
Abstract
Pulmonary arterial hypertension is a complex disease with multiple etiologic factors. PDLIM5, a member of the Enigma subfamily of PDZ and LIM domain protein family, contains an N-terminal PDZ domain and three LIM domains at its C-terminus. We have previously shown that overexpression of PDLIM5 prevents hypoxia-induced pulmonary hypertension (PH), and deletion of PDLIM5 in smooth muscle cells enhances hypoxia-induced PH in vivo. These results suggest that PDLIM5 may be a novel therapeutic target of PH. In this study, we aim to establish a high-throughput screening platform for PDLIM5-targeted drug discovery. We generated a stable mink lung epithelial cell line (MLEC) containing a transforming growth factor–β/Smad luciferase reporter with lentivirus-mediated suppression of PDLIM5 (MLEC-shPDLIM5) and measured levels of Smad2/3 and pSmad2/3. We found that in MLEC, suppression of PDLIM5 decreased Smad-dependent luciferase activity, Smad3, and pSmad3. We used MLEC-shPDLIM5 and a control cell line (MLEC-shCTL) to screen the Prestwick library (1200 compounds) and identified and validated paclitaxel as a PDLIM5 inhibitor in MLEC. Furthermore, we showed that paclitaxel inhibited Smad2 expression and Smad3 phosphorylation in A549 cells. Our study suggests that this system is robust and suitable for PDLIM5-targeted drug discovery.
Introduction
Pulmonary arterial hypertension (PAH) is a complex disease with multiple etiologic factors without effective treatment.1,2 Although there are many subcategories of PAH, including idiopathic PAH, heritable PAH, and PAH associated with other diseases such as connective tissue diseases, 3 they all share common pathological changes in the pulmonary artery, such as proliferation of pulmonary artery endothelial cells and pulmonary artery smooth muscle cells (PASMC), PASMC migration and contraction, inflammation, as well as fibroblast proliferation, activation, and migration. Numerous factors contribute to the pathogenesis of PAH, including genetic, epigenetic, and environmental factors. 4 In our previous report, we have adopted a quantitative mass spectrometry analysis/proteomics approach for global screening of miR-17~92 targets in human PASMC and have identified PDZ and LIM domain 5 (PDLIM5) as a novel miR-17~92 target. 5 We have shown that knockout of PDLIM5 enhances hypoxia-induced pulmonary hypertension (PH), whereas overexpression of PDLIM5 inhibits hypoxia-induced PH. 5 Thus, PDLIM5 protein plays a role in the development of PH, 5 suggesting a potential to target PDLIM5 for the treatment of PH.
PDLIM5, also named Enigma Homologue protein, is a member of the Enigma subfamily of PDZ and LIM domain protein family and contains an N-terminal PDZ domain and three LIM domains at its C-terminus. PDLIM5 is highly expressed in heart and skeletal muscle6,7 and acts as a signal modulator to influence organ development and disease.8,9 PDLIM5 can interact with α-actinin, protein kinase C, protein kinase D, and ID2, a DNA transcription inhibitor, and has been implicated in heart disease and mental disorders. 10 We have shown that in PASMC, suppression of PDLIM5 induces TGF-β3, TβR1, TGF-β activity, Smad2, and phosphorylated Smad2/3 as well as nuclear localization of Smad2/3. 5 PDLIM5 also negatively regulates the expression of SMC markers, a feature of TGF-β signaling. SMC-specific knockout of PDLIM5 enhances hypoxia-mediated vascular remodeling, and the overexpression of PDLIM5 inhibits TGF-β/Smad signaling and prevents hypoxia-induced PH in vivo. 5 These results indicate that PDLIM5 regulates the development of human disease by modulating TGF-β/Smad signaling. Therefore, PDLIM5 may be a novel therapeutic target for human diseases.
