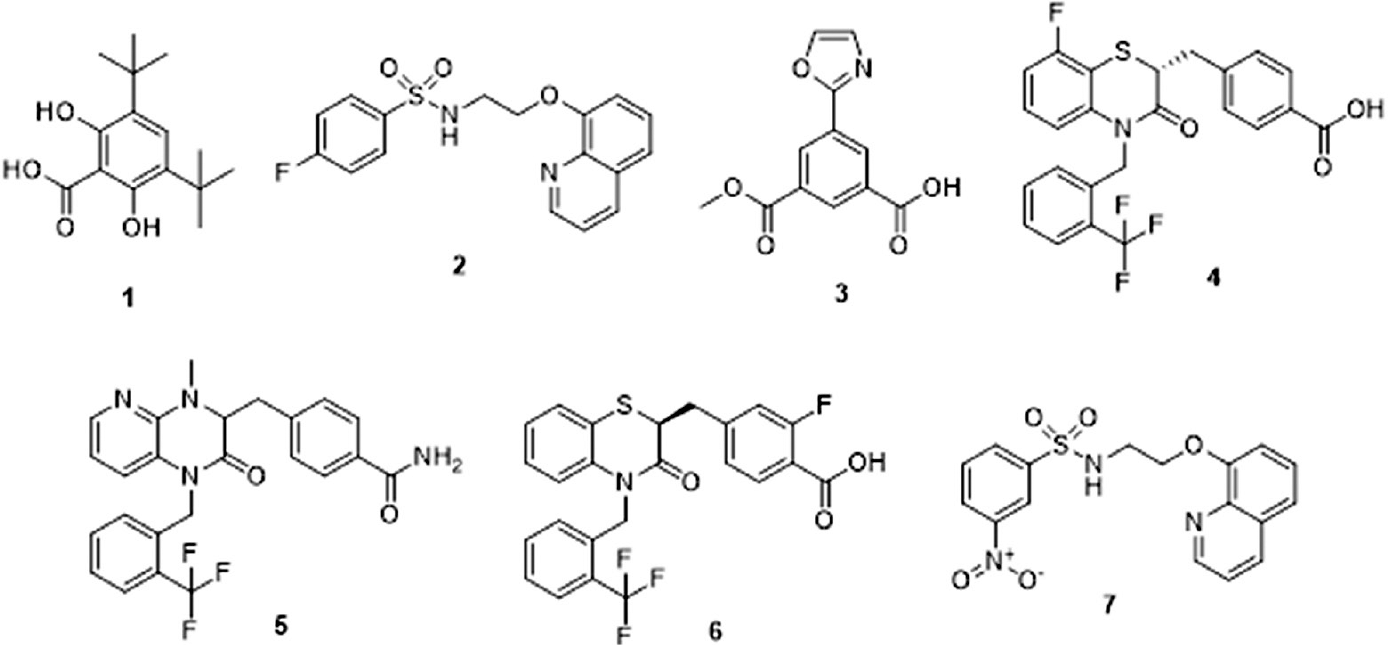
1. 3,5-Di-tert-butyl-2,6-dihydroxybenzoic acid.
2. 4-Fluoro-N-(2-(quinolin-8-yloxy)ethyl)benzenesulfonamide.
3. 3-(Methoxycarbonyl)-5-(oxazol-2-yl)benzoic acid.
4. (R)-4-((8-Fluoro-3-oxo-4-(2-(trifluoromethyl)benzyl)-3,4-dihydro-2H-benzo[b][1,4]thiazine-2-yl)methyl)benzoic acid.
5. 4-((4-Methyl-2-oxo-1-(2-(trifluoromethyl)benzyl)-1,2,3,4-tetrahydropyrido[2,3-b]pyrazine-3-yl)methyl)benzamide.
6. (S)-2-Fluoro-4-((3-oxo-4-(2-(trifluoromethyl)benzyl)-3,4-dihydro-2H-benzo[b][1,4]thiazine-2-yl)methyl)benzoic acid.
7. 3-Nitro-N-(2-(quinolin-8-yloxy)ethyl)benzenesulfonamide.
