
Abstract
Pharmacoperones correct the folding of otherwise misfolded protein mutants, restoring function (i.e., providing “rescue”) by correcting their trafficking. Currently, most pharmacoperones possess intrinsic antagonist activity because they were identified using methods initially aimed at discovering such functions. Here, we describe an ultra-high-throughput homogeneous cell-based assay with a cAMP detection system, a method specifically designed to identify pharmacoperones of the vasopressin type 2 receptor (V2R), a GPCR that, when mutated, is associated with nephrogenic diabetes insipidus. Previously developed methods to identify compounds capable of altering cellular trafficking of V2R were modified and used to screen a 645,000 compound collection by measuring the ability of library compounds to rescue a mutant hV2R [L83Q], using a cell-based luminescent detection system. The campaign initially identified 3734 positive modulators of cAMP. The confirmation and counterscreen identified only 147 of the active compounds with an EC50 of ≤5 µM. Of these, 83 were reconfirmed as active through independently obtained pure samples and were also inactive in a relevant counterscreen. Active and tractable compounds within this set can be categorized into three predominant structural clusters, described here, in the first report detailing the results of a large-scale pharmacoperone high-throughput screening campaign.
Introduction
Vasopressin type 2 receptor (V2R) is a G protein–coupled receptor (GPCR), a seven-transmembrane protein, found in the distal convoluted tubule and the collecting ducts of the kidney. V2R normally responds to vasopressin and stimulates mechanisms that concentrate urine and maintain water homeostasis. Mutation of the V2R results in mistrafficking of the receptor, which in turn results in vasopressin-unresponsive cells. This deficit leads to nephrogenic diabetes insipidus (NDI).1,2 To date, there are no known drugs capable of reversing receptor-mediate misfolding associated with NDI, and current treatment options are limited to alleviating symptoms. 3 Of the 188 reported allelic variants or mutations found in the V2R, at least 70 have been reported to result in traffic-defective receptors. 2 The cell-based assay described here and elsewhere incorporates the use of the L83Q mutation, which results in the misfolded V2R and subsequently loss of receptor function. While other mutants are predicted to work, the choice to use the L83Q mutant for high-throughout screening (HTS) was based on its ability to lead to NDI and also because the level of receptor function, when in the presence of vasopressin, goes from basal (unrescued) to wild-type (rescued) activity when previously treated with the pharmacoperone.
Pharmacoperones are small molecules that enter cells and serve as a “molecular scaffold” to correct the folding of otherwise misfolded proteins. SR121463, a pharmacoperone of V2R, has been demonstrated by many labs to restore a mutated V2R’s response to vasopressin.4,5 Notably, the binding affinity of this molecule to V2R demonstrates incredible selectivity and relatively constant low-nanomolar Ki profiles, regardless of species tested. SR121463 was also previously reported to be active on at least eight NDI-associated V2R mutants. 6 Unfortunately, this compound has the limitation of also being a V2R antagonist, which limits its potential for therapeutic translation. Thus, we need to find V2R pharmacoperones lacking antagonist activity. We have previously reported the development of a 1536-well HTS assay to identify V2R pharmacoperones. 5 We now report the further optimization of this homogeneous no-wash assay, its adaptation to a fully automated robotics platform, and the completion of a large-scale HTS campaign of the Scripps Drug Diversity Library (SDDL) to identify V2R pharmacoperones. Multiple counterscreens and confirmation assays were applied to confirm selective dose-dependent activity. To ensure accuracy, de novo powder samples of the most active and selective compounds were obtained and analyzed to verify activity. The most potent, selective, and chemically tractable V2R pharmacoperones were clustered by structure class and are identified in this report.
