
Abstract
The primary objective of early drug discovery is to associate druggable target space with a desired phenotype. The inability to efficiently associate these often leads to failure early in the drug discovery process. In this proof-of-concept study, the most tractable starting points for drug discovery within the NF-κB pathway model system were identified by integrating affinity selection–mass spectrometry (AS-MS) with functional cellular assays. The AS-MS platform Automated Ligand Identification System (ALIS) was used to rapidly screen 15 NF-κB proteins in parallel against large-compound libraries. ALIS identified 382 target-selective compounds binding to 14 of the 15 proteins. Without any chemical optimization, 22 of the 382 target-selective compounds exhibited a cellular phenotype consistent with the respective target associated in ALIS. Further studies on structurally related compounds distinguished two chemical series that exhibited a preliminary structure-activity relationship and confirmed target-driven cellular activity to NF-κB1/p105 and TRAF5, respectively. These two series represent new drug discovery opportunities for chemical optimization. The results described herein demonstrate the power of combining ALIS with cell functional assays in a high-throughput, target-based approach to determine the most tractable drug discovery opportunities within a pathway.
Keywords
Introduction
Drug discovery today is plagued by high costs of research and development with a low probability of success.1–3 Given the unsustainability of this business model, recent retrospective analyses have focused on identification of the best processes by which to discover novel therapeutics.1,3 Multiple culprits throughout the drug discovery process contribute to this inefficiency. Lack of appropriate biomarkers, inadequate patient stratification strategies, and weak clinical endpoints all may contribute to the high failure rate in clinical development. In the early space, drug discovery programs strive to discover new therapeutics by identifying the intersection between three critical components: molecular target space (target), chemical space (compound), and cellular function or phenotype (therapeutic effect), as illustrated in Figure 1 . The inability to efficiently associate these three functions often leads to failure early in the drug discovery process. Two traditional strategies focused on identifying this intersection are target-based drug discovery (TDD) and phenotypic-based drug discovery (PDD), and each has distinct strengths and limitations. Target-based approaches first focus on the union of chemical matter with a target that has typically been genetically validated agnostic to chemical tractability. TDD in early discovery enables rapid medicinal chemistry optimization of potency and selectivity, while understanding of the target can provide a more transparent path forward into the clinic. However, in the TDD approach, modulation of disease phenotype is independent and subsequent to defining a structure-activity relationship (SAR) between the target and chemical matter and is frequently the rate-limiting step or point of attrition where targets fail.4,5 Taking into context the significant investment in medicinal chemistry resources upfront, TDD can be viewed as a resource-intensive process that limits opportunities to investigate other targets with similar phenotypes that may be more chemically tractable.3,6 In contrast to the TDD approach, phenotypic-based approaches (PDD) first define the intersection between the chemical matter and cellular function ( Fig. 1A ) in a more direct translational model of disease pathophysiology and in this manner identify all potential chemically tractable points of therapeutic intervention. Target deconvolution in PDD may be performed consequent to the identification of chemical matter with the desired phenotype and is generally considered to be the major limitation in the PDD approach.4–9 Although target deconvolution is not necessarily required for optimization of chemical matter, having some understanding of the molecular mechanism of action can greatly enable chemistry and, as discussed earlier, has a significant impact on developing appropriate biomarker readouts and patient stratification strategies, two factors associated with failure in the clinic.5,9
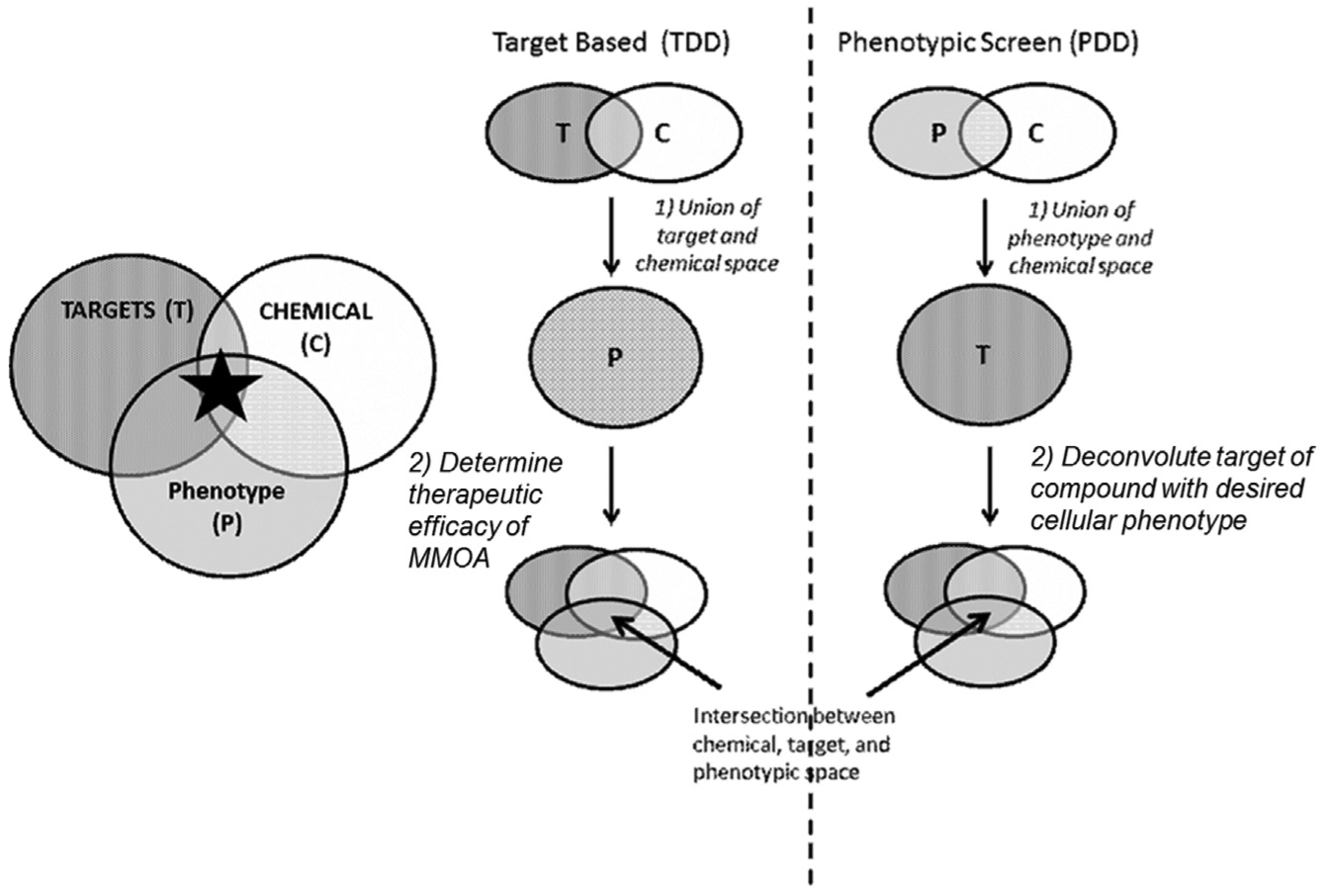
Schematic diagram comparing target-based and phenotypic-based approaches to drug discovery. Drug discovery strives to find new therapeutic entities by exploring the intersection between target space (T), chemical space (C), and phenotypic/cellular function space (P). MMOA = molecular mechanism of action.
