
Abstract
Evidence of adaptive immune responses in the prevention of cancer has been accumulating for decades. Spontaneous T-cell responses occur in multiple indications, bringing the study of de novo expressed cancer antigens to the fore and highlighting their potential as targets for cancer immunotherapy. Circumventing the immune-suppressive mechanisms that maintain tumor tolerance and driving an antitumor cytotoxic T-cell response in cancer patients may eradicate the tumor or block disease progression. Multiple strategies are being pursued to harness the cytotoxic potential of T cells clinically. Highly promising results are now emerging. The focus of this review is the target discovery process for cancer immune therapeutics based on affinity-matured T-cell receptors (TCRs). Target cancer antigens in the context of adoptive cell transfer technologies and soluble biologic agents are discussed. To appreciate the impact of TCR-based technology and understand the TCR discovery process, it is necessary to understand key differences between TCR-based therapy and other immunotherapy approaches. The review first summarizes key advances in the cancer immunotherapy field and then discusses the opportunities that TCR technology provides. The nature and breadth of molecular targets that are tractable to this approach are discussed, together with the challenges associated with finding them.
Introduction
It has long been known that adaptive antitumor immune responses arise during the development of cancer and that they may play a role in restricting the progression of the disease (reviewed in Jessy 1 and Ribas 2 ). Lung cancer and melanoma have been observed in immunosuppressed recipients of organs donated from former cancer patients, even where donors had been in remission for decades and where the transplanted organ was unrelated to the site of the original disease.3,4 The discovery of tumor-infiltrating lymphocytes (TILs)5,6 and of specific antitumor T-cell responses in melanoma patients7,8 in the late 1980s and early 1990s highlighted the role of T cells in the immune response to cancer. Stimulation of patient blood or TIL with their own tumor cells can give rise to T cells with increased antitumor activity toward their own tumor cells compared with their activity against the patient’s nontumor cells.9,10 Subsequently, it was shown that specific T-cell clones could be expanded from lymphocytes derived from melanoma patients when stimulated with tumor cells from the same melanoma patients. 11 This type of natural tumor immunity arises when cancer cells express viral proteins, accumulate somatic mutations, or suffer epigenetic shifts that result in expression and immune presentation of mutated and/or inappropriately expressed proteins.
Peptides from the tumor-associated antigens (TAAs) that arise in this way are presented to TAA-specific T cells by antigen-presenting cells (APCs). In addition, TAAs are presented on the tumor cell surface as peptides in complex with human leukocyte antigen (HLA) class I and II molecules12–15 ( Fig. 1 ) and are recognized by TAA-specific T cells. There are three major types of HLA class I (A, B, and C) that are further divided into subtypes and allelic variants of the subtypes. Different HLA variants have diverse affinities for different peptide sequences, thus ensuring diversity of epitope presentation at the cell surface. A number of recent reviews discuss the role of HLA class I antigen presentation in cancer.16,17
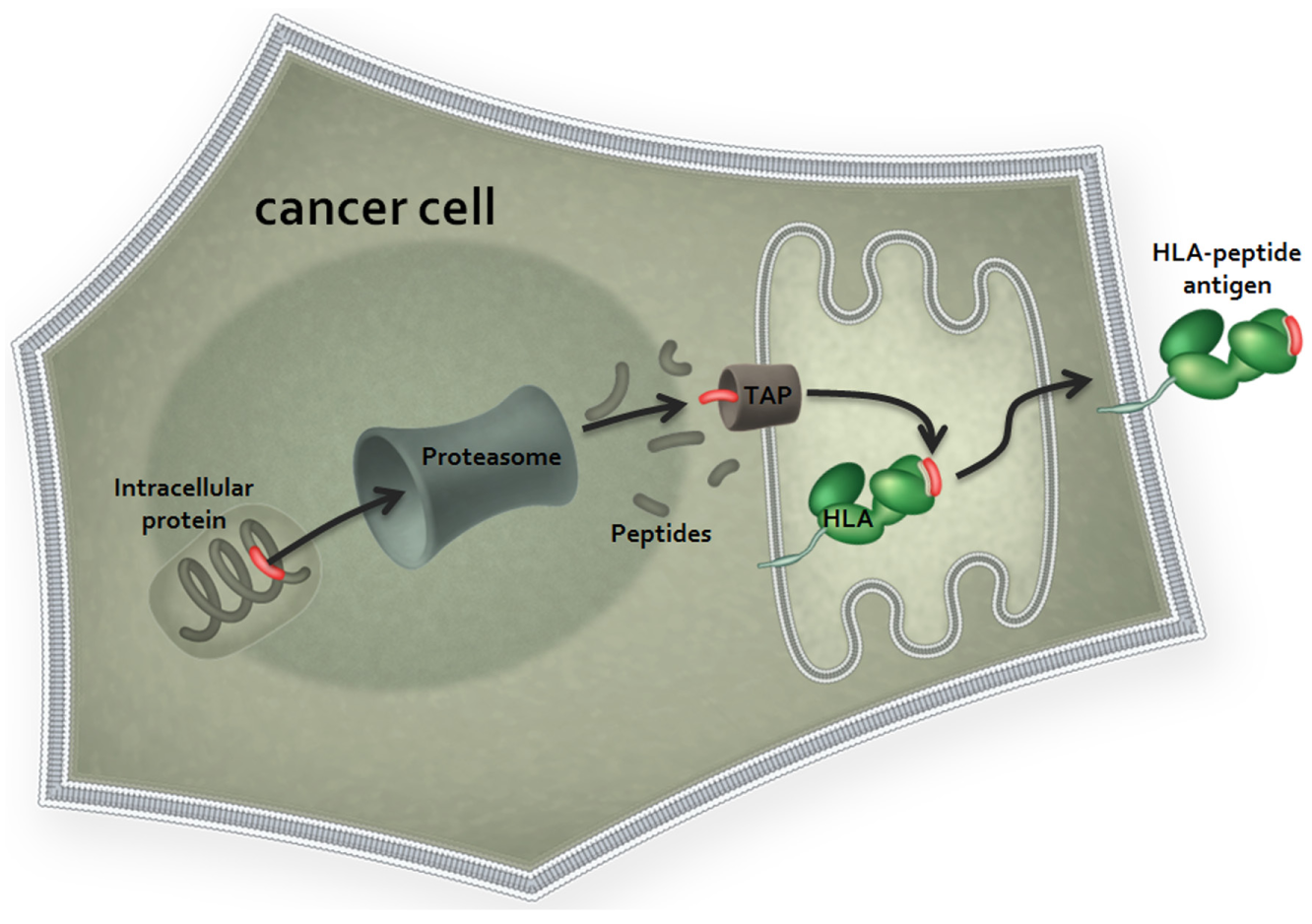
Major histocompatibility complex (MHC) class I antigen presentation. Cancer antigens are proteolytically processed via the proteasome into peptide fragments. Antigen-derived peptides are imported into the endoplasmic reticulum (ER) via the transporter associated with antigen processing (TAP) complex, where they associate with MHC class I complexes in an affinity-dependent manner. Each cell may express up to six class I HLA alleles, two HLA-A alleles, two HLA-B alleles, and two HLA-C alleles that individually determine the peptide binding selectivity of the MHC complexes with which they associate. For simplicity, only one of these complexes is shown. MHC-peptide complexes are exported from the ER via the Golgi to the cell surface, where they are presented as epitopes for T-cell receptor (TCR) binding.
While TAA peptide presentation is common in cancer, truly effective immune responses against tumors appear to be relatively infrequent. There is evidence to suggest that immune responses are important in slowing the progression of the tumor and also influencing the response to therapy. A number of studies suggest that patients with evidence of effective adaptive immune responses also have an improved prognosis in a number of indications.18–24 Moreover, natural adaptive responses also may contribute to the clinical success of other treatments such as neoadjuvant chemotherapy and antibody therapy.25–27 The goal of cancer immunotherapy is to boost the effectiveness of the host immune response, such that the disease is not merely slowed but may be controlled or even reversed.