Materials and Methods
Cell Culture and cAMP Assay
SR121463, a V2R peptidomimetic antagonist, was used in the current study as a known pharmacoperone drug, was generously provided by Dr. Claudine Serradeil at Sanofi-Aventis, and was used as received. The V2R pharmacoperone assay was modified compared to our previous report 5 with the following changes: cells were cryogenically frozen in a 9:1 ratio of serum to DMSO in order to maintain a large batch of uniform cells to be used in different time periods in the screening effort. On the day of screening, cells were thawed and added to 1536-well white, solid-bottom, tissue culture–treated plates (part 789173-F, Greiner Bio-one, Monroe, NC) (3 μL/well, 2000 cells/well) in growth media. This was followed by a 25 nL pin-tool addition of test compounds and controls (positive control is 200 nM SR121463; negative control is DMSO only). The plates were incubated for 17 h at 37 °C, 5% CO2, and humidified conditions prior to the addition of 5 μM vasopressin (final) in 1 μL of stimulation buffer (growth media without fetal bovine serum (FBS) plus 2.5 mM IBMX). After stimulation, plates were reincubated for 2 h prior to their removal to equilibrate at room temperature (10 min), followed by addition of 1 μL of 5× cAMP-Glo detection reagent (Promega Corp., Madison, WI). After incubation at room temperature for 19 min, 5 μL of 1× Kinase-Glo (Promega) was added to each well to quantitate residual ATP levels through the production of a luminescent endpoint. Plates were incubated for 10 min at room temperature and then luminescence was measured using a PerkinElmer ViewLux plate imager (PerkinElmer Lifesciences, Waltham, MA). The optimized counterscreen was identical to the primary screen described, except that cells were cultured in the presence of 1 μg/mL doxycycline (Dox) for 36 h prior to plating and during all phases of the experiment. Dox treatment turns off the expression of the mutant transcript of V2R and leaves the cells devoid of the receptor.
The final HTS protocol is summarized in Figure 1 .
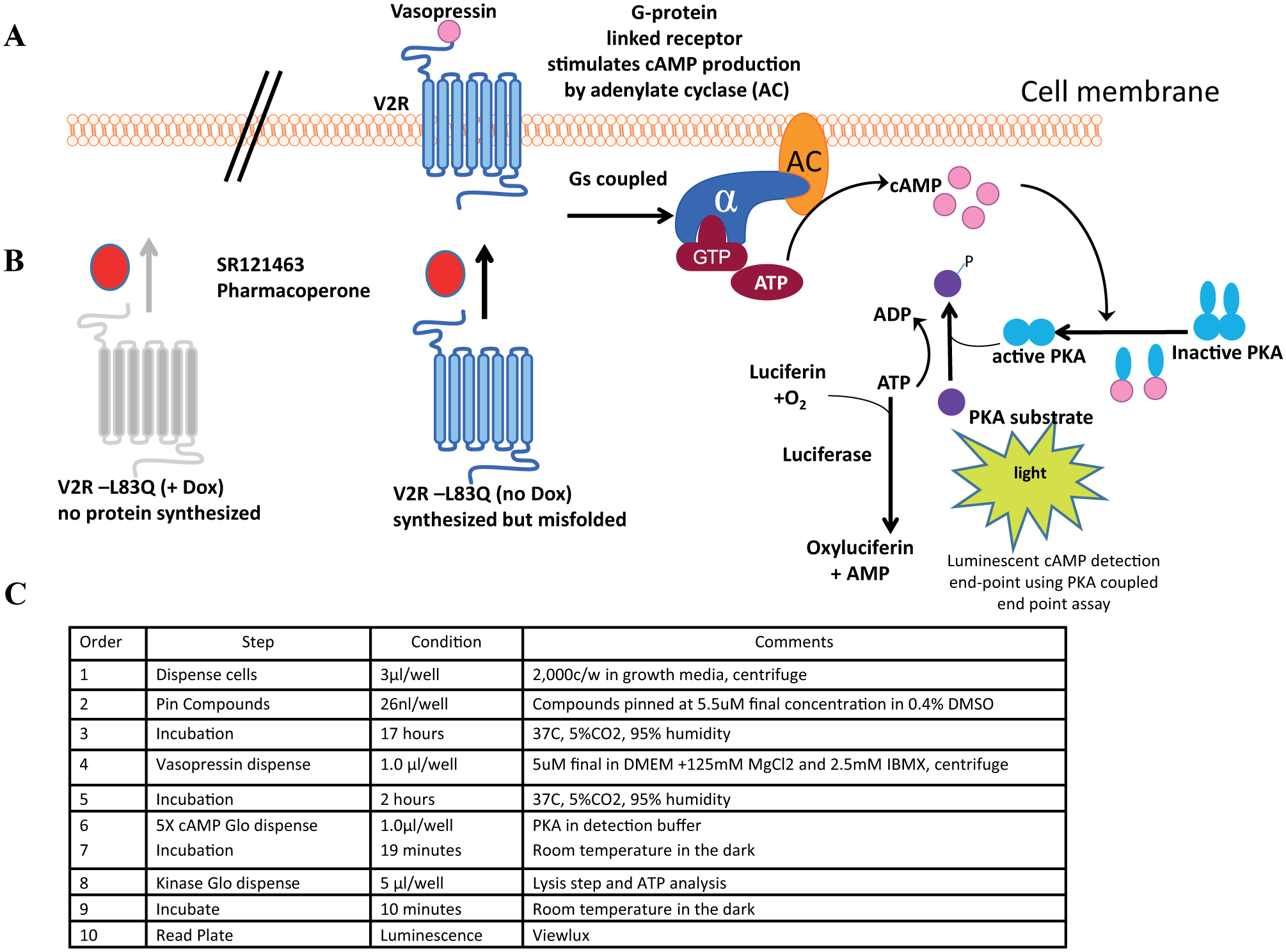
Schematic diagram of the V2R cAMP detection luminescence reporter assay. (
Screening Library