By associating drug, target, and phenotypic space in two separate and distinct stages ( Fig. 1 ), both TDD and PDD require substantial chemistry resources prior to pharmacological association and validation of a target-driven phenotype. By combining the strengths of TDD and PDD in an integrated approach that surveys the druggable target space within a defined phenotype, a new viewpoint is provided on the most biologically and chemically tractable targets within a pathway. This approach offers a simple and efficient means to rapidly advance early drug discovery, especially if the method can be accomplished without the substantial upfront investment of medicinal chemistry resources. Leveraging the flexibility of affinity selection–mass spectrometry (AS-MS) screening, it is possible to analyze both target and chemical space to rapidly evaluate a clinical pathway of interest. AS-MS identifies small molecules binding to targets of interest10,11 in a high-throughput manner agnostic to binding site and/or function. It also requires limited method development and can be broadly applied to different classes of biomolecular targets. AS-MS has been reported recently to confirm the selective target interaction of well-characterized clinical compounds across a large panel of proteins (>100) and also identify potential off-target interactions. 12 The Automated Ligand Identification System (ALIS) is an AS-MS–based methodology built on two-dimensional chromatography coupled online to high-resolution mass spectrometric detection. ALIS is a fully automated process that relies on full-scan high-resolution–accurate mass mass spectrometry and has been used to address multiple challenges in target-based drug discovery.10,13–17 Because this technology relies on liquid chromatography–mass spectrometry (LC-MS), it allows for interrogation of any small molecule that can be assayed in this format. Given the high throughput and flexible method development of ALIS, the technology is uniquely positioned to rapidly screen large collections of compounds to identify ligands to multiple proteins in parallel, such as a pathway of clinical interest.
This study focuses on the NF-κB pathway, as it has been implicated in multiple disease indications, such as chronic inflammation and cancer, and the majority of proteins in this pathway and their functions are generally well characterized. 18 There are two primary NF-κB–mediated cellular pathways, the canonical and noncanonical pathways.18,19 The canonical pathway is activated via ligand interaction with cytokine receptors, such as tumor necrosis factor receptor 1 (TNFR1) and interleukin 1 receptor (IL1R). Signal transduction of the canonical pathway is dependent on IKKβ and NEMO and results in the nuclear translocation of RelA (p65) containing heterodimers. The noncanonical pathway uses a largely different set of proteins within its signaling cascade than those of the canonical pathway. A specific set of cytokines within the TNF superfamily, including CD40, LTβ, BAFF, and TWEAK, are responsible for inducing noncanonical signaling. 20 Transduction of this alternative pathway is dependent on IKKα and ultimately results in the processing of NF-κB2/p100 to NF-κB2/p52 and the nuclear translocation of NF-κB2/p52–RelB heterodimers.18,19,21
Despite extensive validation of these two branches of the NF-κB pathway and their associated targets, considerable efforts within the pharmaceutical industry and academia have failed to produce therapeutics with clinical efficacy.18,19,21,22 These efforts have primarily adopted target-driven approaches, focusing mainly on IKKβ and IKKα because of their central functions within the NF-κB pathway in the context of immunity. 23 Although a number of IKK inhibitors have been developed and shown to exert antitumor effects in experimental models, none so far have demonstrated therapeutic utility in the clinic. The failure to successfully develop an NF-κB modulator for the treatment of either inflammatory or oncology indications may be due to the complexity of the regulation of NF-κB signaling, failure to target the most sensitive regulatory proteins within the pathways, inability to identify appropriate patient populations, or a combination of these issues. 23
In this report, the ability of the ALIS technology to simultaneously screen a large collection of compounds against multiple targets with known function within the NF-κB pathways provides the ability to identify a focused small-molecule library. Hits with selective binding are then assessed in several cellular phenotypic assays that interrogate known biological functions of the potential NF-κB targets. By integrating ALIS with cellular functional assays, compounds observed to modulate the NF-κB pathway are associated with a target and phenotype prior to de novo chemical synthesis. The chemical space of cell-active hits can then be confirmed in a target-driven manner by evaluating structural analogs of the original hit to establish that target binding is required for the observation of the desired cellular phenotype. This approach to determining the intersection of chemical, target, and phenotypic space provides a unique perspective to identify the most tractable entry point(s) for medicinal chemistry based on pharmacological validation and preliminary SAR determination. This opportunistic triage strategy relies on data-driven attrition and, as such, is not intended to identify all potential targets within the pathway. Instead, it is to be considered an efficient methodology to survey new drug discovery opportunities that are both pharmacologically and biologically tractable within a pathway of interest.
Materials and Methods
Production of NF-κB Proteins
Cloning and Expression
All constructs were encoded full-length human versions of each target gene, codon optimized for insect expression, and cloned into pBAC1. All constructs contained either a human rhinovirus 3C (HRV3C) protease cleavable C-terminal His8 tag, N-terminal FLAG tag, HRV3C-cleavable N-terminal His8-FLAG tag, or HRV3C cleavable N-terminal His8-HALO tag. Gene accession numbers and respective purification conditions used are described in the supplemental material.
ALIS
ALIS has been well described previously. 10 Briefly, affinity selection in ALIS was accomplished via an online two-dimensional LC-MS system that incorporated a size exclusion chromatography (SEC) column (2.1 mm I.D. × 50 mm length, packed with proprietary gel filtration media; running buffer: 700 mM ammonium acetate, pH 8.0, flow rate [F] = 300 µL/min, 4 °C column temperature) and a reverse-phase chromatography (RPC) column (Higgins Analytical, Mountain View, CA; Targa C18, 0.5 mm I.D. × 50 mm, 5 µm packing material; mobile phases: water: acetonitrile with 0.2% formic acid, 0%–90% B gradient in 2.5 min, F = 20 µL/min, 40 °C column temperature). The eluent from the chromatography separation was analyzed directly by high-resolution mass spectrometry to identify small-molecule ligands. Each ALIS system is composed of a combination of isocratic and capillary binary chromatography pumps, two variable wavelength detectors, a chilled microplate autosampler (Agilent Technologies, Wilmington, DE), and a custom-built valve box incorporating four switching valves. This allowed for direct coupling of the SEC and RPC separations and column switching in the RPC chromatography step for each subsequent injection, which increased the overall throughput by 40% over previous versions of the ALIS system. Exactive Orbitrap mass spectrometers (Thermo Scientific, San Jose, CA) were used for detection, providing high-resolution (100,000 resolving power) and high-accuracy m/z detection (mass error <5 ppm without internal calibration). Each ALIS sample consisted of mass-encoded compound mixtures (~500 compounds; each present at approximately 0.5 µM) combined with the target of interest (5 µM) in physiological, aqueous buffer with 2.5% (v/v) residual DMSO. To be detectable under standard ALIS screening conditions, compounds need to be soluble in aqueous conditions up to 1 µM, retain on the reverse-phase LC column, and be detectable by mass spectrometry. Targets need to be soluble and capable of binding small-molecule ligands in the ALIS buffers. After incubation at room temperature for 30 min, the samples were cooled to 4 °C and loaded into the autosampler for subsequent injection and analysis. Screening was accomplished in an iterative manner, with two rounds of mixture-based screening followed by confirmation of the ligand of interest as a single, pure compound. Invertase was used to measure nonspecific binding. Compounds yielding reproducible MS signals in each subsequent experiment with the specific target and not producing signal in the invertase counterscreen were considered ALIS hits.
