
Abstract
Drug candidates exhibiting well-defined pharmacokinetic and pharmacodynamic profiles that are otherwise safe often fail to demonstrate proof-of-concept in phase II and III trials. Innovation in drug discovery and development has been identified as a critical need for improving the efficiency of drug discovery, especially through collaborations between academia, government agencies, and industry. To address the innovation challenge, we describe a comprehensive, unbiased, integrated, and iterative quantitative systems pharmacology (QSP)–driven drug discovery and development strategy and platform that we have implemented at the University of Pittsburgh Drug Discovery Institute. Intrinsic to QSP is its integrated use of multiscale experimental and computational methods to identify mechanisms of disease progression and to test predicted therapeutic strategies likely to achieve clinical validation for appropriate subpopulations of patients. The QSP platform can address biological heterogeneity and anticipate the evolution of resistance mechanisms, which are major challenges for drug development. The implementation of this platform is dedicated to gaining an understanding of mechanism(s) of disease progression to enable the identification of novel therapeutic strategies as well as repurposing drugs. The QSP platform will help promote the paradigm shift from reactive population-based medicine to proactive personalized medicine by focusing on the patient as the starting and the end point.
Present Focus on Target-Centric and Phenotypic Discovery: A Need for Innovation
Over the past decade, the standard approaches to drug discovery and development have been the application of target-centric and phenotypic methods.1,2 The basic steps involved in these approaches are outlined in
Figures 1
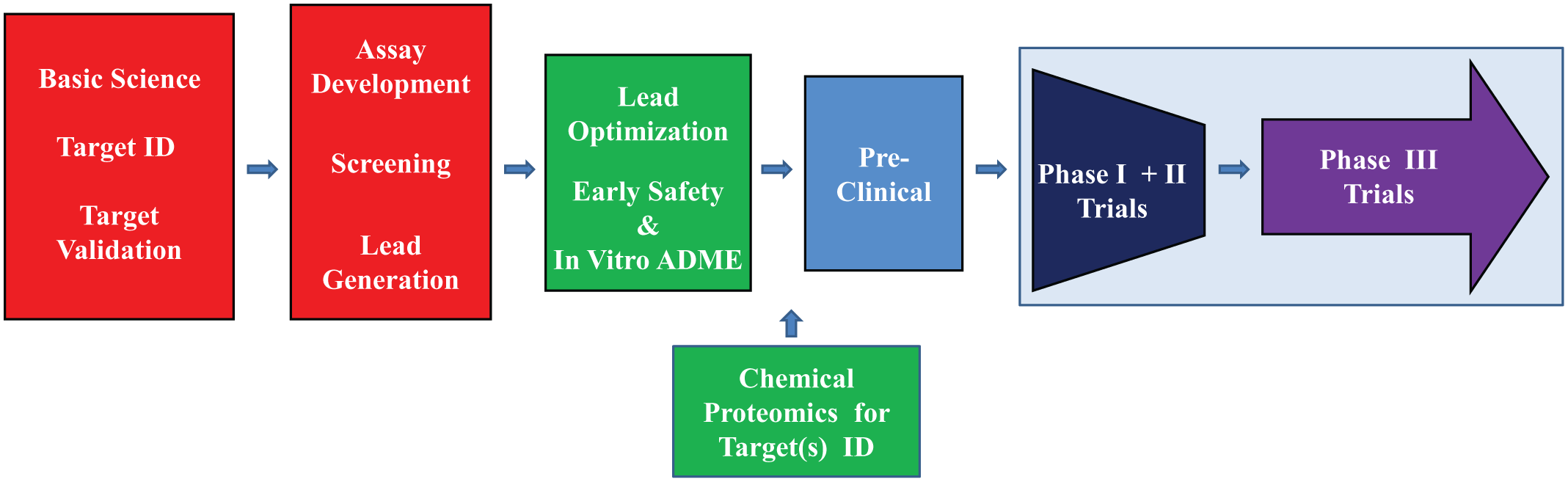
Target-centric drug discovery and development. The basic target-centric approach involves steps that begin with the investigation of the basic science. Although a holistic approach to the disease has not usually been applied, over the past few years additional information has been gleaned from patient data and network analyses as part of basic scientific information. The next step includes the identification (ID) of a druggable molecular target (i.e., a protein or gene associated with the disease), followed by the validation of this target by showing that modulation of the target has the ability to regulate disease-specific biological processes in in vitro and in vivo disease models. The next step involves the development of an assay measuring target activity, screening of selected libraries, and then the generation of leads from the validated hits. The optimization of leads to increase potency and to build in target specificity follows, and increasingly, the application of early safety and absorption, distribution, metabolism, and excretion testing in vitro. The application of target ID techniques such as chemical proteomics is increasingly being employed to identify additional on-target and off-target interactions. The next step involves preclinical testing with animal models for efficacy, toxicity, and pharmacokinetics. The next step involves phase I to III clinical trials.