Manual Patch Clamp Characterization of PAMs
Following triage of HTS hits using the IonFlux HT assay, the seven active compounds were further characterized using manual patch clamp. In addition, a further three HTS hits (compounds 8, 9, and 10) that failed to confirm activity in IonFlux HT were also profiled using manual patch clamp to establish the integrity of IonFlux HT as a triage for genuine PAM activity. Using manual patch clamp, the compound activity at 10 µM was quantified as the potentiation of 50 µM glycine response (EC5–10; see Materials and Methods). Consistent with the IonFlux HT data, the most efficacious PAMs from the HTS hits were compounds 1 and 2, with representative traces shown in Figure 4A and C , respectively. Also consistent with IonFlux HT studies, only compound 1 (at 10 µM) exhibited a small, but reproducible agonist activity in the absence of glycine (data not shown). Subsequently, a lower concentration of compound 1 (1 µM) was assessed and found to have no detectable agonist activity while retaining robust PAM activity at α3-GlyR ( Fig. 4C ). Figure 5F and Table 1 summarize potentiation for all the HTS actives quantified using manual patch clamp, with mean efficacies ranging between 136% and 2710%. Furthermore, the HTS hits that showed no activity in IonFlux HT were confirmed as false positives with no potentiation detected using manual patch clamp ( Fig. 5E ). Unlike the standard compounds, DTBP and DH-CBD, none of the HTS hits demonstrated any time dependence for maximal potentiation (results not shown). The potency of compound 2 was also investigated using the manual patch clamp technique ( Fig. 4B ). For these experiments, an approximate EC50 of 8 µM was determined, a value that is only twofold different from the EC50 determined using IonFlux HT (3.9 µM).
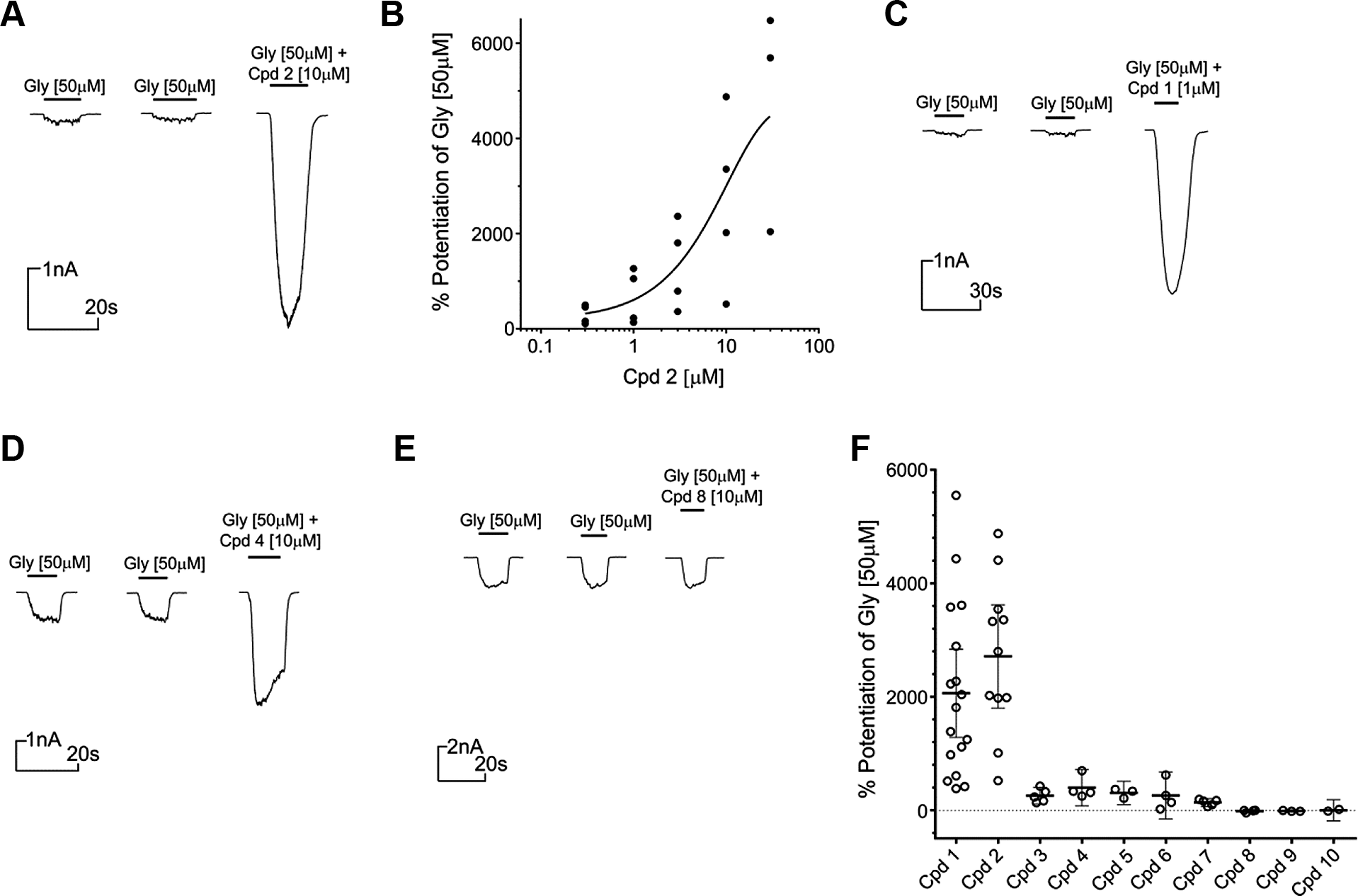
Manual patch clamp characterization of confirmed α3-GlyR HTS hits. (
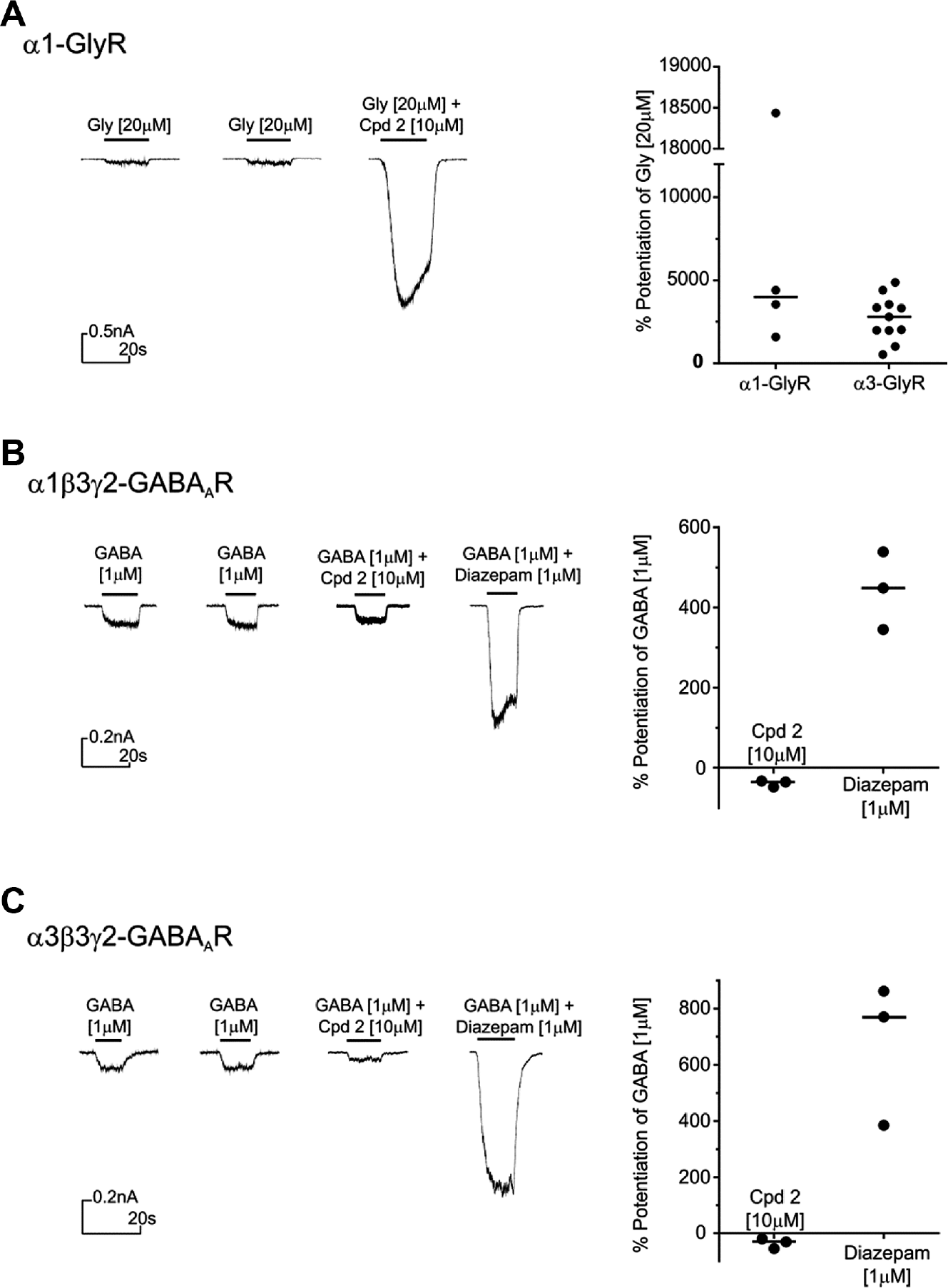
Compound 2 potentiates GlyRα1 but not α1/3β3γ2 GABAARs. (
For these genuine PAMs, activity translated through the screens with notably good agreement between the medium-throughput IonFlux HT and low-throughput patch clamp assays. The compound structures and overall correlation of compound activity through the α3-GlyR screening assays are shown in Table 1 .