Beta-Lactamase Transcription Reporter Assays
TNFα NF-κB Beta-Lactamase THP1 Assays
Reporter gene assays were run according to the manufacturer’s instructions. Briefly, CellSensor NF-κB beta-lactamase (BLA) THP-1 cells (Life Technologies, Carlsbad, CA), containing a stably integrated BLA reporter gene under control of the NF-κB response element, were recovered from liquid nitrogen in reduced serum medium (RPMI 1640 with 0.5% [v/v] heat-inactivated fetal bovine serum [FBS]) overnight. The following day, compounds were preincubated with 32 µL of cells in serum-starved media (10,000 cells/well) in black-wall, clear-bottom, 384-well assay plates (CoStar, Corning, NY) for 1 h in 0.1% (v/v) final DMSO %. After this time, cells were incubated for an additional 3 h at 37 °C/5% CO2 with 1 ng/mL of TNFα (R&D Systems, Minneapolis, MN) to induce BLA expression.
TET CHO Assay
A tetracycline-inducible BLA reporter assay in CHO cells was generated using the Jump-In T-REx System (Life Technologies, Carlsbad, CA), a mammalian expression system that uses regulatory elements from the Escherichia coli Tn10-encoded tetracycline (TET) resistance operon. 24 TET BLA CHO cells were maintained in DMEM with GlutaMAX-I (high glucose) with 10% dialyzed FBS (Life Technologies). Assays with TET BLA CHO cells were run in black-wall, clear-bottom, 384-well assay plates (CoStar) with 25,000 cells per well in 32 µL of reduced serum medium (RPMI 1640 with 0.5 % [v/v] heat-inactivated FBS).
BLA Detection
Detection of BLA expression in both the TNFα NF-κB BLA assay and the TET CHO BLA assay was performed using the ToxBLAzer-FRET B/G substrate (Life Technologies). Prior to substrate loading, assay plates were equilibrated to room temperature. The ToxBLAzer-FRET B/G substrate was added to each well at a final concentration of 200 nM, and assay plates were incubated for an additional 2 h at room temperature in the dark. The signal was read on a Flex Station III (Molecular Devices, Sunnyvale, CA), with an excitation wavelength at 409 nm. The ratio of fluorescence emission values collected at 460 nm and 530 nm was determined. All assays were quantified using the same protocol.
Nuclear Translocation Assays
Translocation of RelA (p65) or NF-κB2/p52 was quantified in A549 cells (ATCC, Manassas, VA). A549 cells were maintained in F12K (Kaighn’s) Medium (Life Technologies) with 10% (v/v) FBS. Assays were run in 384-well collagen-coated, black imaging plates (BD Biosciences, Franklin Lakes, NJ) with 8000 cells per well in low serum (0.5% [v/v] FBS) F12K (Kaighn’s) Medium. Compounds were preincubated with cells for 1 h before addition of cytokine. The final DMSO concentration was 0.5% (v/v). Nuclear translocation of RelA (p65) in A549 cells was induced with 1 ng/mL of either ΤΝFα or IL1β (R&D Systems) and incubated at 37 °C, 5% CO2 for 1 h. Nuclear translocation of NF-κB2/p52 in A549 cells was induced with 100 ng/mL of TWEAK/TNFSF12 (R&D Systems) and incubated at 37 °C, 5% CO2 for 5 h. After induction of RelA (p65) or NF-κB2/p52 nuclear translocation, cells were fixed with 3.7% paraformaldehyde in Hank’s Balanced Salt Solution (HBSS, GE Healthcare, Boston, MA) including magnesium and calcium for 30 min at room temperature. Cells were subsequently permeabilized with 0.5% Triton X-100 in HBSS including magnesium and calcium for 30 min. Once cells were fixed and permeabilized, assay plates underwent immunostaining protocol described in the supplemental materials.
Image Acquisition
Images were captured at 10× magnification in two fluorescent channels, DAPI/DAPI (350/390) and FITC/FITC (475/511), on an IN Cell Analyzer 2200 (GE Healthcare) at two frames per well. The laser autofocus adjusted Z-height at every frame.