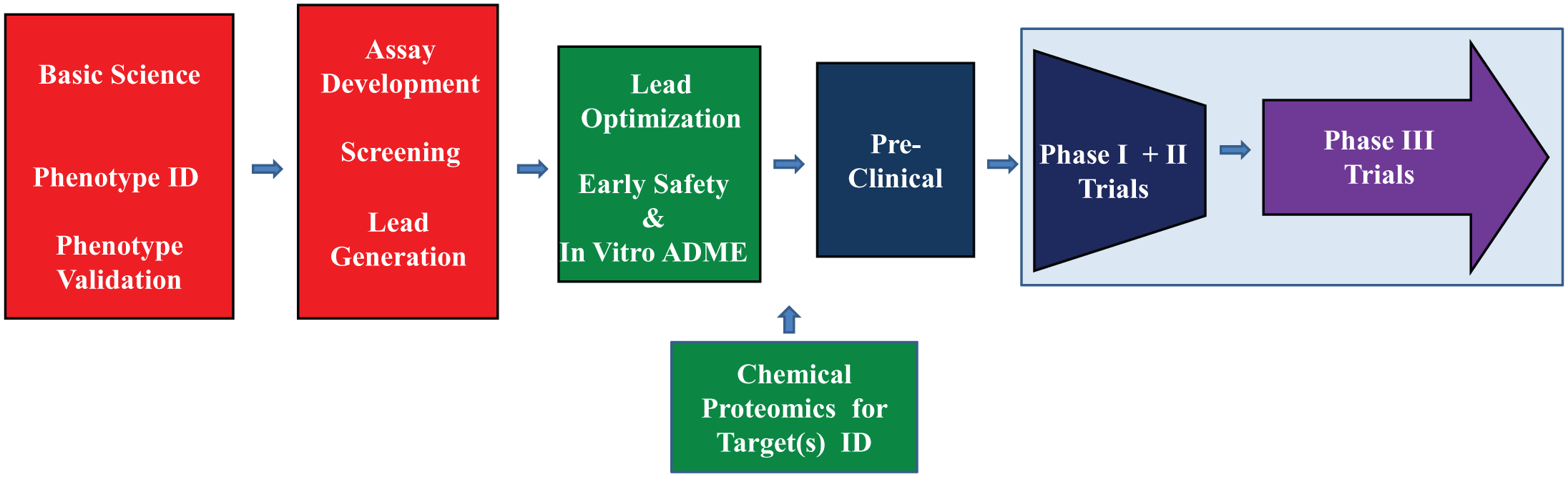
Basic steps in phenotypic drug discovery and development. The basic phenotypic approach involves steps that begin with the biology of the disease. Although a holistic approach to the disease has not usually been applied at this point, additional information has been gleaned from patient data and network analyses as part of basic scientific information. The next steps include the identification (ID), recapitulation, and validation of a disease-relevant phenotype in cell-based and/or organism-based models. The next step involves the development of a phenotypic assay that can be one of two major types: (1) unbiased modulation of a specific pathway identified in the validation step 3 or (2) an unbiased modulation of a disease phenotype that may involve any or multiple pathways in which the goal is to modulate the disease phenotype to a more normal phenotype 4 followed by screening of selected libraries and then the generation of leads from the validated hits.5,6 The next step involves the optimization of leads and, increasingly, the application of early safety and absorption, distribution, metabolism, and excretion testing in vitro followed by the use of methods such as chemical proteomics and RNAi knockdowns to identify on-target and off-target interactions of the leads. If molecular targets are identified during target ID studies, it is possible to shift to the target-centric approach for structure-activity relationship. The next step involves preclinical testing with animal models followed by phase I to III clinical trials.
A key feature of systems biology is the integration of computational and quantitative experimental methods that lead to the creation of formal mathematical models of biological processes and the discovery of emergent properties not identified in the investigation of individual biological components. 13 A major goal of QSP is to reinvent traditional pharmacology and replace the “one-gene, one-target, one-mechanism” hypothesis 14 with an in-depth understanding of the complex networks that are responsible for normal human and disease physiology using iterative quantitative experimental and computational methods.
Biological pathways have been studied for many years in biology and can be described as a series of reactions among molecules that result in changes in a cell such as the events in signaling, metabolism, cell cycle, and other biological processes. Pathways have been hand curated from the published literature in searchable databases, such as the KEGG PATHWAY Database, 15 that have been used to map molecular data sets (e.g., genomics and transcriptomics) to assist in understanding higher-level systems functions. In addition, pathways can be inferred from “omics” analyses of biological samples.16,17 In particular, samples that are blood based (i.e., liquid biopsies) can easily be collected longitudinally and correlated with disease progression. 18
Biological networks involve the interaction(s) among multiple pathways. 19 In systems biology and QSP, biological networks are mathematically modeled for the quantitative analysis of the interactions (i.e., edges) among the components (i.e., nodes) of the network. In network analysis, it is possible to identify the upstream and downstream interactions for a specific node that can lead to an understanding of normal biological regulation and pathophysiology. The creation of formal mathematical models at multiple temporal and spatial scales (molecules, cells, tissues, organs, organisms, and patients) of the networks involved in disease progression (pathology of the disease as it progresses) will be used to explain and to ultimately predict the effects of drug action on network functions and disease physiology. 17