Vaccination strategies against cancer antigens comprised the first line of approach to cancer immunotherapy, and a large number of vaccine trials are currently ongoing. Despite technological advances in the vaccine field, there has been very little evidence of clinical efficacy in most cancer patients thus far.28–31 Only one cancer vaccine, Sipuleucel-T, has been approved by the Food and Drug Administration (FDA) to date for the treatment of refractory prostate cancer. 32 The disappointing results of vaccine trials have highlighted the need to focus on two key areas affecting the efficacy of the anticancer immune response, which together enable tumor cells to hide from immune detection. First, tumors have in themselves developed strategies to effectively evade the host immune response, achieved at least in part by a downregulation of antigen presentation, a shift in the balance of checkpoint pathways to a suppressive phenotype, and the downregulation of surface HLA.17,33 Second, most tumor antigens are by definition “self-antigens,” and thus thymic selection effectively removes the most potent T-cell receptors (TCRs) from the repertoire. Indeed, it has been demonstrated that the binding affinities of natural TCRs against self-antigens are several orders of magnitude less than of those raised against antigens derived from pathogens.34–36 In recent years, it has also been discovered that cancers actively maintain an immune-tolerant local environment, which enables them to persist and to evade immune ablation. The tumor microenvironment is enriched in regulatory T cells, myeloid immune-suppressor cells, and anti-inflammatory cytokines such as interleukin (IL)–10 and transforming growth factor (TGF)–β.37,38 As a result of all of these factors combined, patient-derived antitumor responses are typically lacking in sufficient potency to eradicate the disease.
Several other strategies have been pursued in an attempt to harness the power of naturally occurring TILs and to make them into effective antitumor cells. Systemic delivery of cytokines, including IL-2 and interferon, has been used to nonspecifically enhance systemic T-cell activation, and in fact this remains an FDA-approved treatment for melanoma. Much more recent work has focused on the development of monoclonal antibody-based checkpoint inhibitors, spurred by the clinical success of agents such as the anti–PD-1/PD-L1 and anti-CTLA4 therapeutic antibodies. These agents act to block immune-suppressive interactions at the tumor cell–lymphocyte interface. 33 Among the most recent clinical successes with checkpoint inhibitors are ipilimumab,39,40 which targets CTLA4, and nivolumab 41 and pembrolizumab, 42 which both target the molecular interaction between PD-1 and PD-L1. Sustained responses have been observed in melanoma using ipilimumab, with around 20% of patients treated gaining a long-term survival benefit. Pembrolizumab has also recently reported efficacy in non–small cell lung cancers, with 45% of patients who express PD-L1 showing prolonged overall survival and progression-free survival benefit. 43 However, even when preselected for target expression, it is clear that only a subpopulation of patients responds to treatment with these agents, and this may reflect the low potency of many natural T-cell responses. Furthermore, as these inhibitory pathways are not restricted to tumors, administration of these agents comes with the risk of indiscriminate inflammatory activation, and adverse inflammatory events have been observed in the colon and skin.39–41,44,45
In addition, natural TILs have been directly isolated from patients, activated ex vivo with high-dose IL-2, and re-infused. Several early phase studies have been completed and robust, durable responses observed in advanced melanoma.46–50 Impressively, more than a decade on from treatment, many patients remain completely tumor free. Studies suggest that most of the TIL epitopes are rare mutational “neo-epitopes,” among which only a fraction may be responsible for tumor cell killing.51,52 Melanoma is an indication that exhibits a high mutational load,53,54 and while the reports of efficacy of this approach to melanoma have been striking, it is not yet clear how this approach would tackle tumors with a lower mutational load, including colorectal, ovarian, and renal cancers. 53 The approach represents a truly bespoke therapy that requires significant in-patient preparation. There remains a need to balance these therapies with more widely applicable “off-the-shelf” cancer immunotherapy treatments.
A more tumor-targeted approach may sidestep the potential risk of widespread immune activation, and to this end, several recent strategies have employed antibody technology to direct T cells to a target TAA on the surface of tumors. Given that antibodies can be engineered to high affinity, the approach also circumvents the limitations imposed by thymic selection on self-antigen recognition. In the adoptive transfer of chimeric antigen receptor (CAR) T cells, autologous T cells are engineered ex vivo to express an extracellular antitumor antibody fused to the intracellular signaling domains of a TCR, and thus the resulting engineered T cell has the affinity of a therapeutic antibody for its target antigen, coupled to a fully effective cytotoxic T-cell response. 55 Similarly, soluble antibody-based approaches have also shown clinical promise. These include blinatumomab, a bispecific antibody (BiTE), targeting CD19 on B-cell leukemia cells while also engaging T cells with an anti-CD3 effector end,56,57 and catumaxomab, which is a trifunctional antibody simultaneously engaging the EpCAM antigen on tumor cells, the CD3 antigen on T cells, and Fcγ receptors on diverse immune accessory cells, including natural killer cells, dendritic cells, and macrophages. 58 The use of such antibody technology to direct T-cell targeting can be effective; however, the approach limits the target antigen pool to cell surface antigens, which represent only around 10% of the potential target pool of all possible tumor antigens. Given that antibodies must necessarily act in the extracellular space, targeting may be further limited by restricted epitope accessibility. As a result of these factors, the majority of potential TAAs are intractable to these technologies.
Affinity-matured TCRs recognize intracellular antigens via derived peptides presented in complex with HLA proteins on the target cell surface. As TCR-mediated modalities are not constrained to surface-expressed antigens, the use of TCRs gives access to a wider range of previously “nondruggable” intracellular targets, provided that their expression is restricted to the target tumor. Several groups have shown that it is possible to generate TCR-like antibodies against purified recombinant HLA-peptide complexes folded in a native conformation. 59 This approach, however, has proved challenging since the scaffolds of antibodies have evolved very differently from those of TCRs. While TCRs recognize a very short, linear, and two-dimensional epitope, antibodies recognize a three-dimensional tertiary structural epitope.59,60 Given that most of the proteins that provide a selective advantage to the tumor during oncogenic progression are also intracellular, it follows that the development of TCR-based therapies to target these pathways is an attractive prospect.