Image Analysis
Images acquired from IN Cell 2200 were analyzed by CellProfiler (Broad Institute, Cambridge, MA). In brief, a nuclear region was defined by DAPI signal. The cytoplasmic region was defined by subtracting the area of the nuclear region from the area of the whole cell body. The intensity of the FITC channel in both the nuclear and cytoplasmic regions was determined, and the ratio of nuclear intensity to cytoplasmic intensity was calculated as a measure of translocation of either RelA (p65) or NF-κB2/p52 from the cytoplasm to the nucleus (see
Results and Discussion
Identification of Target-Specific Ligands by ALIS
In this study, ALIS was first used to identify compounds in the screening collection that bind to NF-κB pathway proteins (
Figs. 2
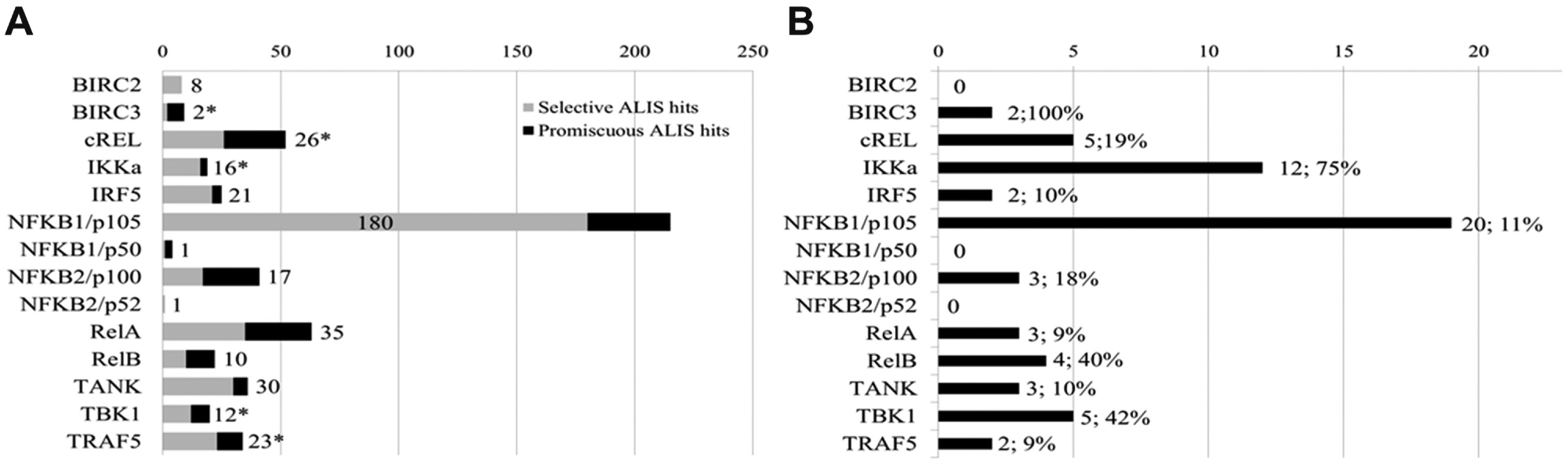
Target-selective compounds identified by the Automated Ligand Identification System (ALIS). (
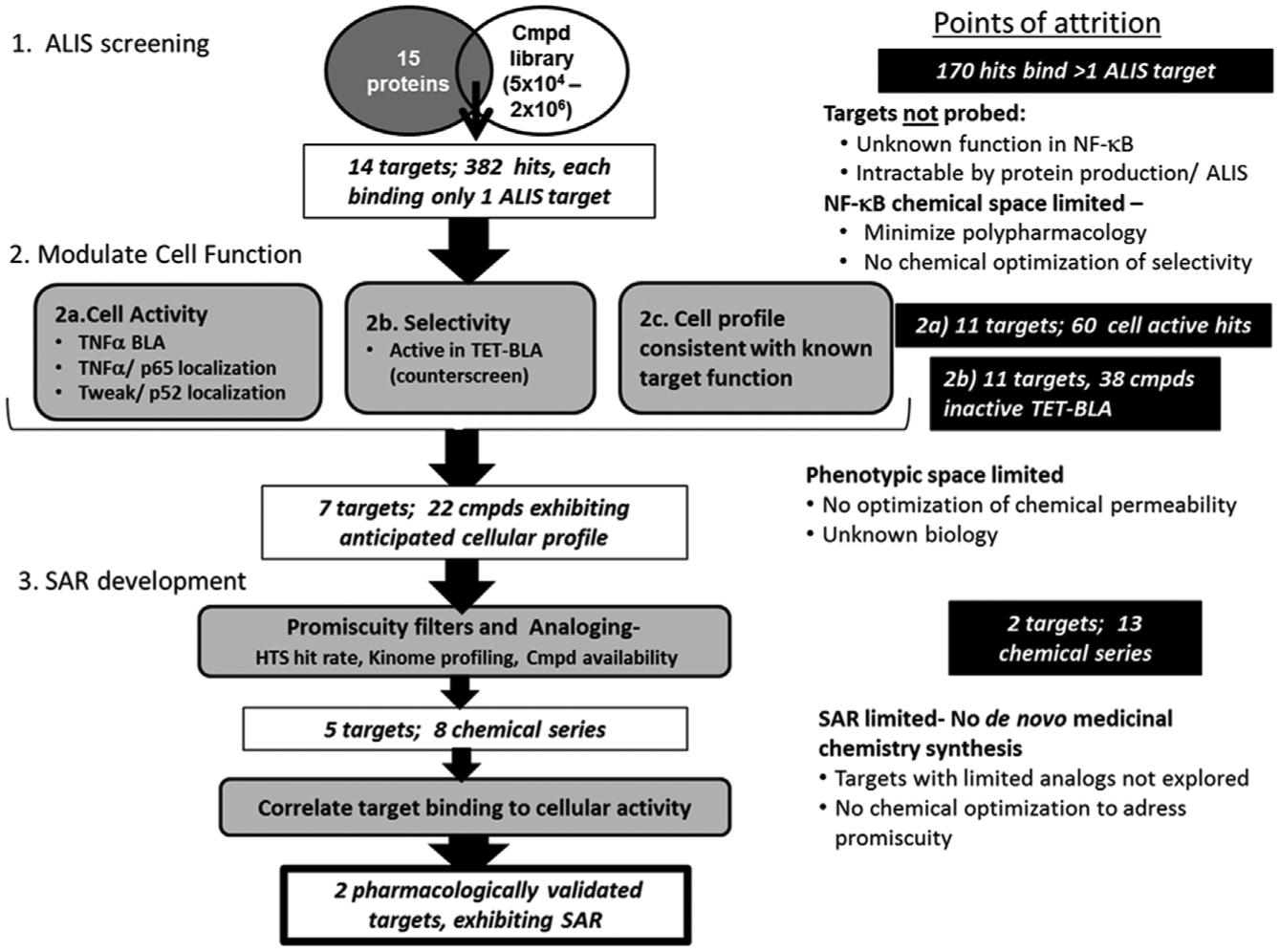
Flow chart detailing the hit reduction process and selection criteria. Points of attrition identify each point at which potential targets and mechanisms are deprioritized from further follow-up in this triage strategy.
Screening the 15 proteins resulted in total of 552 hits, with all but one target (IKKβ) yielding ligands as detected in ALIS. These small-molecule–protein binding events were either termed selective (binding to only 1 of the 15 targets) or reoccurring/promiscuous (binding to 2 or more of the 15 targets tested), as illustrated in Figure 2A . When screened individually in ALIS, each protein exhibited a unique hit rate. No efforts were undertaken to minimize the list or to normalize the library of compounds based on an overall hit rate. The only requirement for compound inclusion in follow-up triage was that the binding was selective for only 1 of the 15 targets. This resulted in a library of 382 selective ALIS binding compounds, encompassing 14 of the 15 targets screened ( Fig. 3 ). Using a target-binding method such as ALIS upfront to define potentially relevant chemical space eliminated the ability to identify novel biology or new targets within the pathway. The requirement for advancement that a compound bind selectively to a single target in ALIS causes one to overlook compounds with polypharmacology as well as those compounds whose selectivity could be further optimized by de novo medicinal chemistry efforts. This triage strategy instead focuses on the most tractable opportunities to associate compound binding to a singular target driving a defined cellular phenotype.