A Definition of Quantitative Systems Pharmacology
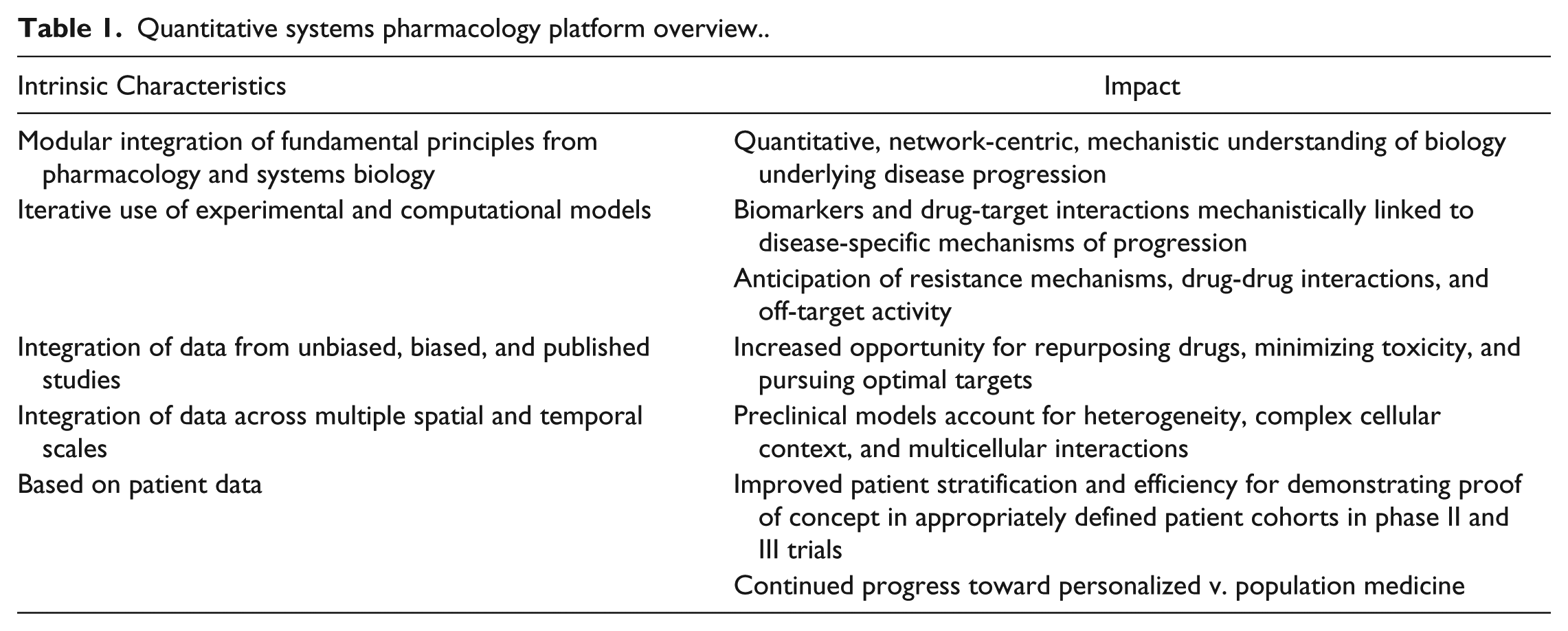
QSP is still a relatively new field, and its definition continues to evolve based on the emphasis of the applications within academia and industry (e.g., network analyses v. pharmacokinetics).20,21 An extensive definition is presented in the white paper that was prepared after the second NIH workshop. 12 We have based our initial efforts in building a complete platform to practice QSP on a more focused definition: “Determining the mechanism(s) of disease progression and mechanism(s) of action of drugs on multi-scale systems through iterative and integrated computational and experimental methods to optimize the development of therapeutic strategies.” Our goal in applying QSP is to improve the efficiency in the development of therapeutics through innovation and collaboration that will support the paradigm shift from reactive population-based medicine to prospective personalized medicine (Table 1).22,23
Quantitative systems pharmacology platform overview.
The Rationale for Implementing a QSP Platform
Multidisciplinary technological advancements in the postgenomic era have fueled the generation of significant data. Integration and analyses of these data are essential to gain new knowledge, allowing us to associate readily observable human traits and disease phenotypes with variations at the molecular level. Our understanding of disease pathophysiology is now increasing as we discover the underlying pathways and their coupling into cellular networks.24–33 Understanding the molecular basis of disease progression will enable the development of therapies optimally designed for genetically defined cohorts of patients, a key driver for the present practice of personalized medicine. Success has been demonstrated by a number of drugs (e.g., Kalydeco, crizotinib, vemurafinib, dabrafenib, tremetinib) approved in conjunction with genetic-based companion diagnostics. 34
Nevertheless, it became clear that both for disorders that are primarily monogenic but especially for complex diseases, merely identifying the defective genes involved is not sufficient. It is also necessary to determine the functional interrelationships among these genes and their products, including epigenetic modifications, and to understand the biological underpinnings that result in the disease phenotype. 35 Traditional drug discovery approaches predicated on the “one-gene, one-target, one-mechanism” hypothesis have been inefficient in developing therapeutics.7–9 Therefore, it would be valuable to explore the application of comprehensive, unbiased strategies such as QSP that are intrinsically network centric and that can integrate, analyze, and validate large, complex data sets with state-of-the-art computational and experimental technologies to more fully understand the disease biology. It is feasible to apply QSP thanks to advances in molecular-to-cellular analyses, algorithmic developments, and increased computational power.20,36–40 The technical approaches and the application to drug discovery and development are rapidly developing.