We sought to establish a high-throughput screening (HTS) platform for PDLIM5 modulators, using a TGF-β/Smad–based luciferase reporting system. Mink lung epithelial cells (MLEC) were stably transfected with a luciferase reporter plasmid driven by a truncated plasminogen activator inhibitor type I (PAI-1) promoter, which contains Smad-binding elements. 11 We generated a stable MLEC-Luc cell line with lentivirus-mediated suppression of PDLIM5. We found that suppression of PDLIM5 decreased Smad-dependent luciferase activity without affecting cell proliferation. By screening the Prestwick library (1200 compounds) in the presence or absence of TGF-β1, we identified and validated paclitaxel as a PDLIM5 inhibitor in MLEC. Thus, this assay system can be used to discover novel PDLIM5 inhibitors via HTS.
Materials and Methods
Materials
TGF
Cell Culture
MLEC were stably transfected with a luciferase reporter plasmid containing a truncated PAI-1 promoter, which contains Smad-binding elements.11,12 MLEC were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin and were used for TGF-β reporter assay. Human non–small-cell lung adenocarcinoma cell line A549 was purchased from American Type Culture Collection (Manassas, VA) and was cultured in DMEM media as described above. All cells were maintained in a humidified incubator with 5% CO2 at 37 °C.
Establishment of Stable Cell Lines with Suppression of PDLIM5 (A549-shPDLIM5 and MLEC-shPDLIM5)
PDLIM5 shRNA lentiviral particles and control shRNA lentiviral particles were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). We transduced A549 and MLEC cells with a mixture of complete medium with polybrene (final concentration 5 µg/mL) and lentiviral particles as we reported previously. 13 We cultured cells in media containing puromycin dihydrochloride (1 µg/mL) to select for stable single clones, which were collected and expanded. The expression of PDLIM5 was analyzed by Western blot analysis.
TGF-β Reporter Assay
MLEC, MLEC-shCTL, and MLEC-shPDLIM5 were plated into 24-well plates at a density of 105 cells/mL/well and cultured overnight. After 48 h incubation with TGF-β1 (5 ng/mL) or phosphate-buffered saline (PBS), MLECs were rinsed with PBS and lysed in passive lysis buffer at 120 µL/well (Promega). Cell lysates were cleared at top speed for 2 min at 4 °C. One hundred microliter supernatants of each sample were transferred to new polypropylene tubes and read with 100 µL luciferase assay buffer (Promega) on a GloMax-96 Microplate luminometer (Promega). The protein concentrations were determined using Bio-Rad protein assay solution (Bio-Rad, Hercules, CA). The relative luciferase activity was calculated by normalizing total luciferase activity to the amount of protein. Each assay was done in triplicate and repeated at least three times.
Western Blotting
We measured protein levels by Western blot analysis as we described previously. 14 Briefly, after three washes with PBS, cells were lysed in mRIPA buffer (50 mM Tris pH 7.4, 1% NP-40, 0.25% deoxycholate, 150 mM NaCl, and protease inhibitors) on ice for 30 min. The cell lysates were cleared by centrifugation at 13,000 g for 10 min, and protein concentrations of the supernatants were determined using Bio-Rad protein assay solution (Bio-Rad). Cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred to BA85 nitrocellulose membranes (PROTRAN, Whatman, Dassel, Germany), and probed with primary and secondary antibodies. Proteins were detected with SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific, Waltham, MA). The following primary antibodies were used in this study: α-tubulin, β-actin, SMAD2/3, pSMAD2 (ser465/467), pSMAD3 (ser423/425), and PDLIM5 (Sigma-Aldrich and Protein Technologies, Inc.). Anti-mouse, anti-rabbit, and anti-goat IgG-HRP conjugates were purchase from Bio-Rad. The density of the protein bands was quantified with ImageJ software.15,16
Bromodeoxyuridine (BrdU) Cell Proliferation Assay
MLEC and A549 cells were plated in 96-well plates at a density of 5 × 103 cells/100 µL/well and incubated overnight. BrdU was added to the culture medium at a dilution of 1:10,000, followed with another 16 h incubation. Cell proliferation activities were detected using a BrdU cell proliferation assay kit (Calbiochem, Gibbstown, NJ) on a GloMax-96 Microplate luminometer (Promega) at the wavelength of 450 nm according to the user manual.