Triaging of ALIS Hits in the NF-κB Cellular Assays
Cell-based assays that monitor NF-κB pathway signaling were used to mechanistically study the 382 target-selective ALIS hits prior to any further optimization of the chemical matter. Four cellular assays, composed of transcriptional reporter and high-content imaging readouts, were designed to interrogate both the canonical and noncanonical pathways. TNFα mediated induction of BLA expression with the CellSensor NF-κB BLA reporter assay in the THP1 monocytic cell line had an average signal-to-background (S/B) of 5.3 (range, 3.9–10.7) and an average Z′ of 0.7 (range, 0.6–0.8, n = 15). In addition, a TET-inducible BLA reporter assay in CHO cells was incorporated to identify compounds that nonspecifically affected transcription and/or translation events or the BLA function itself. This counterscreen assay had an average S/B of 11.2 (range, 8.1–15.0) and an average Z′ value of 0.8 (range, 0.7–0.9, n = 14). Screening in an antagonist format, compounds were considered cell active in these BLA transcriptional reporter assays if they exhibited >50% inhibition at a screening concentration of 10 µM or lower.
A readout of the noncanonical pathway was required to profile targets within this pathway as well as a counterscreen for the canonical NF-κB pathway. Receptor ligands of the noncanonical pathway (CD40, LTβ, or TWEAK) were not observed to activate the THP1 NF-κB BLA reporter cell line, leaving a gap in profiling this NF-κB phenotype. To address this discontinuity, nuclear translocation assays were developed as orthogonal readouts of both canonical and noncanonical NF-κB pathway activation. Image-based analysis of the immunostaining signal associated with each transcription factor directly quantified the activated transcription factor within the nucleus and the inactive transcription factor within the cytoplasm in response to ligand stimulation in wild-type cell lines. Nuclear translocation was calculated as the ratio of mean fluorescence intensity within the nucleus over the mean fluorescence intensity within the cytoplasm. The A549 lung adenocarcinoma cell line exhibited optimal assay windows for translocation of both the canonical (RelA/p65) and noncanonical (NF-κB2/p52) transcription factors in response to TNFα and TWEAK ligand stimulation, respectively. The TWEAK (Fn14 receptor ligand) nuclear translocation assay had an average S/B ratio of 2.1 (range, 1.7–2.5) and an average Z′ value of 0.6 (0.4–0.8, n = 15). The TNFα nuclear translocation assay had an average S/B ratio of 3.3 (range, 2.5–5.7) and an average Z′ value of 0.6 (range, 0.5–0.8, n = 15). Compounds were initially screened in both the canonical and noncanonical translocation assay at either 50 µM or 25 µM, respectively, and were considered to be cell active if they exhibited >50% inhibition in either assay (
In total, 60 (16%) of the 382 ALIS hits tested exhibited cellular activity in at least one of the cell-based assays (
Fig. 2B
). The small percentage of cell-active compounds identified in ALIS could be due to multiple factors. Because all available binding sites on a protein are interrogated in ALIS, compound binding may not affect protein function. Alter-natively, the compound may have limited cell permeability. Directly screening ALIS hits in cell-based assays efficiently narrowed down the original 15 NF-κB targets to 11 targets with selective, cell-active, chemical matter that could be further progressed into additional pharmacological validation studies prior to any medicinal chemistry optimization (
Figs. 2B
Triaging of Cell-Active ALIS Hits Exhibiting Phenotype Consistent with Target Function
Stringent criteria to evaluate the relationship of a compound’s cellular profile to the expected function were used to triage targets for additional pharmacological validation studies. Twenty-three compounds that inhibited the TET-BLA assay (>50% I) were not considered for additional follow-up studies because of their activity in this counterscreen. These activities are independent of the TNFα induction of BLA expression via the NF-κB reporter element and may include mechanisms such as DNA binding, chelating metal ions, and so forth that prohibit defining a direct link between target interaction and cellular phenotype (
Fig. 3
;
Requiring the observed cellular profile to be consistent with apparent target function, 7 of the 14 potential targets were deprioritized, although they had selective ligands identified in ALIS. This rapid triage and mechanistic deprioritization allowed for further focus to be placed on the remaining seven targets of interest. The ligands associated with three of the deprioritized targets (BIRC2, NFKB1/p50, NFKB2/p52) exhibited no cellular activity in any cellular readout. In addition, all of the ligands to IKKα, BIRC3, NF-κB2/p100, and RelB exhibited a phenotype inconsistent with known target function and were not considered for further follow-up (
Twenty-two hits, selectively binding to seven unique targets, exhibited the anticipated cellular phenotype ( Table 1 ; Fig. 3 ) in the NF-κB screening platform. Four of these seven targets, NF-κB1/p105, cRel, RelA, and IRF5, are transcription factors of the canonical NF-κB pathway. Based on the primary role of these four targets to regulate gene transcription, only compounds that exhibited selective inhibition of the TNFα transcriptional reporter assay were considered to have the appropriate expected functionality.18,19,21,25 Impeding nuclear translocation was not considered the only mechanism to disrupt transcription; therefore, inhibition in the TNFα nuclear translocation assay was not considered an essential confirmatory phenotype of NF-κB1/p105, cRel, or RelA. Cellular activity was observed only in the TNFα transcriptional reporter assay for ALIS hits that selectively bound cRel, RelA, and IRF5 ( Table 1 ). Twelve of the selective NF-κB1/p105 ALIS hits inhibited the TNFα BLA reporter assay consistent with the expected phenotype. A subset (5/12) of these hits also inhibited TNFα–mediated nuclear translocation while having no effect on the TWEAK-mediated nuclear translocation of NF-κB2/p52, providing additional insight into the compound’s potential mechanism of action. A single ALIS hit to NF-κB/p105 that selectively inhibited the TNFα transcriptional reporter assay but exhibited nonselective inhibition across the canonical and noncanonical nuclear translocation assays was not considered for follow-up because of this inconsistent cellular phenotype. An additional four NF-κB1/p105 compounds displayed cellular activity only in the TNFα nuclear translocation assay but not the reporter assay and as such did not possess the cellular profile expected of an inhibitor of this transcription factor.