Here, we present the rationale for a drug discovery strategy and platform based on our definition of QSP. Components of the proposed platform (
Fig. 3
) have been and are being applied in the standard drug discovery approaches, including in target-centric and phenotypic approaches (
Figs. 1
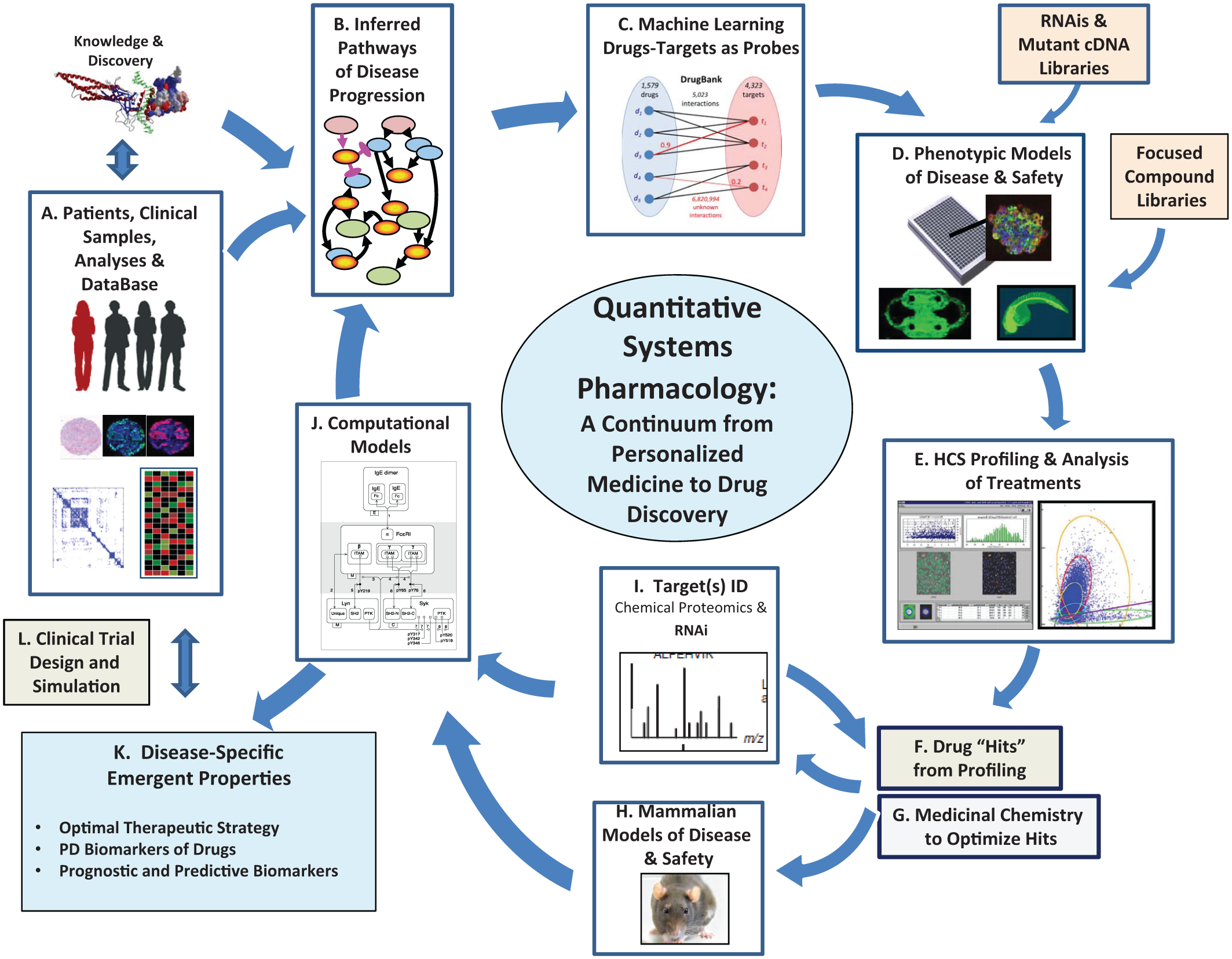
Quantitative systems pharmacology platform for drug discovery and the advancement of personalized medicine being implemented at the University of Pittsburgh Drug Discovery Institute. Details are outlined in Table 2.
Quantitative systems pharmacology platform for drug discovery and the advancement of personalized medicine being implemented at the University of Pittsburgh Drug Discovery Institute. a

This table is in support of Figure 3. QSP = quantitative systems pharmacology; HCS = high-content screening; ID = identification.
As summarized in Table 2 (which supports Fig. 3 ), the QSP platform is capable of addressing the inherent heterogeneity of genotypes and phenotypes in populations of patients with the same disease and identifying distinct treatments for each subpopulation. 34 Implementing the QSP platform requires teamwork among investigators spanning the fields of clinical medicine, genetics, molecular biology, medicinal chemistry, pharmacology, ADME-TOX, computational biology and chemistry, systems biology, engineering, mathematics, and bioinformatics.
QSP and personalized medicine share a focus on the patient at both the start and end point (step A in Fig. 3 and Table 2 ). Personalized medicine currently uses patient data, including standard clinical tests as well as a growing list of “omics” analyses obtained from tissue samples, to select the optimum treatment from existing options. Advances in longitudinal patient sampling (e.g., liquid biopsies for circulating tumor-derived DNA 18 and circulating tumor cells 73 have enabled the monitoring of disease progression. QSP works to expand the existing options for therapies through a combination of an iterative cycle of experimental and computational analyses aimed at identifying disease-specific emergent properties, such as new therapeutic strategies, pharmacodynamic biomarkers, and prognostic and predictive biomarkers ( Fig. 3 , steps A–K; Table 2 ).
The process can be initiated at any step, depending on the availability of validated data for prior steps, with the goal of extracting and integrating information from each step to inform subsequent experimental and computational design and analyses steps in the iterative cycle. The ultimate step is the return to the patients with innovative clinical trial designs and verification of their effectiveness (step L).