HTS
This pilot screening was carried out in white 384-well plates and assembled on a PerkinElmer (Waltham, MA) Automated Workstation (JANUS/Envision). Briefly, we seeded MLEC-shCTL and MLEC-shPDLIM5 at the density of 2000 cells/well and incubated them overnight. The following day, we aspirated old media and added 20 µL of fresh media with or without TGF-β1 (10 ng/mL) to each well, then 20 µL of fresh media containing 25 µM of compounds. The final compound concentration was 12.5 µM and for TGF-β1 was 5 ng/mL. Each 384-well plate contained 320 test wells that received compounds and 32 control wells that received DMSO vehicle. Half of the control wells were cultured in the absence of TGF-β1, whereas the other half was cultured in the presence of TGF-β1 for the calculation of z′ factors. Following a 48 h period of incubation, the luciferase activity was measured following the manufacturer’s instructions (Neolite luciferase substrate, PerkinElmer). The coefficient of variation was calculated with R programming (R Development Core Team).
The z′ factor was determined from experimental data that were derived from the measurements of the assay signal in the presence and absence of TGF-β1 using the following equation:
Statistical Analysis
All experiments were repeated at least three times independently (most were five experiments). t Tests were performed using GraphPad Prism 4 (GraphPad, San Diego, CA) when applicable. The significance level was set at 0.05 and 0.01.
Results
PDLIM5 Is Required for TGF-β/Smad Signaling in Alveolar Epithelial Cells