Soluble TCR Therapeutics: ImmTACs
Immune-Mobilizing Monoclonal TCRs Against Cancer (ImmTACs), developed by Immunocore (Abingdon, UK), are a novel class of soluble bispecific therapeutic molecules, consisting of a monoclonal TCR linked to an anti-CD3 single-chain variable fragment (scFv) effector ( Fig. 2 ).61–64 ImmTACs are precision engineered to possess very high affinity for target tumor antigens overcoming the problem of low affinity to self-derived tumor antigens, thus restoring reactivity to self-antigens that are removed by thymic selection. Wild-type TCRs generated by cloning antigen-responsive T cells from donor peripheral blood mononuclear cells (PBMCs) have low affinities for their HLA-presented peptide antigens, typically in the micromolar range. Affinity maturation of wild-type TCRs can generate TCRs with affinities in the picomolar range using a very similar process to that which is commonly applied to antibody engineering. While phage display is used to generate high-affinity TCRs for ImmTAC development, other techniques (such as yeast display and rational design) have also been used to generate high-affinity TCRs.65,66 Mutations are introduced into the six complementarity determining regions (CDRs) to increase both the affinity for the antigen and its dissociation half-life.67,68 This process has been applied across a number of TCRs to date covering a variety of HLA subtypes,62,63,69–71 and crystal structural studies demonstrate that the three-dimensional architecture of the affinity-matured TCR interaction with the major histocompatibility complex (MHC) remains similar to that of the wild type.65,72 The high affinity of the TCR portion of the ImmTAC for its cognate target drives binding to tumor cells both in vitro and in vivo, subsequent to which polyclonal T cells are recruited from the circulation to the tumor site via the lower affinity anti-CD3 effector ( Fig. 3 ). In this respect, the 1000-fold affinity differential between the TCR and the effector is essential to the effective pharmacodynamic properties of the molecule. Once a sufficient density of the ImmTAC is achieved at the tumor cell surface, a functional immune synapse is formed and a potent cytotoxic T-cell response is initiated. 62 The high affinities of the TCR achieved by precision engineering enable TCRs to recognize low copy numbers of theh HLA-peptide complex.69,71
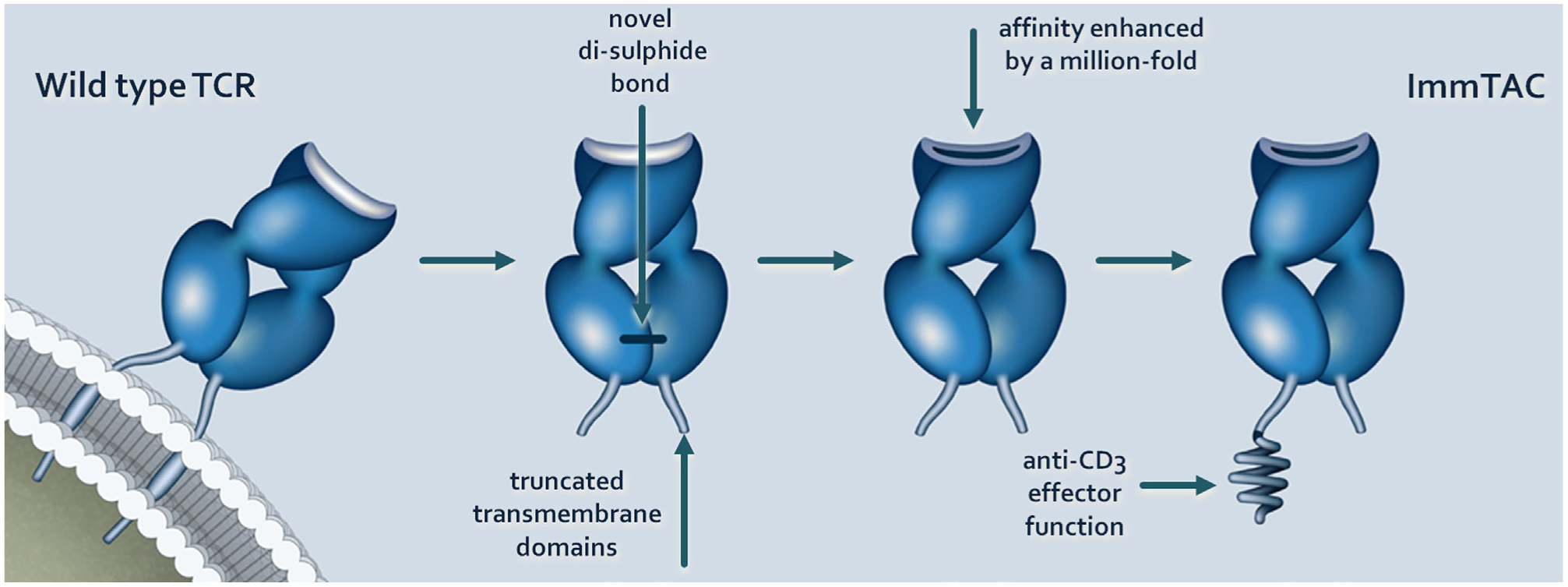
Generation of a soluble Immune-Mobilizing Monoclonal TCR Against Cancer (ImmTAC) from a cloned wild-type T-cell receptor (TCR). Parental TCR molecules are derived from antigen-specific T-cell clones expanded in vitro from donor peripheral blood mononuclear cells (PBMCs). The TCR is made soluble by the removal of the transmembrane domains of the TCR and the introduction of novel cysteine residues to create a disulfide bond and stabilization. Affinity maturation of wild-type TCRs using phage display techniques similar to those applied to antibody engineering generates TCRs with affinities in the picomolar range. T-cell recruitment is driven by a nanomolar affinity anti-CD3 single-chain variable fragment (scFv).
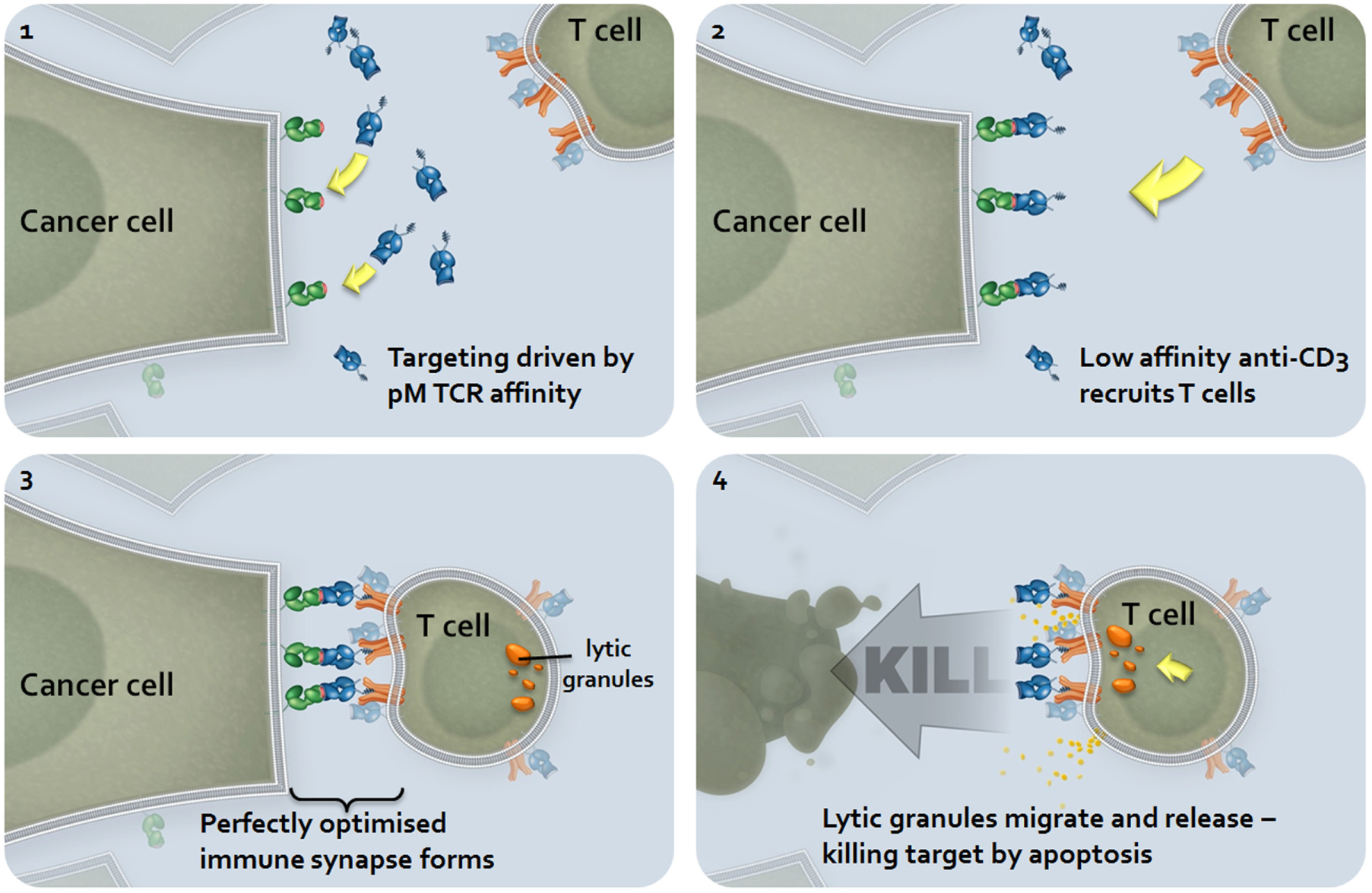
Immune-Mobilizing Monoclonal TCR Against Cancer (ImmTAC) mechanism of action. Systemically circulating ImmTACs are recruited to the site of action via the high-affinity recognition of the peptide-MHC epitope. Circulating patient T cells are then recruited to the site via the lower affinity anti-CD3 single-chain variable fragment (scFv). The difference in affinity between the T-cell receptor (TCR) and the anti-CD3 scFv is an important feature of the molecule, driving the epitope-specific targeting of the tumor. The length of the linker between the TCR and the scFv is critical to enabling the formation of a perfectly optimized immune synapse and the activation of the recruited T cell. Once activated, the CD8+ T cell mediates target cell killing via apoptosis.