Systems-Level Approach Needed for a Mechanistic Understanding of Disease Phenotypes
For several genetically well-defined (i.e., Mendelian) diseases in which the criteria for causality are akin to Koch’s postulates having been satisfied, 35 the successful development of therapies has often depended on a detailed mechanistic knowledge of how particular genotypes give rise to disease phenotypes. In Marfan syndrome, for example, the identification of fibrillin-1 mutations 74 per se was insufficient to identify therapies without the concomitant understanding of the pathophysiology.75,76 In a series of elegant studies employing Marfan syndrome mouse models that recapitulate many of the clinical manifestations of the disease, 76 including a predisposition for aortic aneurysm, angiotensin II–dependent canonical and noncanonical transforming growth factor (TGF)–β signaling was shown to be the responsible pathway for this disorder.77–79 Selective blockade of the angiotensin II type 1 receptor, although sparing angiotensin II type 2 signaling, led to the successful repurposing of the antihypertensive losartan for this indication. 76 Such a network-centric approach implemented in conjunction with clinically relevant models is intrinsic to QSP ( Fig. 3 ; Table 2 ). Because diseases often share pathways linked to their progression, as suggested by the prevalence of comorbidities, 80 we posit that a network-centric QSP approach for understanding pathophysiology will likely identify more opportunities for drug repurposing, in addition to guiding the development of novel therapeutics.
Akin to Marfan syndrome, the genetic defect leading to Huntington’s disease (HD) has been known for more than 20 y (i.e., expanded CAG repeats in the HTT gene, which leads to aggregate-prone extended polyglutamine stretches in the protein product huntingtin). But the function of HTT and the biology of disease progression remain poorly understood. Mutant huntingtin expression is difficult to modulate directly with small molecules, displays pleiotropic gains of function, 81 and induces neuronal subtype–selective cell death, with striatal medium spiny neurons being the most vulnerable. As part of a QSP approach to identifying the pathogenic pathways, 82 knock-in models that recapitulate several clinical features of HD have identified several dysregulated pathways (e.g., cadherin, TGF-β, and caspase signaling) that are also evident in patient postmortem tissue. Transcriptome analysis shows that these same pathways are also dysregulated in medium spiny neurons differentiated from induced pluripotent stem cells (iPSCs) derived from HD patients but not from normal individuals. However, when the HTT mutation is corrected by homologous recombination, the signaling of these dysregulated pathways is normalized, and the disease phenotype (i.e., susceptibility to stress-induced cell death) is rescued. 82
Once dysfunctional pathways have been inferred from the systems-level analysis, as in the examples above (step B in Fig. 3 and Table 2 ), the next step in the QSP platform is to distinguish those that are mechanistically linked to disease progression (i.e., pathogenic) from other dysregulated pathways that may represent an epiphenomenon or a disease-ameliorating compensatory effect. 83 To accomplish this task, iterative use of experimental and computational approaches is essential (steps B–J). The goal is to modulate and quantitatively assess how individual pathways affect disease phenotype. High-content profiling that includes analysis of heterogeneity of cellular responses can then help us determine the role of a particular pathway in the pathophysiology (step E). 56 However, drug-induced normalization of a single pathway may not be sufficient to rescue disease phenotype. Specific proteomic, transcriptomic, and metabolomic data84,85 can then be analyzed and integrated using computational and pharmacodynamic systems analysis tools 86 to construct mathematical (computational) models of pathogenic pathways and their interrelationships (networks; Step J).69–71 This information is in turn used for predicting optimal drug combinations and companion biomarkers 86 (step K) that can then be experimentally validated (or invalidated) and further refined through additional iterations of the QSP cycle prior to clinical trial design.
QSP-Driven Phenotypic Approaches to Drug Discovery in Complex Disease
The great challenge for drug discovery in complex diseases relates to their variations at often hundreds of gene loci, phenotypic heterogeneity, and multifactorial patterns of inheritance. Variants at any component locus may not be necessary or sufficient to result in the disease phenotype, and they may be relatively common in healthy individuals and occur in noncoding regions. The pathogenic consequences of variants depend on cellular and organ context, functional interactions with other gene variants and their products, and associated environmental factors. For example, mutations within the cystic fibrosis transmembrane conductance regulator (CFTR) that disrupt anion channel activity affect epithelial cell function in the lungs, pancreas, and other tissues as part of the cystic fibrosis (CF) phenotype. Indeed, Ivacaftor, identified through phenotypic discovery, was recently approved by the Food and Drug Administration (FDA) as the first drug to directly target any of 10 rare mutations that disrupt the chloride ion channel function of CFTR.87–89 However, CFTR mutations that disrupt bicarbonate anion channel activity but spare chloride anion channel activity predispose individuals to chronic pancreatitis but not pulmonary dysfunction.90,91 The risk of chronic pancreatitis increases further with the coinheritance of pancreatic secretory trypsin inhibitor variants, which multiplicatively increases the risk of chronic pancreatitis (without CF), suggesting gene (product)–gene (product) interactions and a corresponding high-risk pathway. 91 These particular observations with chronic pancreatitis appear to be generalizable to other complex diseases. Probabilistic genetic modeling indicates that gene-gene interactions, especially among common variants underlying Mendelian disorders, appear to make major contributions to complex disease risk and as such are significant determinants of disease phenotype. 80
Thus, focusing on disease phenotype may represent a complementary strategy to individual gene (or protein) targeting. Several reviews reinforce the value of phenotypic screens and their success relative to target-focused approaches, particularly for complex disease.1,2,5,6,56,57,92 In particular, high-content screening (HCS) is a powerful phenotypic screening tool for analyzing the temporal-spatial dynamics of multiple cellular function parameters in both cell-based56,61,93 and experimental organism 94 models. As an example, HCS conducted on a genome-wide scale using RNAi or cDNA overexpression combined with in silico drug-target discovery strategies has been used to repurpose preclinical drugs for both common and rare diseases, such as alpha1-antitrypsin deficiency and acute megakaryocytic leukemia (AMKL).62,63
In the case of AMKL, 62 the disease phenotype is well known (immature megakaryocytes cannot exit from a highly proliferative cycle to achieve polyploidization and platelet differentiation), but the genomic data per se were not sufficient to focus on a small set of candidate pathways or targets. Thus, the QSP cycle was initiated at step D with HCS profiling ( Fig. 3 ; Table 2 ) using small-molecule chemical probes and comprehensive RNAi libraries in a cell line derived from a patient AMKL blast to identify mechanisms of AMKL progression. Dimethyl fasudil was identified as a rare hit that selectively induced both polyploidization and other markers indicative of platelet differentiation in megakaryocytes but not in cells from other lineages (step F). This kinase inhibitor demonstrated efficacy in a murine AMKL model that directly correlated with its in vivo induction of polyploidization in tumor cells (step H).