Characterization of Compound 2 Selectivity
We were particularly interested in compound 2, which was structurally distinct from previously described GlyR PAMs. It demonstrated very high efficacy of potentiation and showed early evidence of a structure–activity relationship (SAR). The selectivity of compound 2 was therefore determined against both α1/3β3γ2 GABAARs and GlyRα1 using manual patch clamp. At 10 µM, compound 2 was found to be selective over the GABAAR isoforms but not over GlyRα1. Figure 5A shows the potentiation of current evoked by 20 µM glycine (~EC5–15) at CHO-α1-GlyR (median potentiation of 3974%) compared with that of CHO-α3-GlyR (median potentiation of 2797%). At the same concentration, this compound showed no significant effect on α1/3β3γ2 GABAAR currents evoked by 1 µM GABA (~EC10; Fig. 5B , C ), but it should be noted that there is a potentially small negative allosteric modulator (NAM) effect from compound 2 at the GABAA receptors tested.
Hence, we have used a fluorescent membrane potential HTS with downstream electrophysiological characterization to successfully identify at least one new efficacious small-molecule potentiator of GlyRα3 with selectivity over common GABAAR isoforms.
Discussion
The ligand-gated chloride channel, GlyRα3, is a promising drug target in the pain field. Research to date has demonstrated that this receptor plays a major role in regulating nociceptive input to the dorsal horn while also exhibiting a restricted expression profile.9,10,13 The evidence is therefore suggestive that small-molecule potentiation of GlyRα3 activity would be analgesic and have reduced potential for unwanted adverse effects relative to other spinal mechanisms, such as GABAA.
Despite the growing evidence for the potential druggability of GlyRα3, including a recently published cryo-EM structure, 20 researchers currently lack high-efficacy GlyR-selective tool PAMs with which to test the analgesic potential of this mechanism. In part, this is likely due to the challenge of developing and applying a translatable high-throughput-scale screening strategy, which is generally required for a successful medicinal chemistry campaign in the absence of any known small-molecule starting points. This target class, a multisubunit fast ligand-gated chloride channel, presents high technical hurdles to overcome in order to develop high-throughput assays that are predictive and translate through to lower-throughput electrophysiology assays. 21 In addition, the lack of knowledge for this target concerning distinct allosteric small-molecule binding sites and the corresponding absence of any suitable existing tool compounds eliminated the possibility of prosecuting early hit discovery through use of competition binding assays. The remaining options were limited, and hence we set out to validate the use of a fluorescent membrane potential assay as a high-throughput means to identify PAMs from a screening campaign. The membrane potential assay format has been used previously to characterize activity of known modulators at the cys-loop receptor target class,21-23 but to our knowledge, there are no reports that describe its use for investigating GlyRα3 activity or as a means to prosecute hit identification at an HTS scale. Coupled with early triage of hits in a medium-throughput electrophysiology assay prior to characterization by manual patch clamp, we have been able to apply a membrane potential–based screening strategy that has successfully identified new small-molecule PAMs of the GlyR family and represent chemical starting points for development of more potent and selective tool compounds.