The first ImmTAC (IMCgp100) to be clinically tested (clinical trial number NCT01211262) has completed a phase I safety and tolerability trial in patients with advanced stage melanoma and has recently entered into a phase IIa study. The drug is designed to target cells expressing the melanocyte differentiation marker, gp100, and it appears to be well tolerated by patients in addition to showing encouraging signs of clinical efficacy (unpublished data). In addition to the potent and durable responses observed in cutaneous melanoma, IMCgp100 has also shown clear signs of efficacy against ocular melanoma (unpublished data), an indication that has previously proven refractory to other immunotherapeutic approaches. 73
Adoptive Therapy Using High-Affinity TCRs
Adoptive T-cell transfer therapy defines a therapeutic approach whereby lymphocytes are extracted from a patient and engineered ex vivo to impart enhanced antitumor cytotoxicity before being reintroduced back into the original donor. The approach includes the CAR T-cell strategy described above. A number of groups have studied the use of viral vectors for the delivery of native TCRs to autologous T cells with the aim of reprograming polyclonal T cells to recognize intracellular tumor antigens. A growing number of clinical trial results have been reported, and significant antitumor responses have been observed.74–76 Engineering T cells with affinity-enhanced TCRs to selected tumor antigens and their application to adoptive T-cell transfer methods is a highly promising strategy with clear benefits similar to those ascribed to ImmTACs, described above ( Fig. 4 ). Affinity enhancement overcomes the restrictions imposed by self-tolerance, and thus these agents are not limited to targeting rare mutational epitopes and can be applied more broadly than the adoptive approaches that depend on extracted TILs or on TCRs cloned from TILs. Highly encouraging and sustained responses mediated by affinity-matured adoptively transferred TCRs targeting the NY-ESO-1 tumor antigen have recently been reported, both in multiple myeloma 77 and synovial sarcoma (Robbins et al. 76 and unpublished data). Importantly, the treatment is well tolerated, without any evidence of auto-immune toxicity, and the reinfused cells demonstrate effective trafficking to the tumor, expansion, and persistence in vivo. No evidence of T-cell exhaustion (PD-1, LAG-3, TIM-1 expression) is observed in these studies, with any relapses being attributed instead to loss of either the target antigen or transduced T cells. In the synovial sarcoma study, evidence for the establishment of a memory stem cell population in vivo is observed (unpublished data). In line with CAR T cells, the adoptive cells persist in the recipient, to date extending up to and possibly beyond 2 years.
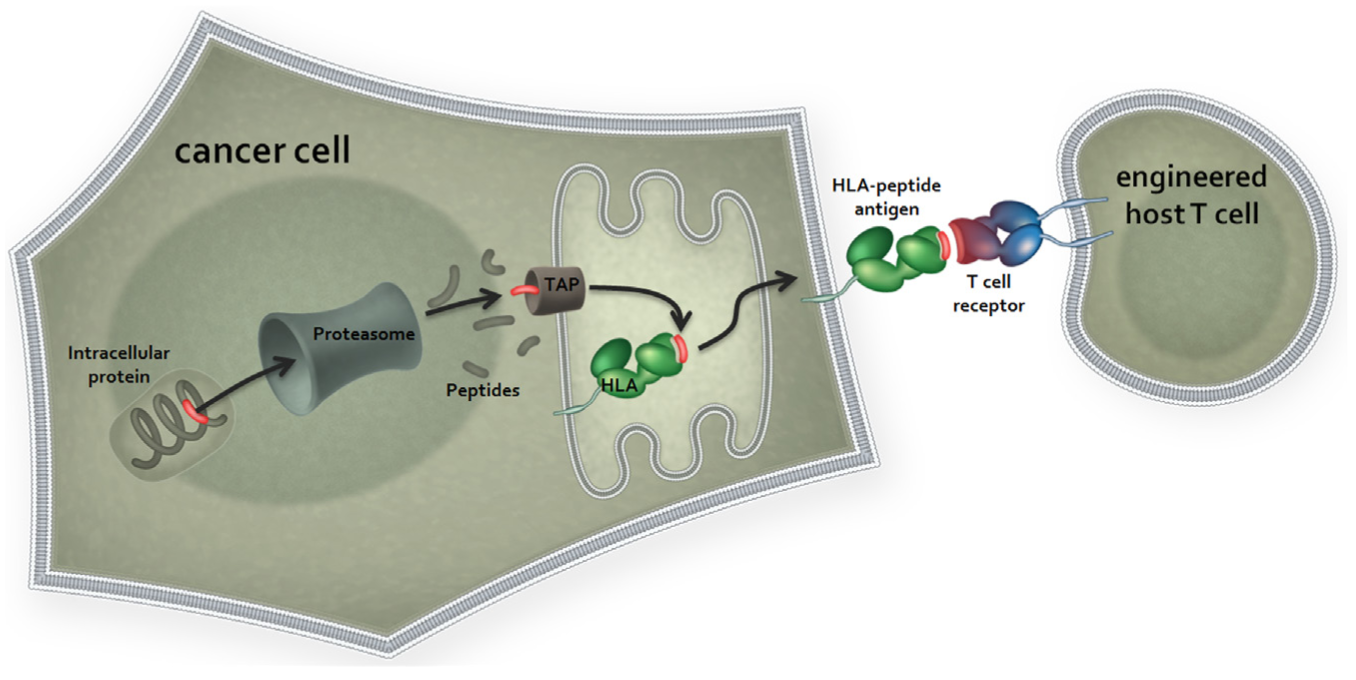
Adoptive transfer of affinity matured T-cell receptors (TCRs). High-affinity TCRs are introduced into patient autologous T cells via means of lentiviral expression vectors. The transduced TCRs are processed and assembled as wild-type TCRs and expressed at the cell surface, forming fully functional signaling complexes. Human leukocyte antigen (HLA)–peptide antigen recognition of tumor epitopes and subsequent T-cell activation and tumor cell killing can then proceed. TAP, transporter associated with antigen processing.
When increasing the affinity of a TCR to a given TAA, there is the potential to also decrease its target cell specificity. 78 The potential risks of cross-reactivity have been reviewed79,80 but are beyond the scope of this current review, the focus of which is antigen selection for immunotherapies based on affinity-enhanced TCR technologies. It is important that any affinity-enhanced TCR is subjected to rigorous preclinical tests, and this is discussed in more detail later in this current review.
Target Discovery: Candidate Antigens
The success of any TCR-based therapy is critically dependent on the selection of the best target antigens. The ideal TCR target is one that is present on tumor tissues at levels that are sufficient for TCR recognition but at the same time is either absent from all adult normal tissues or at least present at levels below those required to elicit a TCR-mediated response. These so-called clean target antigens may, for example, arise from viral oncoproteins,81,82 where the target is absent from the patient genome, or from tumor-specific point mutations, fusions, and frameshifts, where the amino acid sequence is changed from wild type.83–87 Viral oncoproteins are rare in most patient populations, however, and while cancer-specific somatic mutations (often referred to as neo-antigens) may seem attractive targets, 88 they are not generally sufficiently frequent within a patient population to make the approach commercially viable for an engineered TCR-based approach. Moreover, in the case of somatic mutations, it is also necessary that the target mutation lies within the sequence of a presented peptide. By far the most tractable approach is to select targets from self-antigens that are either aberrantly expressed or highly overexpressed in tumor tissues relative to normal tissues. Such antigens can be broadly classified into groups according to their biology and distribution.