The SDDL, used for this HTS campaign, currently consists of 644,951 unique compounds, representing a diversity of druglike small molecules that are chemically relevant to traditional and nontraditional drug discovery biology. 7 The SDDL has been curated from more than 20 commercial and academic sources and contains more than 40,000 compounds unique to Scripps. SDDL compounds are selected based on scaffold novelty, physical properties, and spatial connectivity. In its current state, the SDDL has several focused sublibraries for screening popular drug discovery target classes (e.g., kinases/transferases, GPCRs, ion channels, nuclear receptors, hydrolases, and transporters), with diverse chemistries (e.g., click chemistry; pan-assay interference compounds [PAINS]–free collections; Fsp3-enriched, covalent inhibitors; and natural product collections) and with desirable physical properties (“rule of five,” “rule of three,” polar surface area, etc.).8–13 All samples in the SDDL were confirmed for purity via liquid chromatography–mass spectronomy (LC-MS) and/or nuclear magnetic resonance (NMR) to provide adequate quality assurance/quality control (QA/QC) after completion of an HTS campaign.
Screening Data Acquisition, Normalization, Representation, and Analysis
All data files were uploaded into the Scripps institutional HTS database (Symyx Technologies, Santa Clara, CA) for plate QC and hit identification. Activity for each well was normalized on a per-plate basis using the following equation:

where “High Control” represents wells containing cells with SR121463 at EC100 and a 5 µM vasopressin stimulus, while “Low Control” represents wells containing just DMSO with the 5 µM vasopressin stimulus, and finally, the “Test Wells”contain test compounds with the same vasopressin stimulus dose. The Z′ and signal to basal ratio (S/B) were calculated using the High Control and Low Control wells. In each case, 24 wells/control were used to generate these values and a Z′ value greater than 0.5 was required for a plate to be consider acceptable (
Results
Assay Principle and Screening Strategy
The V2R HTS assay was miniaturized into 1536-well plates and read using the ViewLux to measure the level of luminescence detected correlating to the amount of cAMP generated upon addition of the potential pharmacoperone. Pharmacoperones will allow V2R to fold properly and shuttle to the surface, allowing the vasopressin ligand to bind, driving the production of cAMP, which then activates protein kinase A (PKA). These events will reduce the ATP-to-ADP ratio within the cell. Using the pathway where cAMP activates PKA, the cAMP-Glo Max luminescent detection system detects cAMP by using PKA as a substrate. The activated PKA is phosphorylated by the ATP and converts it to ADP. The Kinase-Glo detects the ATP levels remaining in the well ( Fig. 1 ). This results