Next, chemical proteomics and kinase and kinome-shRNA screening (step I in Fig. 3 and Table 2 ) identified the mechanistic target of dimethyl fasudil as Aurora kinase A (AURKA), both a negative regulator of endomitosis and platelet differentiation and a targetable AMKL dependency (step K). This finding was corroborated in vivo (step H) both genetically (AURKA knock-out animals) and pharmacologically using Alisertib, an Aurora kinase A inhibitor under clinical development for other oncology indications.
Thus, a QSP-driven, mechanism-focused, phenotypic approach led to the repurposing of Alisertib for the novel differentiation-inducing treatment of AMKL (ongoing clinical trial). Computational modeling in the same study (step J in Fig. 3 and Table 2 ) led to insights regarding five kinase pathways that regulate endomitosis and could suggest additional experiments designed to inform the rational design and testing of combination therapies, at which point a subsequent QSP cycle would commence at steps B and C. Indeed, the mechanistic studies that have led to an AMKL indication for Alisertib have now been extended to patients with another megakaryocyte-based neoplasm, primary myelofibrosis, with the prospect of implementing combination therapeutic strategies involving the Janus kinase inhibitor, ruxolitinib. 95
QSP as a Means to Address Resistance to Targeted Cancer Therapy
A promising application of QSP is in the development of precision cancer therapy designed to achieve sustainable remission.96,97 Functional genomic analyses of tumor mutations can identify targetable tumor dependencies (e.g., BCR-ABL, EGFR, BRAF) that guide the selection of inhibitors capable of improving progression-free survival without the side effects of traditional chemotherapy. However, resistance emerges through the clonal evolution of tumor cells with mutations that escape the initial selective inhibitor. Maximizing treatment response requires the a priori rational combination of synergistic therapies96,98 in anticipation of the mechanisms of resistance likely to emerge. 97 This can be accomplished through iterative use of the QSP cycle, starting at step A to identify the patient genotypes and phenotypes that can be engineered into phenotypic models (step D), to identify those pathways with the potential to confer resistance. For example, mutant cDNAs representing core nodes in 17 pathways that are frequently implicated in cancer cell proliferation, survival, differentiation, and cell death can be expressed to activate or inhibit their respective pathway. 55 Pathways conferring resistance to particular drugs were identified using these cDNAs, and when these resistance-conferring pathways were themselves inhibited with a second drug, sensitivity to the original targeted drug was restored, thereby suggesting a potential polypharmacologic strategy for addressing resistance. Mechanisms of resistance involved either the activation of a compensatory pathway to bypass the one inhibited by the drug or the activation of pathways that alter the cell state to be less dependent on the drug-targeted pathway for viability. 55
Secondary mutation of the target itself represents another important strategy for the tumor to reactivate the oncogenic signaling pathway in the presence of a drug. These mutations confer resistance by several possible mechanisms, such as direct or allosteric prevention of drug-target interaction, enhanced binding of a competitive substrate, or the conversion of a receptor antagonist to an agonist. 97 Examples include BCR-ABL mutations that confer imatinib resistance in chronic myeloid leukemia 99 and the F876L mutation in the androgen receptor that confers resistance to enzalutamide for the treatment of castration-resistant prostate cancer.100,101 A holistic QSP approach can take into account the emergence of resistance, including its molecular mechanisms, 102 at the outset of a drug development program and thus inform the design of polypharmacologic strategies that effectively trap emerging resistance to enable more sustainable remission and cure rates.