Cancer Testis/Cancer Germline Antigens
Cancer testis antigens (CTAs)/cancer germline antigens 89 comprise genes where expression in normal tissues is linked only to germline lineages or other similarly immune-privileged tissues. This class includes the first human tumor antigen to be identified, MAGEA1, which was identified in the early 1990s by panning a melanoma CTL clone against a melanoma complementary DNA (cDNA) expression library. 8 Over subsequent years, many more CTAs have been identified, including other members of the MAGE family.90–93 In addition to autologous typing methods, a technique known as SEREX, which uses bacterial libraries to screen sera from cancer patients, 94 has also been used to identify multiple CTAs from diverse gene families.90–93,95–100 The classic CTA is X-linked and frequently referred to as CT-X antigen. It is specifically expressed in male germ cells and/or in placental trophoblasts, both of which are considered immune-privileged sites on account that they do not produce HLA complexes. 101 This antigen class also includes those that are linked to the meiotic division cycle, the so-called mei-CT antigens, and these tend to be autosomal encoded. 102 Many CTAs are of unknown or poorly defined function. Some studies have suggested a survival function for some of these antigens, which may be consistent with their prevalence in late-stage and drug-resistant tumors,103–105 but there is also increasing evidence that a somato-germline transition process could in itself be an oncogenic driver, and the appearance of CTA in tumors may thus be incidental to this transition.106–110 The aberrant somatic expression of CTA that arises during tumorigenesis results from these epigenetic changes and differs significantly between indications.111–113 CTA expression is common in lung, ovarian, liver, bladder, and breast tumors as well as in cutaneous melanoma, although expression is rare in other indications, including leukemia, colon, renal, and pancreatic tumors.114,115
Like many CTAs, the MAGE genes share high sequence homology and overlapping, although not identical, tumor expression patterns. This high degree of similarity between CTA family members may be therapeutically advantageous in situations where the target peptides are shared by more than one family member with the potential to be recognized by the same TCR. The relatively abundant frequency in tumors coupled with their lack of expression in normal tissue makes this antigen class an important source of targets for TCR-mediated immunotherapy. It is important to remember, however, that many CTAs have been identified by their characteristic expression pattern and by sequence homology to other CTAs but are not necessarily identified on the basis of antigenicity.94,116,117 Some research has suggested that despite their restricted expression pattern, some CTAs, such as MAGE and CTAG1B/NY-ESO-1, may be expressed in the epithelium of the thymic medulla, and therefore naturally derived T cells that recognize these antigens may be subject to the same negative selection as those that recognize any other self-antigen. 118 Furthermore, analysis of publicly available sequencing data from The Cancer Genome Atlas project (http://cancergenome.nih.gov/) have shown that some CTAs, particularly some members of the MAGEC class of antigens, have been shown to carry a heavy mutational load in cancer. 119 Taken together, these data indicate that CTA expression alone may be insufficient to trigger an antitumor response in some cases and that somatic mutations in these sequences may also be required. In this case, artificially engineered TCRs with high target potency may circumvent the constraints imposed by negative thymic selection of some natural TCRs to cancer testis antigens.
A current list of antigens that have been classified as CTAs can be found on the Ludwig Institute CTPedia website (http://www.cta.lncc.br). 116 Of note, not all genes that are known as CTAs appear to be restricted entirely to the germline. Some CTAs are also found expressed at low levels in the CNS (e.g., MAGEA12 and GAGE antigens), while others have low but widespread expression, including many of the autosomal linked CTAs such as CTCFL, SPAG4, and DDX43. 117 This tissue distribution factor may influence the choice of such genes as putative targets, depending on whether the approach is one of adoptive T-cell transfer or soluble TCR biologic, as different stringency criteria may apply. One further challenge that faces CTA targeting is that their expression may be heterogeneous in primary tumors, which may limit their efficacy as targets.120–122 Investigation of promoter methylation of CTAs has revealed that these epigenetic changes coincide with this focal expression pattern, 123 although expression of these genes may become more prevalent as tumors progress.111–113
Alternative TCR Target Antigen Classes
Not all high-affinity TCR antigens necessarily have to be restricted to germline expression or mutational “neo-antigens.” Under certain circumstances, normal somatic tissue expression of a target may be tolerated. A second class of TCR antigens, which may be referred to as a “tissue differentiation or lineage marker” class antigens, describes targets where expression is highly selective for a specific lineage of target cell but where there is no relative change in levels of expression between the tumor cells and the normal cells of the same type. These antigens are attractive targets where expression is associated purely with noncritical tissues or with tissues in which any noncancerous material may have already been removed from patients by prior surgery. Such antigens would include those specific to breast, ovary, and prostate lineages, for example, and would also include melanoma target antigens such as MelanA, gp100, and tryosinase that are each in effect markers of melanocyte lineage. Provided that the expression of the antigen is maintained in the diseased cells, these antigens may still be effectively targeted therapeutically. Such antigens can make attractive TCR targets because their levels of expression in tumors tend to be much higher than those of CTAs and may be retained in up to 100% of patients. On the other hand, many cancer differentiation antigens may be lost in late-stage disease as the tumors become de-differentiated or, in the case of metastases, where cell colonies are established outside of their natural signaling context. In the case of melanoma, T-cell responses to tissue differentiation antigens have been observed in patients, despite the fact that they are also self-antigens,124–131 but in most solid tumors, natural adaptive responses to self-antigens are rare. The advent of affinity-matured TCRs makes therapeutic targeting of these antigens a realistic proposition.
Another class of TCR targets may be termed tumor-selective or overexpressed antigens. 132 This group represents antigens with low, but detectable, background expression across normal tissues but where the difference in expression levels between normal tissues and tumors is sufficient to suggest that a therapeutic window could be achieved in vivo. Based on current understanding of the technology, it is likely that this group comprises the largest pool of potential targets for TCR therapy. It can be challenging to identify and validate such antigens, but in return, these antigens can be highly valuable as they are frequently associated with multiple indications and may be represented at high frequencies in patients. Thus, this antigen class provides certain advantages over both differentiation antigens and CTAs, but with the drawback of a higher risk of on-target toxicity. This group may include antigens such as the Wilms tumor antigen WT1, which is upregulated in cancer but is not entirely absent from normal tissues. 133 While multiple factors may affect the relative efficiency of antigen presentation between cell types (both tumor and normal), our internal observations support a broadly linear relationship between RNA expression of a given antigen and recognition by a specific TCR (Immunocore, internal unpublished data). Quantitative reverse transcription (RT)–PCR and RNA sequencing data can therefore be presumed to be predictive of the rate of translation for a given antigen, which in turn relates to antigen processing and presentation. However, full validation of these targets is necessary as reliably predictive methods of determining therapeutic windows for TCRs are not yet developed. At present, preclinical validation relies heavily on empirical functional testing of candidate antigens with TCRs on cells in culture and on in situ hybridization (ISH) or immunohistochemistry (IHC) studies on tissues.