in an inverse relationship between the amount of cAMP present and the luminescent signal. The control pharmacoperone SR121463 displays a dose-dependent response in the cAMP-Glo Max system and was used to optimize the conditions for the HTS campaign ( Fig. 2 ). Based on data in other reports, the EC50 for SR121463 was in the expected range for the entire campaign (–8.99 ± –9.08 M; N = 22 separate curves). 5
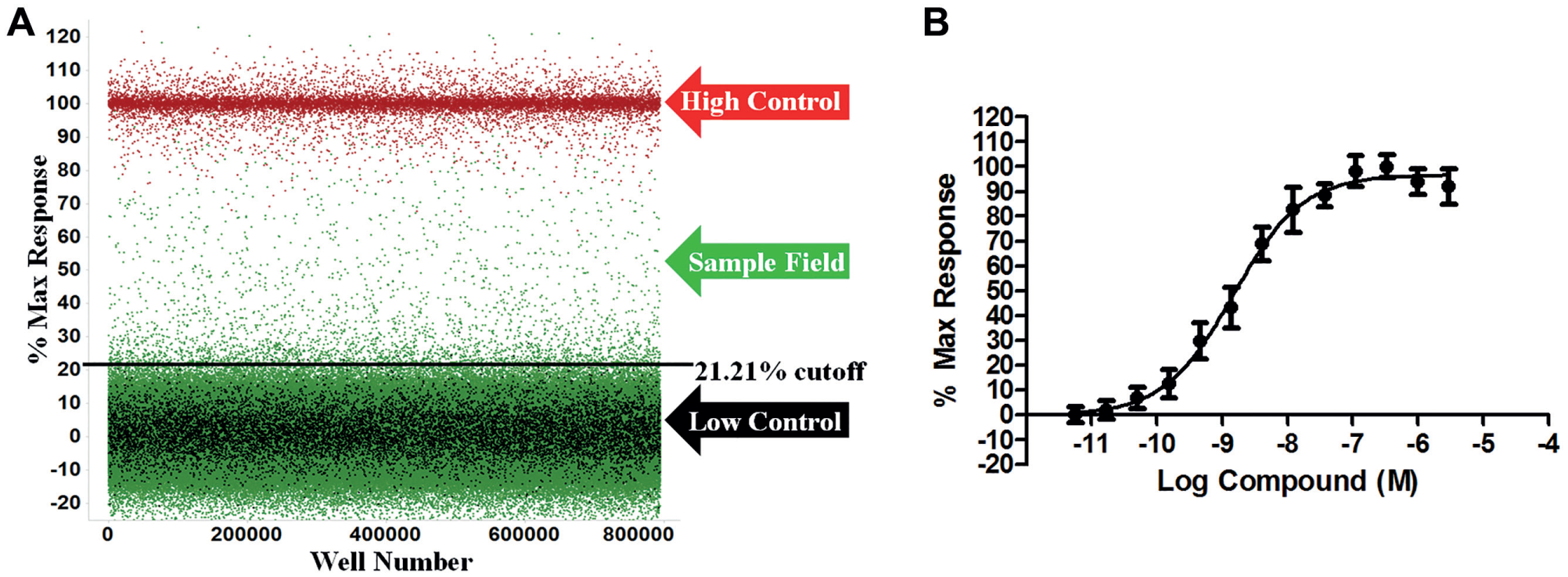
(
The fully automated Kalypsys/GNF robotic platform was utilized in this HTS campaign to accomplish the tasks of dispensing cells and detection reagents, transferring test compounds, and controlling the necessary incubation periods prior to plate measurements by the ViewLux (
1536-Well Format Assay Optimization
The first step in 1536-well assay development was to optimize the cell number per well. The S/B, as calculated by the high control response (EC100 SR121463) divided by the low control response (DMSO only), was not significantly affected by the number of cells plated per well for most seeding densities. However, increasing the cell concentration resulted in suboptimal Z′ values (
Primary HTS
In the primary screen, 644,951 SDDL compounds were tested in singlicate at a final nominal test concentration of 5.22 µM using the optimized conditions described in Figure 1 . The results of the primary screen are shown in Figure 2 , and the statistics can be found in Table 1 . In summary, 3472 compounds were found to be active; that is, they exhibited statistically significant activity, deemed at a cutoff of >21.2%, which was determined using an interval-based mathematical algorithm described previously. 7 Briefly, samples with activity greater than three standard deviations above the high control or less than three standard deviations below the low control were excluded from use only in further calculations; that is, they were not removed from consideration as hits. Two values were then calculated: (1) the average percent activation of all compounds tested in the screen and (2) three times their standard deviation. The sum of these two values was used as the cutoff parameter; that is, any compound that exhibited greater percent activation than the cutoff parameter was declared active.9,10 Day-to-day reproducibility was monitored using the SR121463 dose–response curve. Z′ for the primary screen was found to be 0.73 ± 0.09 with an S/B of 4.8 ± 0.8. The log molar pEC50 of the SR121463 was stable at −8.99 ± −9.08 over 22 plates throughout the primary screen.