Using QSP to Address Heterogeneity in Drug Discovery and Development
Because heterogeneity can be the product of genetic and/or nongenetic processes,57,103 disease genotypes and phenotypes can vary within (intratumor) and between (intertumor) experimental models and individual patients, confounding our ability to develop optimal therapeutics and diagnostics.42,52,58,59,98,104–107 The QSP cycle addresses heterogeneity from the analysis of patient samples (step A in Fig. 3 and Table 2 ) through the development of mathematical models (step J). For example, the application of HCS to a phenotypic model of HD using patient-derived iPSCs demonstrated the ability of correcting the mutant HTT gene with homologous recombination to reverse cell death in response to stress. 82 In the future, iPSCs could be modulated with molecular and/or small-molecule perturbations to identify and quantify any heterogeneity in patient-specific disease models. 56
In cancer, spatial relationships between tumor and stromal cells add another layer of heterogeneity. Spatial heterogeneities underscore the importance of capturing data within the context of cellular and tissue architecture that is otherwise lost through population-averaged methods. 57 The QSP platform takes advantage of new computational pathology methods that use panels of multiplexed fluorescent biomarkers to quantify single-cell intratumor heterogeneity while preserving spatial relationships within the tumor.43,57,108,109 In addition, single-cell proteomics from mass spectrometry imaging can likewise be incorporated into pathway models, 110 as can data from cell-based assays in microplates, 56 kinetic HCS measurements in single living cells, 16 and single-cell genetic analyses. 111 HCS of disease-specific samples (e.g., patient biopsies, patient-derived iPSCs, patient-derived xenograft [PDX] tissue, tissue-engineered models) can define and quantify the heterogeneity for use in the development of both therapeutics43,56,108 and more biologically relevant experimental, phenotypic models of disease.48,49
The QSP process also takes into account nongenetic sources of heterogeneity. Although many diseases have some genetic component, most have environmental or stochastic components, as long demonstrated by twin studies.112,113 Through its integrated computational modeling, QSP can help address environmental and epigenetic influences on health and disease. If particular environmental exposures are suspected as contributing to a disease phenotype, the QSP framework has the potential to test these associations and to probe interactions between particular genetic variations and environmental factors.
Computational Approaches within the QSP-Driven Drug Discovery Process
Computational analyses of complex data sets and computational modeling are central to the effectiveness of the QSP platform.21,69,114 Computations serve three major functions: (1) data integration with a variety of inference analytics, (2) predictions of drug-target and drug-drug interactions, and (3) predictive, computational modeling of the networks involved in the disease progression (see
Fig. 3
and
Table 2
,
The inference of pathways involved in disease progression based on the patient “omics” data from step A is the next task using algorithms16,17 that are continually being improved (step B in Fig. 3 and Table 2 ). Network-based approaches to human medicine are particularly useful for assessing the significance of disease-associated mutations identified by genome-wide association studies mentioned above, building on the wealth of theory and methods that have been developed for network models in general. 37 We note in particular the development of a new disease-module–detection algorithm by Barabasi and coworkers, 117 inspired by the observations that disease-associated proteins interact with each other. For example, the application of network models to asthma proved to be particularly useful in explaining disease heterogeneity and capturing novel pathways. 118
The prediction of approved drugs as probes of the potential molecular targets within the inferred pathways is the next computational task (step C). Computational methods for predicting unknown drug-target associations have been explored for a number of years, particularly for repurposing drugs for additional indications and identifying potential off-target effects.36,44–46,119,120 For example, the probabilistic matrix factorization approach is scalable to large databases such as STITCH and available as a web-based tool applied to DrugBank. 47
In addition, valuable transcriptional drug treatment databases including CMap 121 and LINCS 122 have searchable measurement data for a variety of experimental manipulations using multiple cell types, approved drugs, drug doses, and time points that are publically available. These and other databases have been used to connect genes, drugs, and diseases through changes in gene expression. 123 Approved drugs demonstrated to be effective in one disease can serve as candidates for other diseases exhibiting the same changes in gene expression.124,125 The mining of these types of publically available data sets complements the design and implementation of steps B through D in Figure 3 and Table 2 .
Computational modeling of disease progression networks (step J) starts with creating mathematical models that are consistent with published biological knowledge of the molecular and cellular mechanisms6,69–71,115 (steps A and B) as well as experimental data generated through steps C though I. The computational models make predictions that can be tested experimentally, and the model can be improved in an iterative process. New inferences may then be formulated as experimentally testable hypotheses. A key feature of the QSP platform is the experimental validation (or invalidation) of these hypotheses, which then provide feedback and new data to be incorporated into computational models to further improve their predictive ability. Rule-based languages for modeling,69–71 such as BioNetGen, 72 approach model design with mathematical definitions. Computational models that incorporate HCS data are being developed to address network complexity, cellular heterogeneity, spatial complexity, and multicellular systems56,57 in parallel with advances in causal model discovery. 126 The iterative process used in the platform is essential to learning about underlying mechanisms and designing specific therapies.
QSP and Personalized Medicine Form a Continuum
Because of its comprehensive capabilities and flexibility, the QSP platform will play a critical role as we move medicine from a reactive to a proactive process,22,23 particularly with the increasing penetration of DNA sequencing in the clinic. 127 The future routine collection of “omics” data in the clinic, including transcriptomics, epigenomics, metabolomics, proteomics, and computational pathology (step A in Fig. 3 and Table 2 ), will further enable an unbiased approach to inferring pathways of disease progression as a starting point for developing personalized therapeutics and their personalized companion diagnostics (step B). In particular, the QSP platform will facilitate the identification of pharmacodynamic, diagnostic, prognostic, and predictive biomarkers (step K). All of this information along with pharmacogenomic analysis will then contribute to the efficient design, simulation, and implementation of clinical trials (step L) that permit a focus on personalized rather than population-based medicine. The NCI-MATCH (Molecular Analysis for Therapy Choice) trial 128 is an initiative in that direction, which includes patients with any solid tumor or lymphoma who have one of many genomic abnormalities known to drive cancer. The goal for the QSP-personalized medicine continuum is to expand the options available at step A and to be there to develop new options as diagnostic and therapeutic needs are identified.