In more challenging indications, cell cycle antigens could also be considered. This class represents the plethora of potential antigens derived from proteins whose expression is closely linked to the mitotic cell cycle. Antigens of this class do not necessarily have a direct biological role in the cell cycle machinery or its control, but they comprise antigens whose overexpression may drive the cell cycle or may be overexpressed as an indirect consequence of it. They are notably absent from normal quiescent adult cells but are expressed in some normal proliferative tissues such as the gastrointestinal (GI) tract, female reproductive tract, and skin. The impact of targeting such antigens with TCRs may be considered akin to chemotherapeutic approaches in terms of mechanism of action and potential for on-target toxicity. However, with such a gene-targeted approach, there is some potential for a degree of selectivity, which may not be possible with a more generalized antiproliferative therapy. Many proto-oncogenes and signaling molecules fall into this category, including serine/threonine kinases134–136 and components of the nuclear factor (NF)–κB signaling pathway. 137 Rapidly dividing cultured cells present many peptides derived from these antigens in complex with their surface HLA, and these are often easily identified by proteomic approaches (discussed in more detail below). It should be noted that such antigens present significant challenges in preclinical development of the TCR therapies.
It may also be feasible to use high-affinity TCRs to target peptides that contain specific posttranslational modifications. Deregulation of signaling cascades is a common event in oncogenesis and has the potential to generate peptides with alternative posttranslational modifications that are unique to tumors. Several reports in the literature describe the isolation of such peptides. A panel of more than 60 tumor-selective phospho-peptides were reported to be differentially presented in hematological malignancies, particularly in more aggressive cancers. 138 Interestingly, and unlike the cancer testis antigens, circulating mononuclear cells from healthy individuals were able to mount cytotoxic responses to these peptides, suggesting that the phospho-antigens may be present at low levels in normal tissues. Other posttranslational epitopes have also been reported in other indications, for example, a deamidated tyrosinase peptide139,140 and IRS2 phospho-peptide in a range of tumors.141,142
Target Discovery: Target Identification and Selection
In our experience, we have found that it can be challenging to identify suitable TCR targets from the existing literature. This may be due in part to the diverse methods employed in the literature to investigate antigen overexpression in tumors (qRT-PCR, RT-PCR, IHC), the specificity of the reagents used, and the diverse sample sizes and patient populations in published studies. All of these factors make it difficult to compare and contrast the suitability of potential target antigens. Moreover, and perhaps most important, very few publications investigate antigen expression relative to a broad range of normal adult tissues but focus mainly on the changes of expression in the tumor solely with respect to the tissue of origin. When addressing possible targets for cancer immunotherapy, the literature has historically been largely focused on antigens for vaccine therapy. In 2009, the National Cancer Institute (NCI) published the results of a project aimed at the prioritization of cancer antigens. 143 The project assigned ratings to antigens based on key characteristics, including features such as immunogenicity in a clinical setting. The study assigned high ratings to many cancer vaccine antigens, which included overexpressed antigens such as CEA and Her2Neu. Our internal investigations have suggested that the expression levels of these antigens in some normal tissues are well above the threshold at which high-affinity TCRs may confer activity (Immunocore, unpublished data). Indeed, adoptive T-cell strategies using CAR T cells or T cells with engineered TCRs that target Her2Neu and CEA, respectively, have both been associated with on-target toxicity in the clinic relating to the expression profiles of the target antigens.144,145 Interestingly, the NCI-prioritized antigen list also included EpCAM, the antibody target of the BiTE therapeutic molecule, AMG110, and this target is also expressed to high levels. On account of the potency of high-affinity TCRs, targets for TCR therapy are subject to strict expression thresholds that are much lower than those of traditional vaccine antigens. As a consequence of the highly diverse mechanisms employed by antibodies, vaccines, and high-affinity TCRs, it is most likely that each different therapeutic approach will draw upon very different, if not almost exclusive, target antigen pools.
An example of a target finding flowchart for TCR therapeutics is outlined in Figure 5 . Of increasing value to target identification for TCR-mediated therapy is the rapid emergence of large publicly available “omics” data sets. Next-generation sequencing has revolutionized target identification in many fields and especially for TCR therapy, where a target is largely independent of both its intracellular location and its biological function. Most notably, large RNA sequencing data sets such as the NIH’s Cancer Genome Atlas (http://cancergenome.nih.gov/) are immensely valuable resources and offer expression and mutation information on sample sizes that stand up to statistical scrutiny. As next-generation sequencing analysis techniques evolve, it is now becoming possible to distinguish alternatively spliced transcripts. Thus, some abundantly expressed genes may also become targets for TCR therapy in cases where a cancer-specific splice variant can be distinguished from a more widespread variant by a unique peptide epitope. By comparison to transcriptomics, it is currently more difficult to derive meaningful screening tools for TCR target identification by combining public proteomics data sets. As the use of genomics tools drive more novelty into TCR target searching, it is highly beneficial to access any data that help to build confidence in the existence of the target at the protein expression level. In a recent example, an in silico analysis of cancer expressed sequence tag (EST) data sets resulted in the identification of a significant number of novel CT genes, 146 but little is yet known about the proteins they encode. The human protein atlas (http://www.proteinatlas.org/) is a public resource that combines antibody validation with tissue expression profiling across normal and cancer tissues in addition to cancer cell lines. Databanks of liquid chromatography–mass spectrometry (LCMS) derived protein expression results can also be found, which include sources such as the EMBL-EBI PRIDE archive (http://www.ebi.ac.uk/pride/archive/) and MOPED (https://www.proteinspire.org/MOPED/mopedviews/proteinExpressionDatabase.jsf). Each of these data sets can be used to build a picture of a novel antigen by which to rank its relative suitability. There is undoubtedly a great deal of novelty still to be unearthed with regard to both wild-type self-antigens and new cancer germline genes.
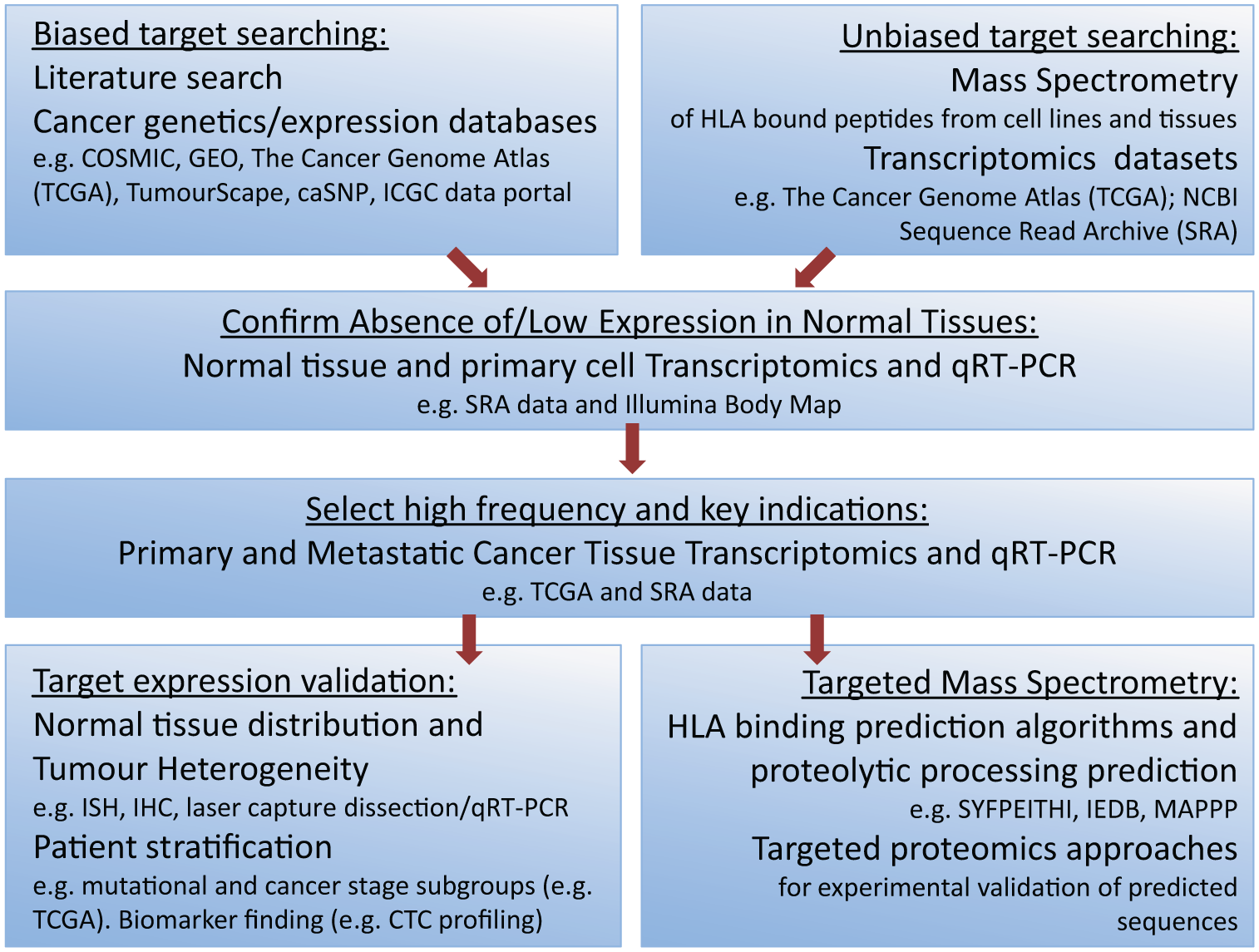