V2R Ultra-HTS Campaign Summary and Results.
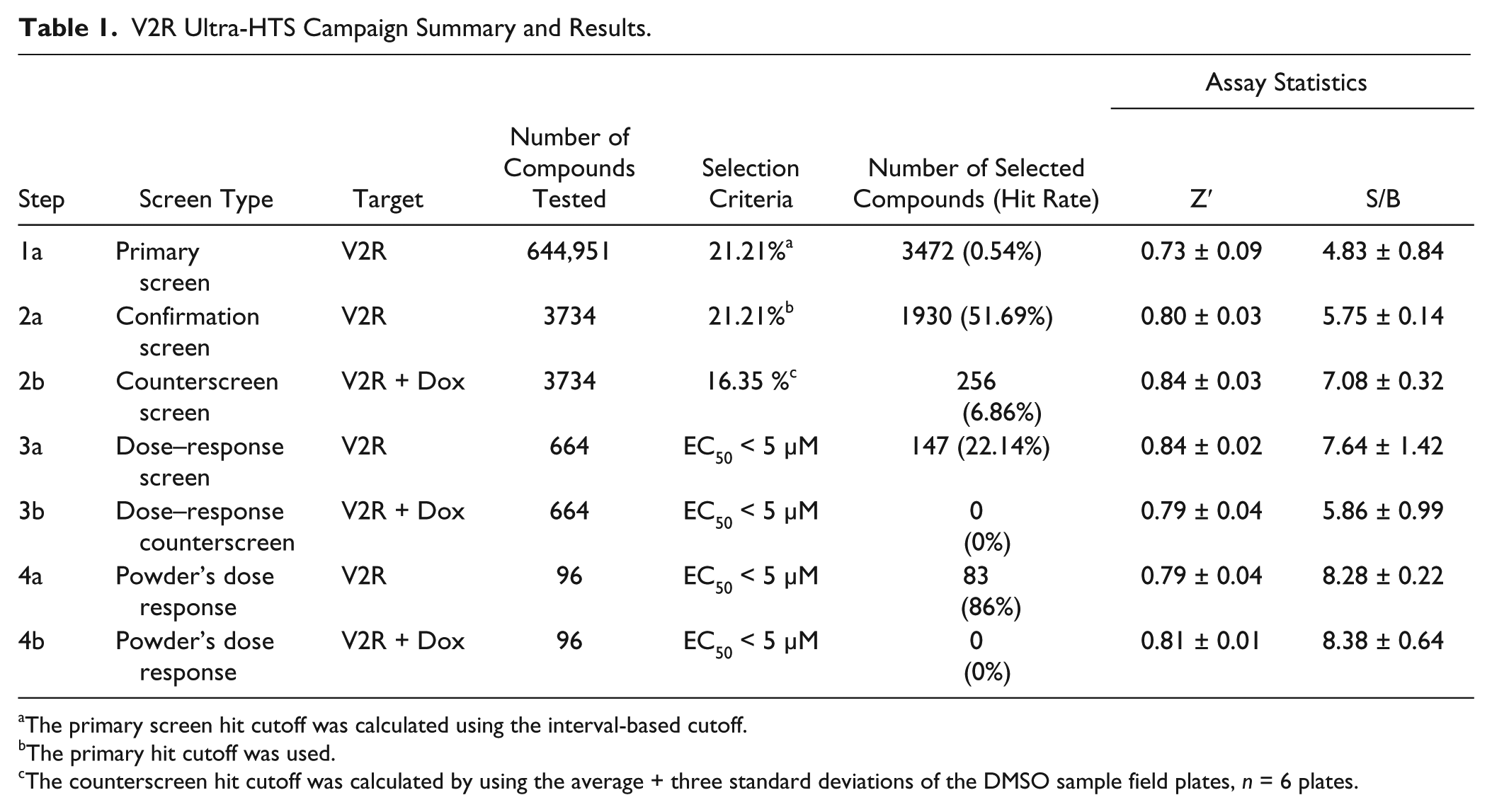
The primary screen hit cutoff was calculated using the interval-based cutoff.
The primary hit cutoff was used.
The counterscreen hit cutoff was calculated by using the average + three standard deviations of the DMSO sample field plates, n = 6 plates.