Challenges Exist in the Implementation of QSP for Developing Therapeutics and Advancing Personalized Medicine
The implementation of the QSP strategy and platform has been initiated to optimize the understanding of the mechanisms of disease progression at the level of preclinical testing before investing in expensive clinical trials where failures have been a major problem. However, there are ongoing challenges to fully develop and implement the computational and experimental methods, although the references listed in Table 2 and the body of this perspective demonstrate a good starting point. It is clear that the optimal implementation of QSP will be an evolutionary process. Furthermore, the training of scientists, engineers, and computational and systems biologists must evolve to support the new approach to developing therapeutics.
Some of the computational challenges include the need to improve the methods used to integrate diverse patient data types, to optimally harness “big data” generated from a growing number of high-throughput “omics” that require sophisticated data storage, analysis, archiving, visualization, and causal discovery. In addition, the development of useful computational models of networks involved in disease progression requires formalizing complex disease processes order to generate hypotheses that can be experimentally tested. Advances in the mathematical methods and systems biology tools, including the capture of heterogeneity in the network structures, need to be developed to fully capture the complexity of the biology.
Challenges also exist for the experimental approaches outlined in our strategy and platform. There is a need for a new focus on developing sophisticated phenotypic models that are more disease relevant. This will involve accepting a slightly lower throughput for phenotypic screening, as well as investing in the use of multicell, three-dimensional, human cell–based models, including the use of patient-derived, iPSCs and/or primary patient samples. This is a departure from the traditional approach of optimizing throughput relative to disease relevance. The best experimental models will also involve temporal analyses of live samples to gain dynamic information. The present generation of disease-relevant models is useful, but further advancements are needed, including developing standard plates and/or chips that provide physiological flow for the models with the maximal possible throughput. Finally, the application of chemical proteomics as part of the approach to identify the molecular targets from phenotypic profiles is powerful, yet still evolving. Validation studies are still ongoing to demonstrate the strengths and weaknesses of the approach.
Moving Forward with QSP and Personalized Medicine
We believe the time has arrived for QSP to become the key approach to determine the biology underlying disease progression and to support the advancement of personalized medicine. Various specific platforms such as the one outlined here are possible, and further innovation is important. 129 Yet the comprehensive unbiased approach, however practiced, has the potential to do more than identify a relatively small set of candidate drug targets that are merely associated with a particular disease: it allows for the informed selection of drug targets and biomarkers mechanistically linked to the pathophysiology. The QSP process also supports improved patient stratification and more quantitative predictions regarding the pharmacologic effects of particular drug-target interactions and thus increases the efficiency of demonstrating proof of concept in clinical trials. Each component of the QSP cycle has its own role in the drug discovery process, and their seamless integration is required for the identification of emergent, disease-specific properties. For example, computationally inferring pathways of disease progression (step B in Fig. 3 and Table 2 ) is facilitated by the analysis of multiple spatially and temporally distinct samples from an individual patient both to account for heterogeneity and to associate these pathway data with clinical observations and treatment outcomes over time (step A).
The priority now is to aggregate and disseminate available toolkits of experimental and computational methods that can comprehensively analyze data and specifically model and modulate the pathways of disease progression for any disease. Such libraries of toolkits would be used to determine the pathway and network dependencies for specific disease phenotypes (steps D, E, H, J), validating computationally derived pathway analyses and distinguishing these from epiphenomena (e.g., driver mutations from passenger mutations). The development and implementation of standardized experimental and computational tools will require strong organizational leadership among academia, industry, and government sponsors and regulatory agencies to establish a culture of multidisciplinary team building across institutions. We believe that recognition of the value of QSP for drug discovery and the development for personalized medicine will motivate the adoption and implementation of this paradigm.
Footnotes
Acknowledgements
The authors would like to thank the members of their laboratories for significant contributions to our thinking. We are implementing full programs in QSP applied to neurodegenerative diseases (starting with HD), metastatic breast cancer, and liver diseases (including hepatocarcinoma and nonalcoholic fatty liver disease). We cannot acknowledge all members of each program team that are drawn from the University of Pittsburgh, Carnegie Mellon University, the University of Pittsburgh Medical Center, as well as external collaborators, but special thanks to Robert Friedlander, Adrian Lee, Steffi Oesterreich, Paul Monga, Ed Prochownik, David Perlmutter, Jaideep Behari, David Whitcomb, Nathan Yates, Bert Gough, Andreas Vogt, Larry Vernetti, Chakra Chennubhotla, Jim Faeder, Tim Lezon, Greg Cooper, Xinghua Lu, Russell Schwartz, Robert Murphy, and Zoltan Oltvai for ongoing discussions. We also acknowledge a valuable collaboration with the National Center for Advancing Translational Sciences screening center at the National Institutes of Health. Special thanks to Michelle Kienholz for critically reviewing and editing the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Our development of the QSP platform has been supported by PA Department of Health, SAP #4100068731 to A. M. Stern, National Institutes of Health (NIH) awards P01 DK096990 and U19 AI068021 to I. Bahar, and an NIH award P30 CA047904 (a subaward for a chemical biology facility), a NExT-CBC RFP S11-030-NCI award, and an NIH 5UH2TR000503 award from National Center for Advancing Translational Sciences (NIH) to D. L. Taylor.