As expected, our screening campaign of a large compound subset yielded a low genuine hit rate and large number of false positives. For the follow-on screen of nearest neighbors, the membrane potential assay was performed at n = 2 (at the expense of the counterscreen), and hence we can use this to infer some information about the source of the overall false-positive rate. Combined, the repeat screen yielded 31 hits at 2xSD with nine compounds being confirmed in both n values. Extrapolating these results, we would estimate that our primary screen yielded a 30% confirmation rate from n = 1 membrane potential assay hits (based on 9 from 31 hits) and a translation rate from membrane potential to IonFlux format also in the region of 30% (based on 3 from 9 hits). Therefore, we estimate that the overall high false-positive rate from the original screen resulted from (in roughly equal proportions) variability in the primary screen, off-target activity (filtered out through the counterscreen), and genuine lack of translation between the fluorescence and electrophysiological assay formats. In addition, it is worth noting that through follow-on screening of nearest neighbors, we identified genuine hits between >2xSD and <3xSD activity thresholds, suggesting that we could have missed additional PAMs in our original screen using the 3xSD activity cutoff. However, despite the high rate of false positives, through use of the medium-throughput IonFlux HT assay, we were able to rapidly identify genuine PAMs with an excellent translation of activity through to manual patch clamp.
Efficacy of potentiation in the IonFlux HT SP assay, but not the membrane potential assay, was predictive of efficacy in manual patch clamp, and hence the membrane potential format was suitable for identifying hit material, but would not have been an appropriate choice for SAR generation. Previously published values describing maximal potentiation of GlyR currents by DTBP (279% ± 109) 14 and DH-CBD (989% ± 171) 12 correlated well with our median calculated manual patch clamp values of 269% and 699%, respectively, giving us confidence in comparing efficacy with our HTS hits using this format. The efficacy of HTS hit PAMs (maximum potentiation ranged from 136% to 2710%) marks a significant advancement in the nonselective compounds available currently. Although of relatively weak potency (µM) and not selective over GlyRα1, the most promising screening hit (compound 2) did not show activity against the structurally related GABAAR, another ligand-gated chloride channel and member of the cys-loop receptor family. Existing tool GlyR PAMs, such as those used as the standards for our assays, also display a lack of potency (DTBP EC50 at GlyRα1 in the presence of 10 µM glycine is 9.4 µM ± 10.2; CBD EC50 at GlyRα1 in the presence of 10 µM glycine is 12.3 µM ± 3.8)14,16 and a lack of GABAAR activity.14,15 However, these compound classes have well-described polypharmacology that confounds their use in understanding the specific role of GlyRs in pain and inflammatory regulation. Specifically, DH-CBD is a well-characterized cannabinoid receptor agonist, while DTBP and its derivatives are described as dual cyclooxygenase and 5-lipoxygenase inhibitors.24,25 Although a more complete understanding of compound 2’s selectivity profile remains to be determined, our internal data suggest a lack of promiscuity (having never been identified as an HTS hit in any other screening campaign and demonstrating no significant activity in Pfizer’s internal promiscuity binding panel; data not shown). This opens up the possibility for use as an early pharmacological tool to further investigate the relative roles of GABAA and GlyRs in controlling nociceptive input to the dorsal horn and, more generally, a means to “boost” glycinergic inhibition without affecting GABAergic drive. Considering this early in vitro pharmacology data and compound 2’s favorable physicochemical properties (e.g., molecular weight 346.4, cLogP 3.6, LogD 2.7), we believe these screening hits represent chemical starting points for further work to develop the SAR and investigate potential for both increased potency and GlyR subtype selectivity.
Previous attempts to develop high-throughput membrane potential assays suitable for identification of PAMs for ligand-gated ion channels have met with mixed success.21-23 In general, the result is an inefficient screening strategy that relies on very low-throughput patch clamp systems to characterize genuine hits from the large number of HTS false positives. Our work has demonstrated that such high-throughput plate-based assays are adequate for identifying hit material when used in conjunction with latest-generation electrophysiological screening platforms to triage and rapidly lead to the identification of compounds with the desired activity. As such, we have described a new set of compounds with GlyRα3 PAM activity that show promise as early tool compounds and potential for the development of potent and selective GlyR PAMs.
Footnotes
Acknowledgements
The authors would like to thank Phil Stanley for guidance with the statistics and data analysis procedures.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.