Example target identification flowchart for affinity-enhanced T-cell receptor (TCR) epitopes. Candidate antigens may be initially identified either from sources of literature (publications and publicly available databases) or in an unbiased fashion by proteomic approaches in cancer cell lines and tissues and bioinformatic interrogation of large public RNA sequencing data sets such as The Cancer Genome Atlas (TCGA) or the NCBI’s Sequence Read Archive (SRA). Early filtering to exclude antigens with expression in normal tissues is advisable, and it is suggested that this process examines both whole tissue and individual normal cell types. Large RNA sequencing data sets can dramatically increase the throughput and success rate of target identification when applied to both normal tissue screening and cancer indication and frequency determination. Mass spectrometry approaches to targeted peptide identification are labor intensive but worthwhile where an antigen has high frequency of expression in tumors and a strictly restricted expression profile in normal tissues. Prediction algorithms for human leukocyte antigen (HLA) binding affinity such as the SYFPEITHI and IEDB matrices can be used to identify the most likely peptides from a given sequence, but experimental validation is essential. IHC, immunohistochemistry; ISH, in situ hybridization; qRT-PCR, quantitative reverse transcription polymerase chain reaction.
Target Discovery: Proteomic Discovery of HLA-Presented Peptides
To generate TCRs to a given antigen, it is also necessary to identify the cognate peptide(s) that are presented on the surface of tumor cells in complex with class I HLA. This is one of the most challenging aspects of TCR-based target discovery and involves mass spectrometry of peptides eluted from immune-precipitated HLA complexes derived from cancer cells.147,148 Experimental validation of peptide presentation is required because expression of a protein in a cancer cell does not necessarily imply that a peptide from any given HLA restriction will be presented. Factors such as sequence diversity, length, and differences in intracellular processing all come into play. Peptide identification may be approached in one of two ways. In the first of these, the screening may be run in an unbiased “discovery mode” in which the most prevalent and easily detected peptides presented on the cells are identified. This approach typically identifies many thousands of peptides on a given cell or tumor type, and such screening can rapidly generate large quantities of peptide data that can be compiled into a searchable database and mined for target discovery. One drawback of this approach, however, is that many peptides may pass undetected on the basis of biochemical properties. In particular, the charge of a given peptide can strongly influence whether the peptide is likely to be effectively eluted during LC-MS purification steps or indeed it can be detected by mass spectrometry. In our experience, many tumor antigens have evaded detection in this manner, and a more targeted searching approach has been necessary. Using prediction algorithms for HLA binding, such as the SYFPEITHI algorithm (www.syfpeithi.de), 149 BIMAS (http://www-bimas.cit.nih.gov/molbio/hla_bind/), 150 and Rank Pep (http://imed.med.ucm.es/Tools/rankpep.html), it is possible to rank possible candidate peptide sequences for a given protein sequence. Such approaches have been used successfully to identify T-cell epitopes from tumors. 151 (SYFPEITHI is the most commonly used algorithm and offers the facility to either identify and rank peptides for a given HLA restriction from a specified protein or alternatively screen a peptide epitope against multiple HLA types to identify its preferred binding partner. SYFPEITHI outputs of greater than a score of 20 indicate stable peptide-HLA complex.) Similarly, in the IEDB online tool (http://tools.immuneepitope.org/mhci/), a lower percentile rank suggests stronger peptide-HLA complex. The ability to predict likely epitopes enables searching for peptides specifically on the basis of a defined charge and mass. These algorithms, however, can only be used as guides and do not generally take into account factors such as intracellular proteolytic processing (i.e., the likelihood that a given protein is specifically cleaved at the identified sites to derive the specific predicted nonamer or decamer sequences). One exception is the MAPPP algorithm (http://www.mpiib-berlin.mpg.de/MAPPP/information.html), which attempts to address this by combining the FRAGPREDICT algorithm for proteolytic processing with the HLA binding matrix of SYFPEITHI. 152 Also, a number of public databases catalogue peptides identified by mass spectrometry, including IEDB (http://www.iedb.org/), SYFPEITHI, and MHCBN (http://imtech.res.in/raghava/mhcbn/).
It is important to remember that a TCR epitope is recognized as a protein complex, bound by an HLA molecule. As mentioned earlier in the review, many tumor cells evade immune detection by evolving to express lower levels of surface HLA, and this can have a significant impact on epitope density. In addition, as each tumor cell presents peptides on up to six different HLA alleles, there is evidence to suggest that some HLA alleles may compete for binding of a given peptide. Since some peptides have overlapping binding affinities for different HLA types, the cell surface epitope density for a given high-affinity TCR may depend on the HLA genotype of the target cell. It is also likely that a number of factors may influence HLA class I peptide presentation in vivo, and the process should be viewed as dynamic rather than static. For example, there is substantial evidence that proteolytic processing changes under inflammatory conditions and also in disease. The so-called immunoproteasome is induced in response to interferon-γ and is known to regulate cytotoxic T-cell responses to viral epitopes during infection. 153 Although its principal biological function is in the regulation of the antiviral response, the fact that activated CTL produces abundant quantities of interferon-γ would suggest that the impact of the immunoproteasome on antigen presentation during TCR therapy could be profound. The tumor microenvironment will also have an important role in the therapeutic response, and changes in cell surface costimulatory and coinhibitory molecules will also come into play.
Target Prioritization and Preclinical Safety
TCR binding is dependent on the allelic variant of the HLA protein with which it is in complex. 154 Therefore, high-affinity TCR target selection restricts and defines the patient population for a given TCR. For example, in the US Caucasian population, the most prevalent HLA allele is of HLA-A*02, which is represented at around 50% of the population. 155 This means that 50% of that population would not respond to an HLA-A2–restricted TCR. In contrast, HLA-A*02 is less common in Japan than it is in other populations, and HLA-A*24 is the most prevalent allele. The most comprehensive and up-to-date information regarding allele frequencies in different populations can be found in the Allele Frequencies Net Database (AFND) (www.allelefrequencies.net). 155 Thus, HLA allele frequencies have implications for the eventual size of the target patient population for a given TCR epitope. To inclusively treat patients across a worldwide population, a panel of HLA-restricted and antigen-specific TCR reagents covering the most common HLA-restricted epitopes would need to be available. While many candidate targets may derive multiple peptides for presentation, each of diverse HLA restriction, it cannot be guaranteed that the most prevalent HLA restrictions will be represented among these peptides.149,150,152 The predictive algorithms discussed earlier may help to rank targets based on the likelihood of deriving such epitopes. High-frequency epitopes are clearly very valuable, but a patient population with a low epitope frequency but a high unmet medical need may also be chosen, provided that potential responders can be easily identified by minimally invasive methods or if relevant biopsy material is readily available.