Previously, we showed that PDLIM5 inhibits TGF-β/Smad in pulmonary artery smooth muscle cells. 5 To investigate the role of PDLIM5 on TGF-β/Smad in alveolar epithelial cells, we transduced a luciferase reporter cell line derived from mink lung epithelial cells (MLEC-Luc; Fig. 1A, B ) with control lentivirus and lentiviral particles containing small hairpin RNA against PDLIM5. After selection with puromycin, we established two stable cell lines, MLEC-shCTL and MLEC-shPDLIM5, and confirmed the silencing of PDLIM5 ( Fig. 1A–C ). We found that suppression of PDLIM5 did not alter cell proliferation ( Fig. 1D ), but it decreased Smad-dependent luciferase activity ( Fig. 1E ) and levels of total and phosphorylated Smad3 ( Fig. 1F, G ). These results suggest that PDLIM5 is a positive regulator of TGF-β/Smad in alveolar epithelial cells,
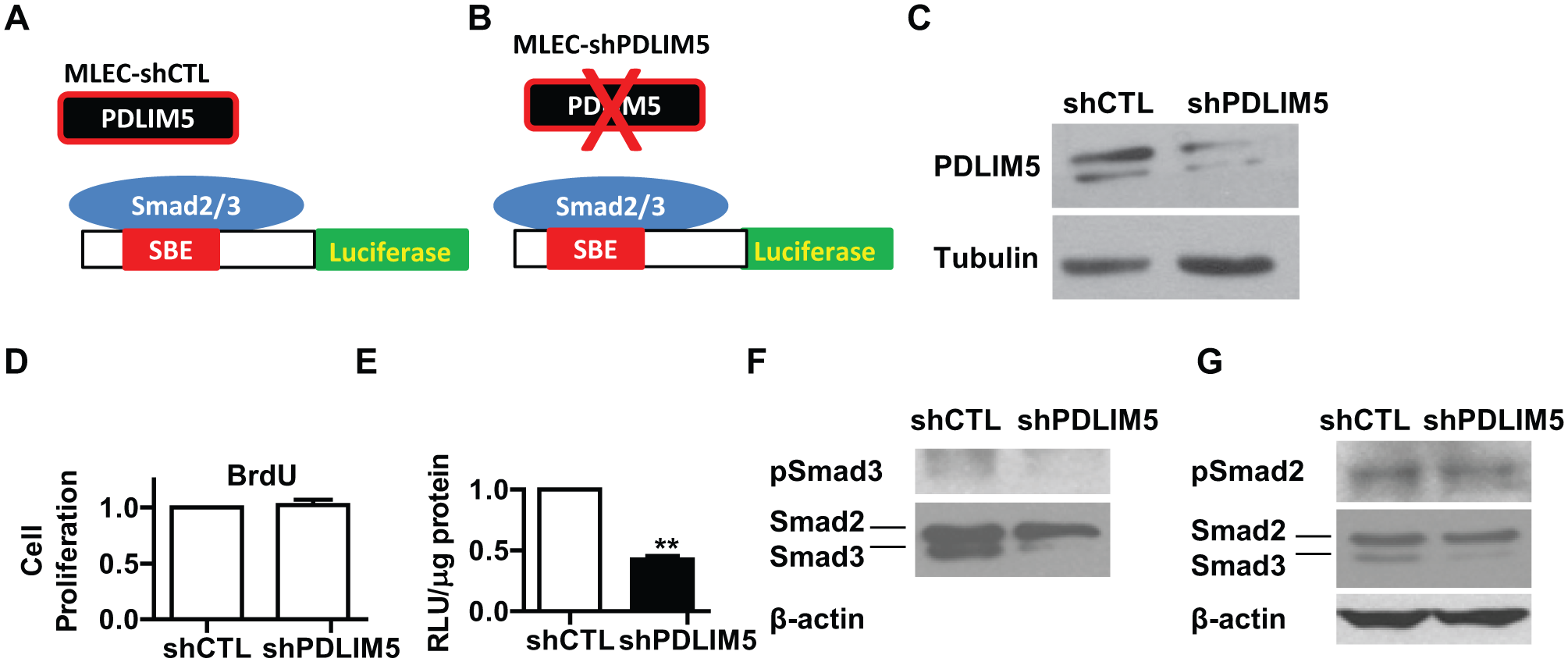
Suppression of PDLIM5 decreases TGF-β/Smad signaling in alveolar epithelial cells. (
Concept of Using MLEC-shCTL and MLEC-shPDLIM5 for HTS to Identify PDLIM5 Inhibitors
Because PDLIM5 is required for TGF-β/Smad signaling (
Fig. 1
), we hypothesize that a TGF-β/Smad-based luciferase reporter system can be used to identify PDLIM5 inhibitors via HTS (
HTS Platform for PDLIM5 Inhibitors
To examine the suitability of this system for HTS, we screened the Prestwick Collection, a library of 1200 FDA-approved drugs. We determined the z′ value in this 1200 compound screening as described previously17,18 and found that the z′ factors of all 16 plates exceeded 0.5, confirming the suitability of this assay system for HTS ( Fig. 2A ).
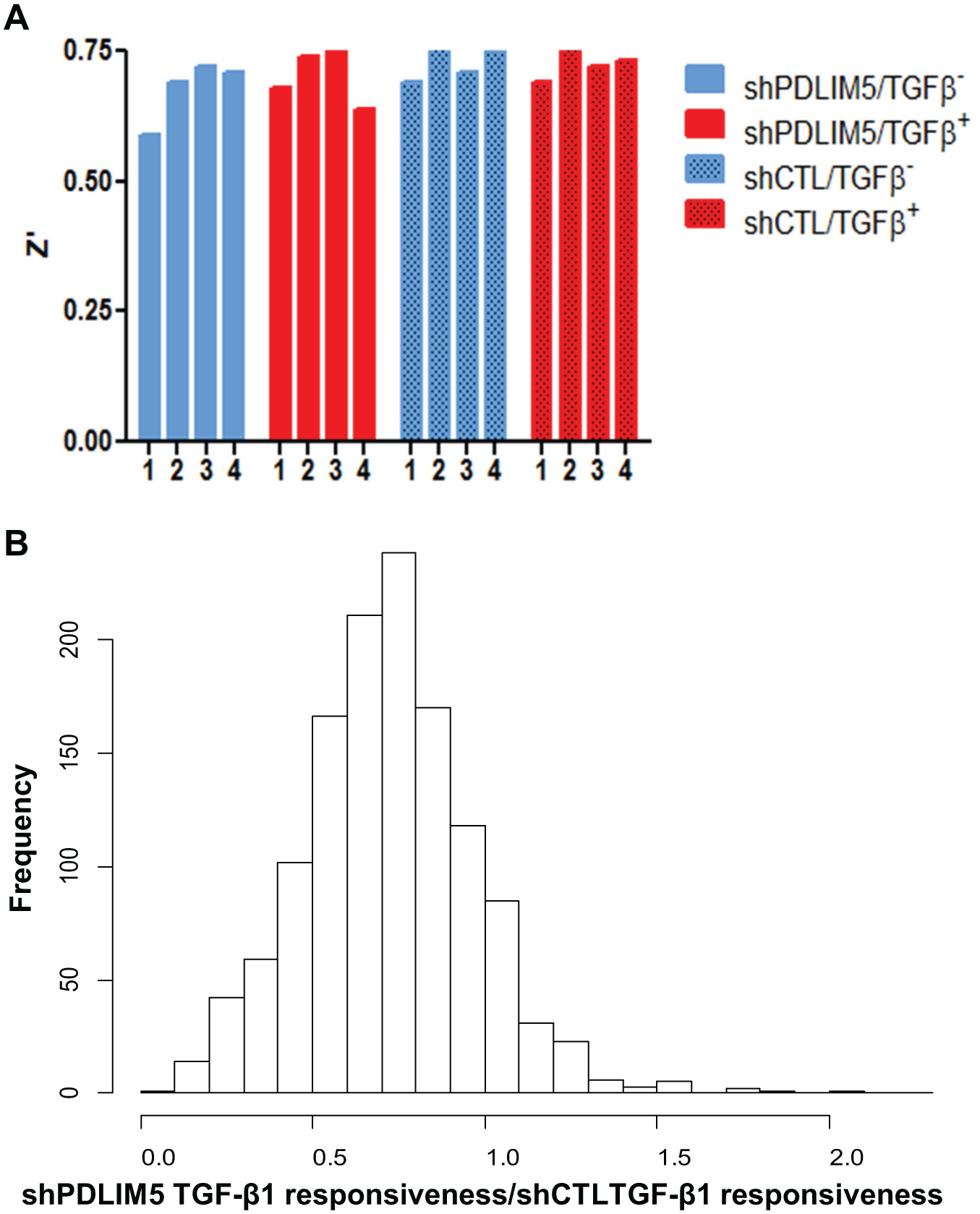
Feasibility of PDLIM5-targeted high-throughput screening. (