Postulated cellular profile of inhibitors to each target in the four NF-κB screening assays and number of compounds that exhibited cellular profile. a
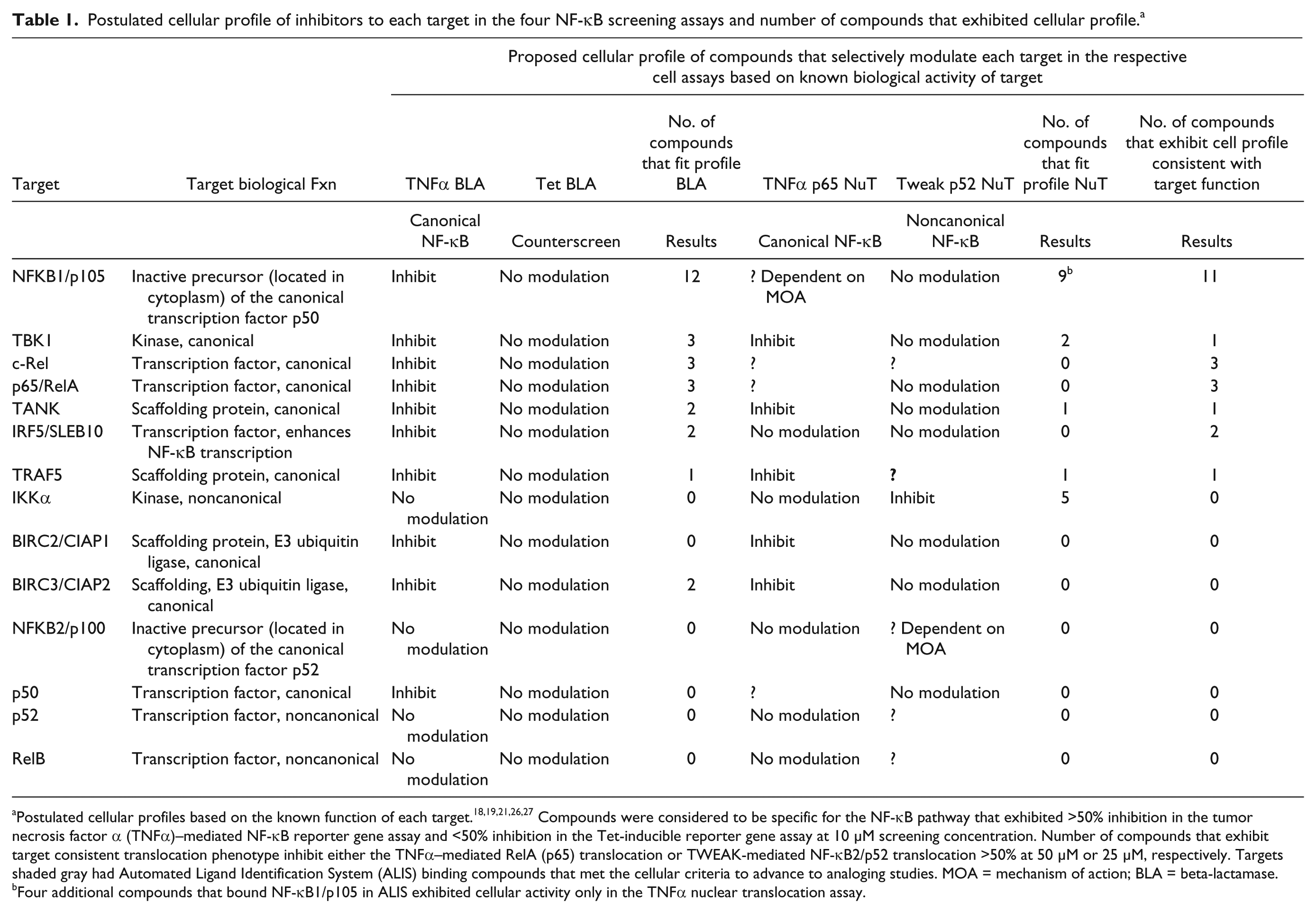
Postulated cellular profiles based on the known function of each target.18,19,21,26,27 Compounds were considered to be specific for the NF-κB pathway that exhibited >50% inhibition in the tumor necrosis factor α (TNFα)–mediated NF-κB reporter gene assay and <50% inhibition in the Tet-inducible reporter gene assay at 10 µM screening concentration. Number of compounds that exhibit target consistent translocation phenotype inhibit either the TNFα–mediated RelA (p65) translocation or TWEAK-mediated NF-κB2/p52 translocation >50% at 50 µM or 25 µM, respectively. Targets shaded gray had Automated Ligand Identification System (ALIS) binding compounds that met the cellular criteria to advance to analoging studies. MOA = mechanism of action; BLA = beta-lactamase.
Four additional compounds that bound NF-κB1/p105 in ALIS exhibited cellular activity only in the TNFα nuclear translocation assay.
Compounds that bound in ALIS to one of three nontranscription factor targets, TANK, TRAF5, and TBK1, selectively inhibited both the TNFα transcriptional reporter and nuclear translocation assays as expected for targets upstream of transcription (see
Table 1
). TANK, TRAF5, and TBK1 are two scaffolding proteins and a kinase, respectively, which make up a functional complex to transduce TNFR1 ligand-mediated activation, upstream of transcription and the IKK signaling complex.
26
In total, six compounds against these three targets exhibited cellular activity in either the TNFα transcriptional reporter assay and/or nuclear translocation assay. However, only three of these seven compounds exhibited a consistent cellular phenotype in both assay formats and were considered for subsequent validation studies (
Binding–Cell Activity Correlation Demonstrates That Specific Target Binding Induces Cellular Phenotype
The goal of this triage strategy was to prioritize targets to be pursued as drug discovery programs based on chemical tractability and pharmacological validation of the targets discovered prior to committing any medicinal chemistry resources. Whereas identification of the seven targets within the NF-κB pathway with chemical matter (22 compounds in total) that displayed a cellular activity consistent with the known function of the respective target took a phenotypic-based approach, pharmacological validation of these targets was conducted in a target-based manner. Structurally similar analogs to the original ALIS hits profiled in both target-binding studies and the NF-κB cell functional assays enabled a causal link between target binding and cellular NF-κB activity to be determined. The data from these compounds, both those that bind and those that do not, provided a means to confirm that target-compound binding was essential for cellular activity. In addition, preliminary SAR could be elucidated from the overall behavior of a chemically related series of compounds. Targets and their respective chemical series were prioritized based on the degree of pharmacological validation that could be ascertained as well as observed structural components contributing to cellular activity as these would be optimal starting points for medicinal chemistry optimization.