Once expression specificity and frequency criteria are met, other biological factors may drive the selection of target antigen for TCR-based therapy. As applied in the NCI’s prioritization of cancer antigens study, ranking criteria such as immunogenicity, oncogenicity, and expression homogeneity are also sound guidance principles for the selection of targets. 143 Antigens for which there is evidence of cellular and humoral responses in disease, such as those CTAs identified by CTL cloning and SEREX, respectively, may prove to be good candidate antigens. In vivo immunogenicity suggests that HLA peptide complexes for some HLA restrictions must naturally exist and builds confidence that it is possible to identify the parental wild-type T-cell clones from which to generate the final affinity-matured TCR. 143 Although the biological function of the target protein may not be of primary relevance, one could envisage that a protein that conveys a selective advantage to a cancer cell would also be a favorable TCR target. It may also be advantageous to select an antigen that is associated with cross-presentation as this could strongly affect the efficacy and duration of the antitumor response. 156 In this scenario in vivo, mature dendritic cells would take up antigen at the tumor site and migrate to the lymph nodes, where they would present to naive T cells, 157 or alternatively, this process may occur in lymphoid structures within tumors themselves. 158 Evidence suggests that both expression levels of the target antigen and nature of the targeted peptide can influence the extent of cross-presentation.159,160 It may be possible to investigate cross-presentation in vitro, but the parameters governing this process may be difficult to incorporate to a level that would enable high-throughput target triaging. There is evidence in the literature for antigen cross-presentation in some cases, for example, ImmTAC targeting of gp100, which can give rise to indirect presentation of MELAN-A/MART1 antigens in vitro. 161
The choice of target will inevitably be influenced by the therapeutic approach to which it is applied. Cell-based therapy is not amenable to a classic phase I dose escalation trial design as cell populations administered are dynamic and persist and expand in vivo. A follow-up study of patients treated with CAR T cells has demonstrated the long-term persistence of the modified cells, estimating a half-life in excess of 16 years. 162 Thus, safety testing of T-cell adoptive therapies for the overexpressed self-antigen class where the activity relies on a therapeutic window becomes more challenging. In a study in which adoptive transfer was used to deliver a high-affinity TCR-transduced T cell targeting the MELAN-A/MART-1 melanocyte antigen for the treatment of melanoma, significant toxicity was seen in the form of uveitis, caused by inflammation of the melanin-rich cells of the retina. 74 In contrast, using a soluble TCR-based therapy where dosing can be more easily regulated, targeting of melanocyte antigens is already known to be well tolerated clinically and does not result in ocular toxicity (unpublished data). On-target dose-limiting toxicities can be dependent on a number of factors, related to expression levels of the target antigen and level of TCR affinity, for example. Some evidence suggests that in cellular therapy, transduced T cells can occasionally induce severe inflammation and neuropathology in the brain, 75 and while the CNS may have a degree of immune privilege, it is not yet clear whether any expression of a target antigen in the brain could be tolerated in either form of affinity-matured TCR cell therapy approach.
While both soluble and adoptive TCR therapies have been shown to be well tolerated in the clinical setting, preclinical safety testing strategies for both modalities need to be extensive and rigorous. Serious and even fatal adverse events have been observed with the use of these powerful agents.75,145,163 On the one hand, preclinical assessment for on-target toxicity is relatively straightforward and is simply a matter of extensive normal tissue target expression profiling by qRT-PCR and histological and laser-capture techniques. However, testing for the potential off-target toxicities that may arise as a result of TCR cross-reactivity is more difficult to address. In one notable case, cardiac toxicity of a TCR directed against the MAGEA3 cancer testis antigen had not been detected preclinically in experiments conducted on in vitro cultured cardiomyocytes. 163 Follow-up investigations revealed that the toxicity observed in this case was the result of TCR recognition of a peptide derived from the cardiac muscle specific protein, titin. 164 These studies have led to key improvements in the way in which off-target toxicities of high-affinity TCRs are evaluated. These include the use of better in vitro culture models for cardiovascular tissues and, crucially, the introduction of novel peptide scanning methods (Alanine Scan; X-Scan), whereby single amino acid substitutions in target peptides are used in vitro to identify the critical residues required for TCR recognition. Using this method, it is possible to identify motifs by which off-target peptide recognition can be predicted.164,165
Future Perspectives
High-affinity TCRs, in their various guises, have shown great clinical promise and the potential to therapeutically circumvent the constraints that self-tolerance and the immunosuppressive tumor microenvironment place upon the natural antitumor immune response. The technology applies a novel modality that for the first time makes the entire proteome accessible to therapeutic targeting. With such novelty, of course, comes the need to develop novel companion diagnostics, in addition to clinical and exploratory biomarker approaches. Without a doubt, biomarker innovations will greatly influence the field of TCR therapy in the coming years, both in terms of the impact these could have on improvements in patient stratification and in terms of our understanding of the molecular markers of successful clinical outcome. For example, it is now widely accepted that tumors possess a circulating stem cell–like subpopulation known as cancer stem cells (CSCs), which have the capacity to seed metastases and which share many of the biological features and markers of normal stem cells. 166 With improved detection methods, CSCs also provide an easily accessible means of antigen expression typing that would circumvent the need for invasive biopsy. Although currently in their infancy, innovations such as single-cell transcriptomics, 167 coupled with microfluidic cellular enrichment techniques, 168 may, in the long term, enable profiling of CSCs in a clinical setting for use both as biomarkers of disease progression and as diagnostics for initial patient stratification. Achieving diagnostic success in this area would mean that “all comers” clinical trials may become feasible for antigens that are present at lower frequencies across a broader range of indications.
New developments in transcriptomics will also become valuable in our understanding of the mechanisms that drive efficacy in adoptive T-cell studies. Such methods are increasingly being used to selectively track artificially engineered cells to understand their trafficking and expansion in vivo, in addition to evaluating cellular trafficking to sites of on-target activity and toxicity. Data accumulated thus far demonstrate that, as may be expected, adoptively transferred T cells migrate throughout the body and accumulate in tissues where target antigen is expressed.83,163 However, efficacy of cellular therapy will depend on a number of factors, including features such as cellular phenotype.169,170 As perhaps may be predicted, evidence suggests that persistence of the infused cells is key to the durability of the response.171–173 Less clear is the role played by T-cell phenotype in the durability of response, with some studies suggesting that a less differentiated phenotype is more advantageous and others suggesting a memory phenotype is key.174–176 Either way, it is highly likely that T cells in situ are subject to significant phenotypic plasticity, and thus there is a need to evaluate the T-cell function at the site of disease.24,177
The results of tumor genome analysis clearly demonstrate that cancer cells are highly heterogeneous, even when derived from the same patient. 178 Therefore, while TCR-based reagents may have promising efficacy when delivered as monotherapies, it is possible that clonal selection may drive the emergence of antigen-negative resistant cells over the long term. 179 In addition, alleviating the suppression of the T-cell response with a checkpoint inhibitor may have its limitations and liabilities in patients where natural T-cell responses are either weak or absent. Combining agents that potentiate antitumor immunity with ones that introduce targeting and specificity will be an effective strategy to drive sustained and powerful clinical responses in cancer, and it is inevitable that there will be an explosion of immunotherapy combination trials in cancer. The observation that PD-L1 levels are increased on tumor cells following long-term ImmTAC delivery (Linette et al. 163 and unpublished data) is a strong rationale to support the notion that relieving checkpoint inhibition would enhance the therapeutic benefit of a soluble TCR.
Footnotes
Acknowledgements
The authors thanks Joanne Oates, Stephen Megit, and Namir Hassan for critical reading of this manuscript and for their helpful advice during the compilation of this manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: E.S.H., M.E.L., and B.J. are employed by Immunocore.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.