We examined the distribution of the overall ratio of the TGF-β1 responsiveness between two cell lines (MLEC-shPDLIM5/MLEC-shCTL). As shown in
Figure 2B
, the distribution of the overall ratios of the TGF-β1 responsiveness exhibited a bell-shape curve that was shifted left, confirming our assumption that MLEC-shPDLIM5 cells are less responsive to TGF-β1 (
Fig. 2
), therefore indicating that this system is useful for the discovery of PDLIM5 inhibitors. We calculated the coefficient of variation for 16 wells of control replicates on each plate and found that they are well below 10% in the TGF+ group or 20% in the TGF– group (
Identification of Paclitaxel as a PDLIM5 Inhibitor by Screening the Prestwick Collection (1200 Compounds)
We then set up the cutoff threshold of the overall ratios of the TGF-β1 responsiveness (MLEC-shPDLIM5/MLEC-shCTL) as 1.5 for the selection of PDLIM5 inhibitors. There were 11 putative PDLIM5 inhibitors ( Table 1 ). We cherry-picked these 11 hits and retested them in the second round of the HTS. In this round, we added 20 µL of media containing 25 µM of compound first, then 20 µL of media with or without TGF-β1 (final compound concentration was 12.5 µM and of TGF-β1 was 5 ng/mL) to eliminate the variation due to the order of treatment. Paclitaxel and disulfiram were confirmed in the second-round screening ( Table 1 ). The facts that PDLIM5 is known to regulate microtubule dynamics 19 and that paclitaxel is a well-known cancer drug that interferes with microtubule stability demonstrate the quality of our HTS system.
List of Small Molecules That Inhibit PDLIM5 in MLEC.