Confirmation and Counterscreen Assays
Active compounds were selected for confirmation screening, which used the same reagents and detection system as the primary screening assay, but tested each of the 3734 compounds at a single concentration (nominally 5.22 µM) in triplicate. The V2R cAMP pharmacoperone confirmation assay performance yielded an average Z′ of 0.80 ± 0.03 and an S/B of 5.75 ± 0.14. Using the primary HTS assay cutoff, 1930 hits were confirmed with activity equal to or above 21.2%, equating to a reasonable confirmation rate of 51.7%.
A counterscreen assay, similar in format to the V2R cAMP pharmacoperone assay, was then performed. This assay exploits the tetracycline (Tet)-responsive promoter and use of the same cell line as the primary assay, but treated with Dox ( Fig. 1 ). In this way, the assay can identify sundry “off-target” hits and compounds that modify cAMP production by other means. The V2R + Dox counterscreen assay performance had an average Z′ of 0.84 ± 0.03 and an S/B of 7.08 ± 0.32. Using a standard method for the counterscreen hit cutoff (the average of the DMSO samples + three standard deviations), which equates to 16.35%, 256 hits were identified. 16 These 256 hits presumably activated an off-target pathway and were not of further interest.
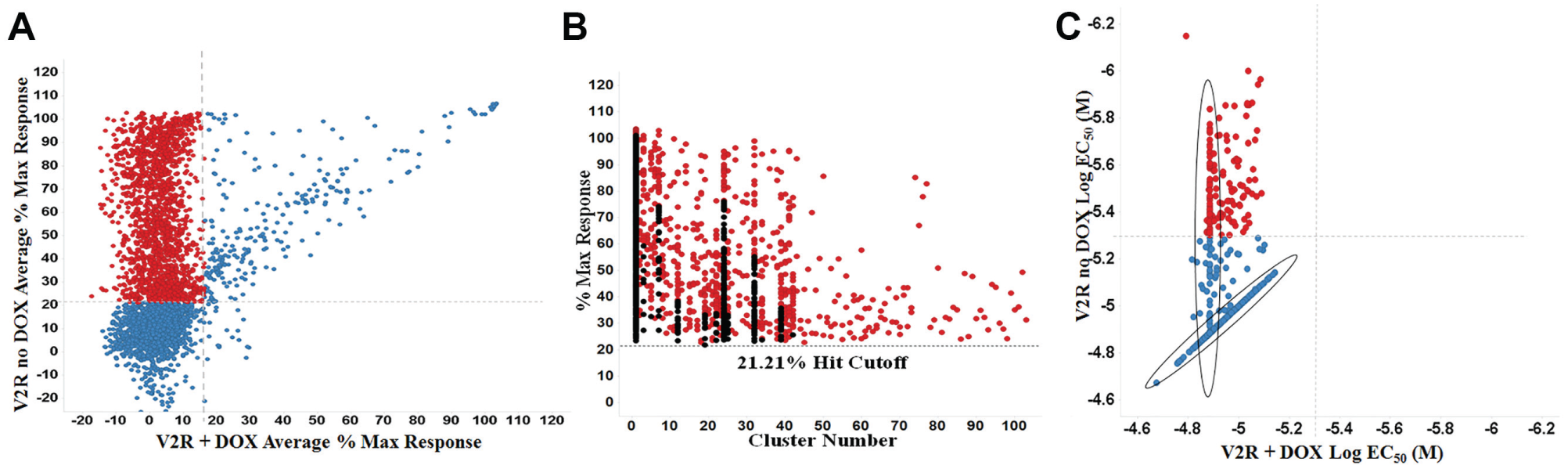
A correlation plot of average maximum percent response for the confirmation screen and counterscreen is shown in
Figure 3A
. Highlighted in red are compounds with activity only in the V2R expressing system. A total of 1694 compounds found active in the primary assay and not in the counterscreen were further investigated using structural cluster analysis. Clustering was based on their maximum common substructures and ranked based on highest activity in each cluster. The top 640 compounds from this cluster ranking are indicated in red (
Fig. 3B
). Using well-established and previously utilized in silico tools such as PAINS and promiscuity filtering, we identified 666 actives that have low promiscuity and were free of PAINS structures.13,17 These 666 compounds were selected to be tested in dose–response studies, of which 664 were available (
(
Titration and Counterscreen Assays
The titration assays were run using the confirmation and counterscreening assay protocols, but with 664 compounds tested at 10 dose levels (threefold dilutions) in triplicate. The V2R cAMP pharmacoperone titration assay performance had an average Z′ of 0.84 ± 0.02 and an S/B of 7.64 ± 1.42, and the counterscreen titration assay had an average Z′ of 0.79 ± 0.04 and an S/B of 5.86 ± 0.99. For each tested compound, percent activation was plotted against compound concentration. A four-parameter equation describing a sigmoidal dose–response curve was then fitted with an adjustable baseline using Assay Explorer software (Symyx Technologies). The reported EC50 values were generated from fitted curves by solving for the x-intercept value at the 50% activity level of the y-intercept value. The following rule was used to declare a compound as “active” or “inactive”: compounds with an EC50 of >5 µM were considered inactive. Compounds with an EC50 of ≤5µM were considered active. A total of 147 compounds were confirmed active in the V2R cAMP pharmacoperone assay while being inactive in the V2R + Dox cAMP counterscreen ( Fig. 3C ). Later, the same 664 samples tested in the titration assays were submitted for LC-MS purity analysis. A total of 647 samples (97.4%) were validated for molecular identity and acceptable purity. As determined by nominal methods (ultraviolet-visible [UV-VIS] spectroscopy, MS, and evaporative light scattering detector [ELSD]), 625 samples demonstrated a purity of >80%.