Analogs to the 22 original screening hits that selectively bound to seven protein targets in ALIS (representing 21 unique chemical series) were selected as described in the supplemental material. NF-κB1/p105 had two cell-active compounds that were identified as analogs to each other based on the structural similarity parameters described. Chemical series were evaluated for their ability to derive a correlation between observed cellular activities in the NF-κΒ screening platform to the direct binding of the compound to its respective ALIS target. Eight series with poor kinome selectivity and/or high HTS hit rate were considered promiscuous, which would prohibit assessing an on-target mechanism to establish direct causality of binding to the observed cellular phenotype that would prevent the correlation of the observed cellular activity An additional five chemical series were categorized as low priority for chemistry follow-up because of either an insufficient number of analogs (<10) available for analog studies or because analog studies determined that target binding was not essential for cellular activity (
Eight series (associated with one of five targets) were considered as potential starting points for medicinal chemistry optimization. Rank ordering these series by observed SAR and pharmacological validation, two series found the union between functional causality and defined initial SAR. The remaining six series had insufficient evidence to either establish causality or define preliminary SAR and are described within the supplemental section. One of the two pharmacologically validated chemical series selectively bound to the transcription factor NF-κB1/p105 in ALIS. This series had few analogs (34) to profile in ALIS binding and NF-κB screening assays (
Pharmacological Validation of TRAF5 and Its Chemical Matter
The TRAF5 chemical series had a large number of analogs available (123) to be screened in the gene reporter assay and nuclear translocation assays. The data obtained on these compounds established a consistent mechanism across the entire chemical series. Gene deletion studies of TRAF5 reported a moderate effect on TNF-induced NF-κB activation.19,26 Consistent with the known function of TRAF5 to interact with TNFR and mediate signal transduction, these compounds selectively inhibited both the TNFα BLA reporter assay and the TNFα/RelA (p65) nuclear translocation assay ( Fig. 4A , B ). A consistent shift in potency was observed between the TNFα BLA reporter assay and the TNFα/RelA (p65) nuclear translocation assay with IC50 values in the BLA reporter assay typically being five- to seven-fold more potent than those determined in the nuclear translocation assay (data not shown). Even with this shift noted, compounds rank ordered consistently in the two assays, thus supporting the hypothesis that the compounds have the same mechanism of inhibition in the two assay formats. Twenty-seven of 31 analogs were confirmed to interact with TRAF5 in ALIS binding studies. All compounds with TNFα BLA IC50 values <10 µM and TNFα RelA (p65) nuclear translocation IC50 <50 µM exhibited binding to TRAF5. Four analogs that did not exhibit binding to TRAF5 were inactive in both the TNFα reporter gene assay and RelA (p65) nuclear translocation assay, further confirming a positive correlation between required TRAF5 binding and the inhibition of the TNFα canonical NF-κB pathway ( Fig. 4B , C ). Images from the TNFα nuclear translocation assay provided further evidence that compounds not binding to TRAF5 did not inhibit RelA (p65) nuclear translocation ( Fig. 5D ), whereas RelA (p65) was entirely localized within the cytoplasm when cells were treated with compounds that bound TRAF5 ( Fig. 5C ).
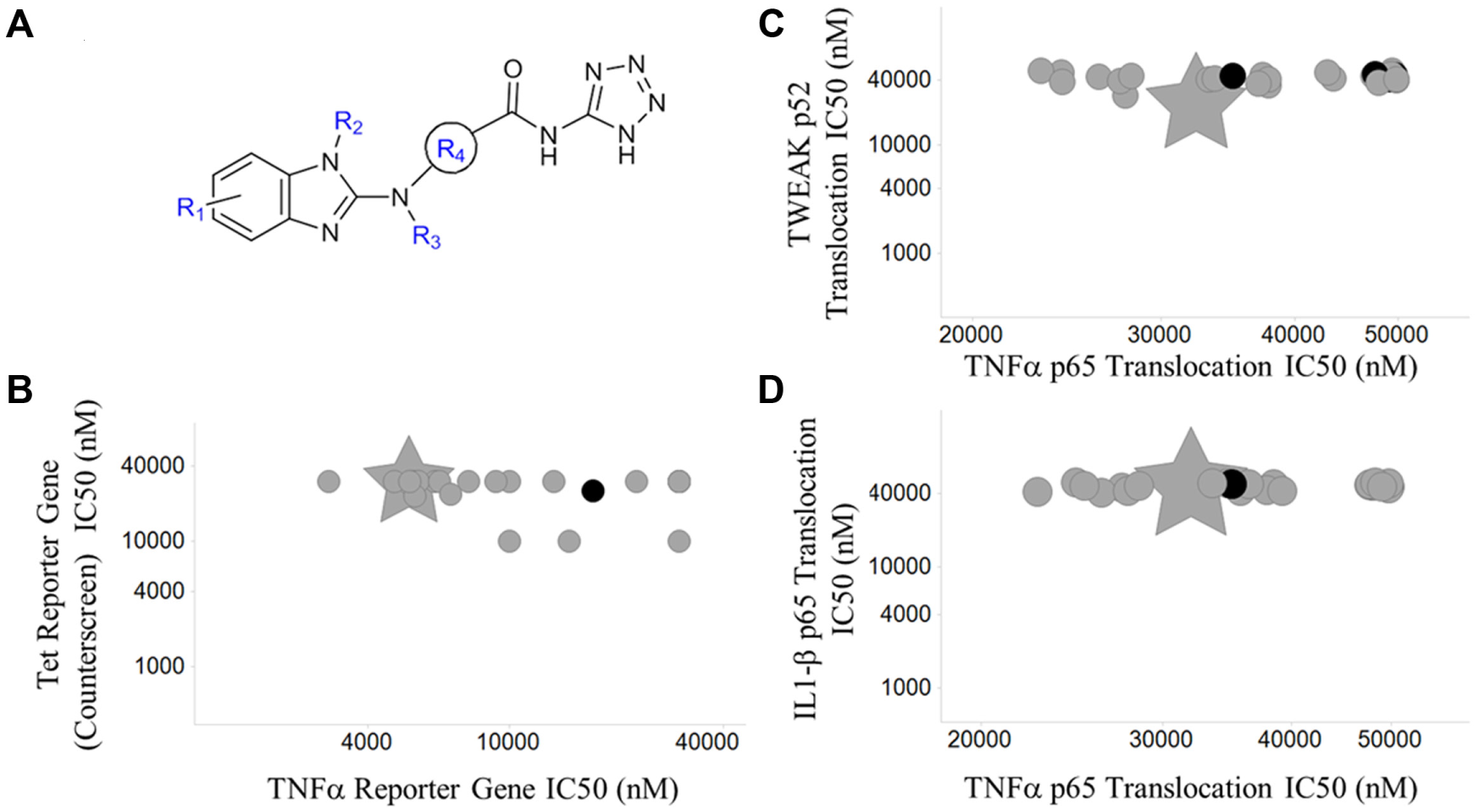
Pharmacological validation of the TRAF5 series. (
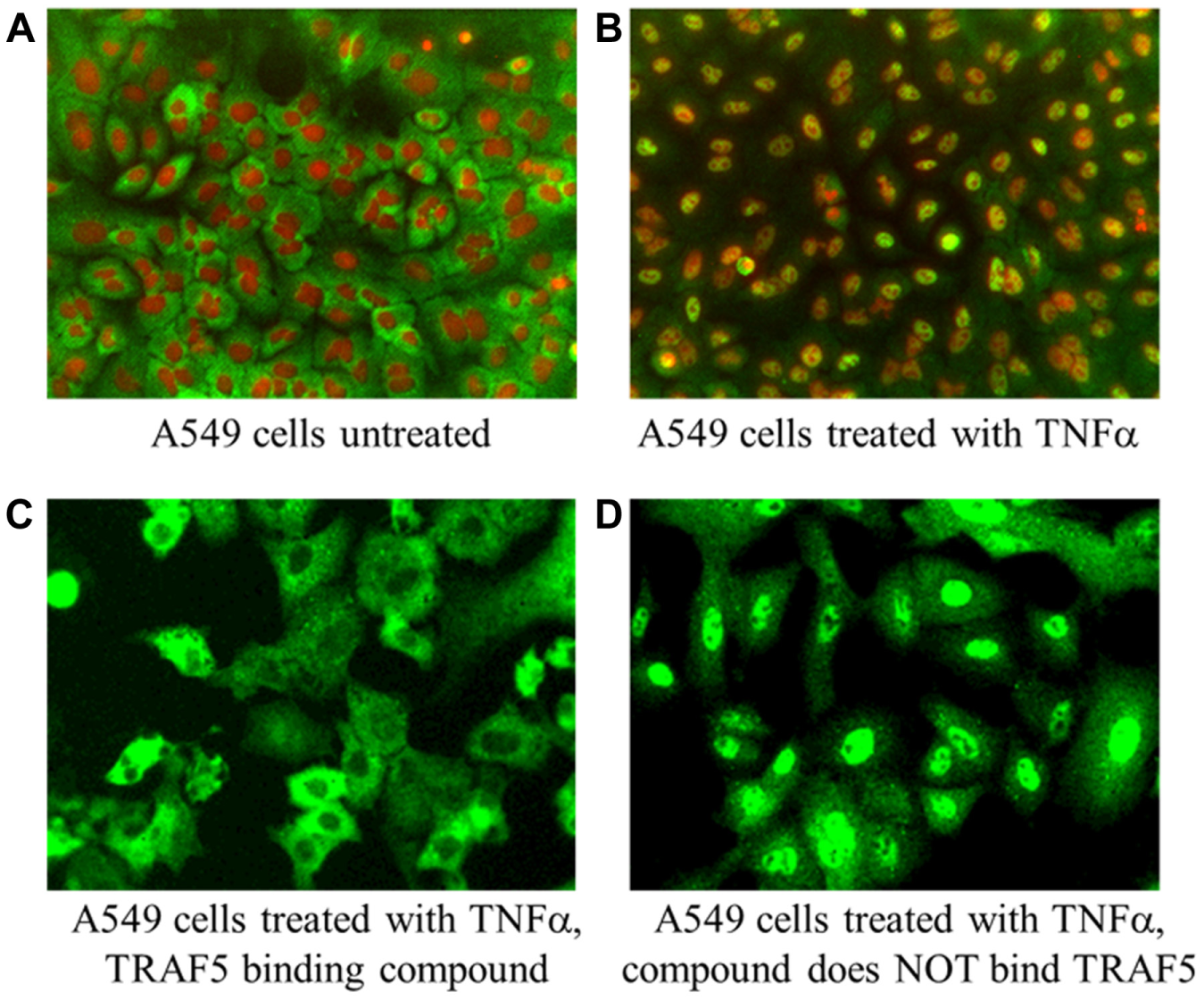
Visual confirmation of TRAF5 causality in the tumor necrosis factor α (TNFα)–mediated RelA (p65) translocation assay. Red masks define nuclei, and green masks define RelA (p65). (
TRAF5 is one of seven isoforms within the TRAF (TNF receptor associated factor) protein family and mediates the signal transduction of multiple receptors within the TNFR superfamily via protein-protein interactions directly with receptors, adaptor proteins, and ubiquitinases (i.e., cIAP1/2).21,26,28 TRAF isoform selectivity using ALIS was not directly assessed in this study because of difficulties expressing TRAF isoforms. Therefore, cellular profiling of the TRAF5 chemical series in NF-κB signal transduction pathways activated by factors other than TNFα, such as TWEAK and IL1β, provided additional evidence that this chemical series modulated TRAF5 in the NF-κB pathway with some selectivity over other TRAF isoforms. In the noncanonical TWEAK/NF-κB2/p52 nuclear translocation assay, in which TRAF5 and other TRAF family members do not function by positive transduction of pathway signaling, the majority of the compounds that inhibited TNFα signaling had no effect on TWEAK-mediated signal transduction ( Fig. 4C ). More importantly, in an IL1β/RelA (p65) nuclear translocation assay in which TRAF6 is essential and the only TRAF isoform involved in IL1R-mediated signal transduction, all TRAF5 compounds were inactive ( Fig. 4D ). Cellular profiling of these structurally similar analogs in TNFα, IL1β, and TWEAK nuclear translocation assays and transcriptional reporter assays provided strong evidence, along with TRAF5 binding studies, that these compounds modulate the NF-κB pathway through inhibition of TRAF5.
These data clearly demonstrated a positive correlation between direct binding to TRAF5 and appropriate modulation of the cellular phenotype. Based on the function and structure of this protein, TRAF5 had previously been considered a nondruggable target by small-molecule modulation.26,28 Identification of TRAF5 as a druggable target validates the power of this integrated approach with AS-MS and cellular functional assays to rapidly triage druggable target space by phenotype and identify the most tractable starting points for medicinal chemistry optimization.
The primary objective of early drug discovery is to associate druggable target space with a desired phenotype. Numerous approaches to improve the efficiency by which new drugs are discovered have been dependent on medicinal chemistry to determine the union of three distinct but equally important parameters, chemical, target, and phenotypic space. This study clearly demonstrates the validity of integrating affinity selection with functional cellular assays to identify and triage the most pharmacologically tractable targets in the NF-κB pathway without the need for de novo chemical synthesis.
This high-attrition triage strategy was intended to determine the most readily tractable starting points for initiating a small-molecule drug discovery program. As discussed earlier, small-molecule inhibitors of the NF-κB pathway are limited to the kinases IKKα and IKKβ, targets known to play a central role within the pathway and considered to have good druggability. This study took an empirical approach to determine which targets with known function in the pathway are the most chemically tractable. Leveraging the high-throughput screening capacity and universal screening capabilities of ALIS, the druggability was assessed of 15 targets with known function in the NF-κB pathway, spanning a range of target classes, irrespective of whether the target was previously considered to have druggable features. Of the 15 targets screened in ALIS, 382 target selective compounds that specifically recognized 1 of 14 targets were identified. Directly integrating the ALIS target binding results into functional readouts of the NF-κB pathway refined the target and chemical space to seven targets with chemical matter (22 compounds) that exhibited a desired phenotype. Empirical evaluation of these seven druggable targets to establish a direct correlation between target binding and cellular phenotype pharmacologically validated two targets, NF-kB1/p105 and TRAF5. Furthermore, this combined data set distinguished TRAF5 as the best drug discovery opportunity that was readily tractable with a biological hypothesis, pharmacological validation, and preliminary SAR. Despite this triage strategy having multiple points of attrition, the identification of chemical series to both NF-κB1 and TRAF5 illustrates the utility of the empirically assessing the druggability of known biological targets. The discovery of a chemical series that modulated TRAF5, a target that has generally been considered to have poor druggability by small molecules, 29 validates the strength of this approach to identify new drug discovery opportunities.
Footnotes
Acknowledgements
We would like to thank Dr. Jeremy Presland and Dr. Xianshu Yang from Department of Pharmacology, Screening and Protein Sciences, Merck & Co., for their support in this work. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.