Of the 147 confirmed active and counterscreen-negative compounds, 96 were available from vendors and de novo powder samples were obtained. These samples were solvated in DMSO and subjected to the same assays in CRC format. Of the 96 compounds evaluated, 83 (86%) were confirmed active in the primary assay exhibiting an EC50 of ≤5 µM, while none were active in the counterscreen. The most active compound identified was SR01000443595, which, upon testing as de novo powder, yielded an EC50 of 70 nM.
Discussion
The HTS campaign was successful at identifying potential V2R pharmacoperone leads. During assay optimization, several issues were resolved. For example, high cell concentrations resulted in higher variability and lower Z′ values (
In summary, the primary screen tested 644,951 compounds, of which 3472 (0.5%) were found to be active. This hit rate is moderately low but not out of a normal range (0.5%–1.5%), as found across >280 HTS campaigns tested at the Scripps Research Institute Molecular Screening Center. In this assay, using the cAMP-Glo detection technology, compounds with activity in the negative direction would be the result of more luminescent signal. In this case, these negative effectors would likely be kinase inhibitors that affect the PKA-coupled detection assay format or ATP analogs, which affect the luciferase activity. Further investigation of the primary screen activity for a false-negative rate found 1398 compounds (0.2%) that exhibited activity in the negative direction below three standard deviations plus an average of the sample field (<–21.89%). Analysis of the compounds with the most negative activity, that is, at or below −50% (190 in total), found they were all known kinase inhibitors, including staurosporine. While these impacted the overall primary hit rate, they should also be ruled out based on lack of V2R target directed activity. It should be noted that the confirmation and counterscreen stage of the HTS campaign tested 3734 compounds, which is greater than the primary actives found. We had increased our library size with additional compounds and added the new compounds to the confirmation and counterscreen assays to determine if any of those compounds could also be active.
At the completion of the titration assays, 147 compounds were identified to selectively rescue the effect of vasopressin on the accumulation of cAMP in cells containing the mutated V2R gene at an EC50 of <5 µM. These 147 compounds were further analyzed by their substructures (
Fig. 4
). Other considerations, such as assay promiscuity, undesirable PAIN moieties, and rule-of-five noncompliance, were used to narrow this set of selectively active compounds (
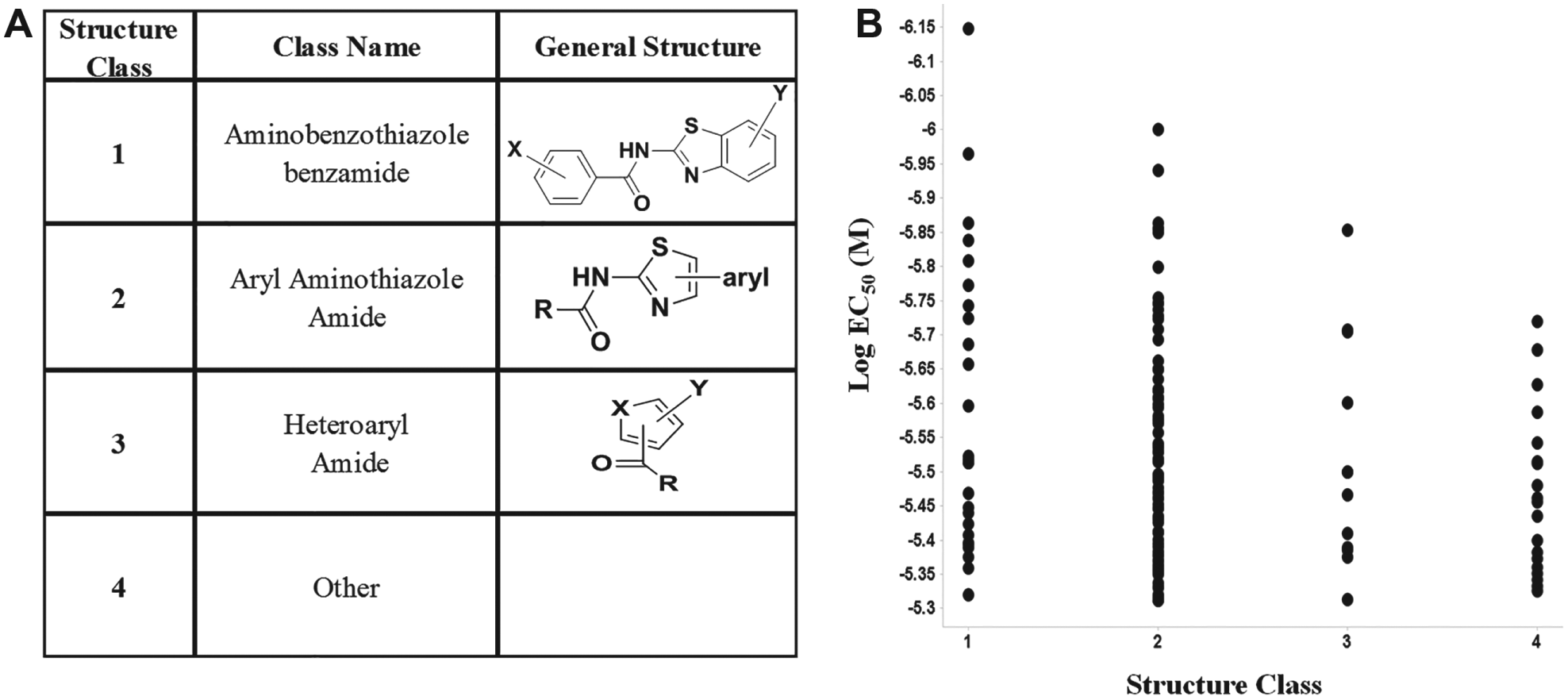
Comparing EC50 to compound structural classes. A total of 147 compounds were confirmed as activators in the V2R pharmacoperone HTS campaign and were inactive in the counterscreen. (
We are unaware of any other reported structures in these chemical classes known to restore activity to misfolded proteins, including, in this instance, rescuing activity of a V2R mutant. To the best of our knowledge, these efforts represent the first large-scale pharmacoperone HTS assay of any kind completed to date. Lessons learned will serve as a guide for other pharmacoperone screening projects, including gonadotropin-releasing hormone receptor (GnRHR), a target we wish to pursue in a future effort.
Conclusion
We have successfully completed an HTS campaign, identifying potential pharmacoperones capable of rescuing V2R activity in cells expressing a mutated V2R misfolded protein. We have shown that in this system, we can restore the function of the V2R by demonstrating vasopressin stimulation of cAMP production in these mutant cell lines in the presence of a pharmacoperone (with multiple test compounds showing efficacy). Leads identified fall into three chemical clusters and appear to set a basis for medicinal chemistry efforts in probe development. Further efforts are underway to determine whether these compounds act as rote agonists or as antagonists in cells expressing the wild-type V2R protein. Those that show no antagonist characteristics could be interesting leads for therapeutic intervention. Through the appropriate selection and application of assays, HTS has proven to be productive in identifying potential pharmacoperones that may be useful in the treatment of diseases caused by protein misfolding and misrouting, including diabetes insipidus.
Footnotes
Acknowledgements
We thank Pierre Baillargeon and Lina DeLuca (Lead Identification, Scripps Florida) for compound management.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health grant 5R01DK099090-04.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.