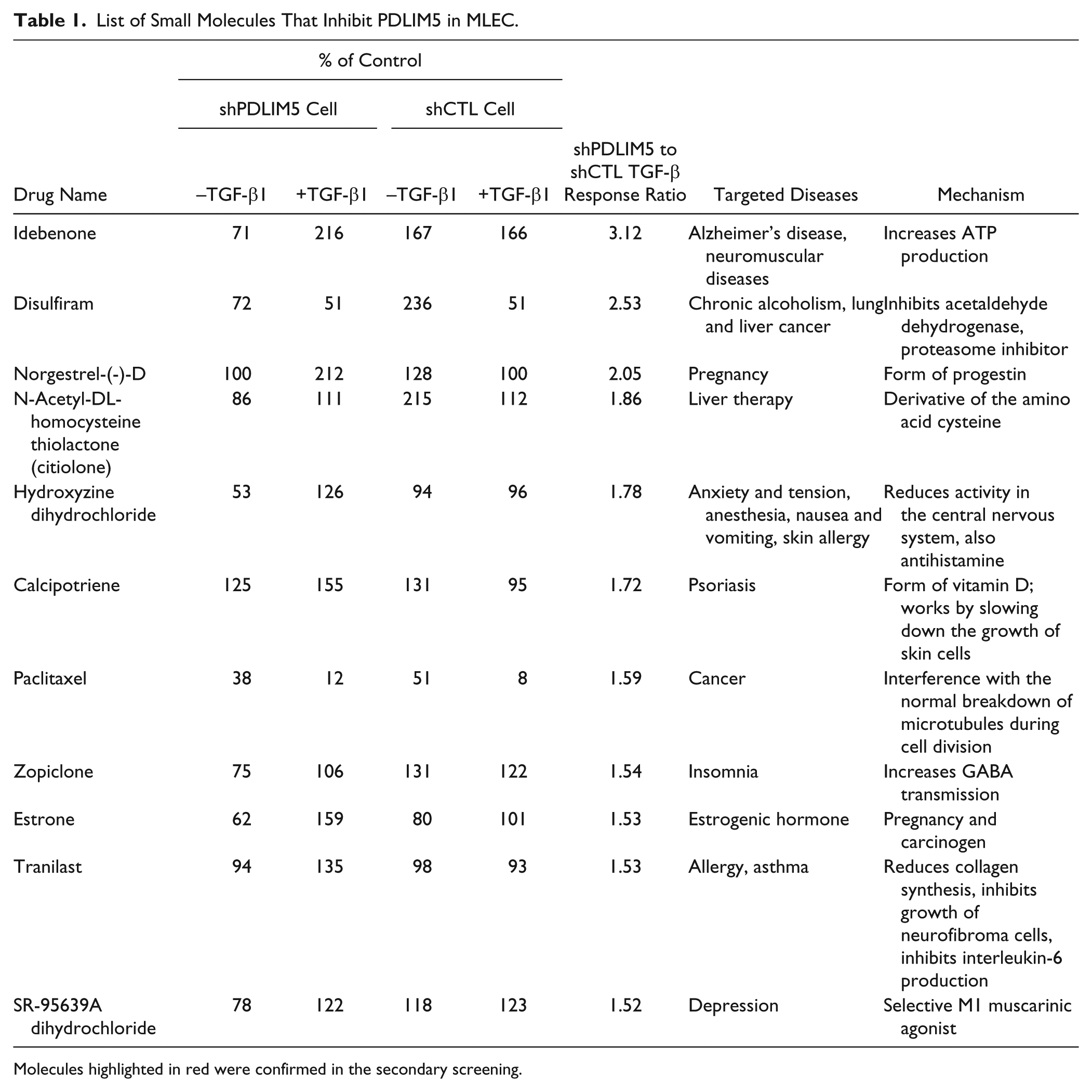
Molecules highlighted in red were confirmed in the secondary screening.
Paclitaxel Inhibits TGF-β/Smad Signaling in Non–Small-Cell Lung Carcinoma (NSCLC) Cell Line
Because PDLIM5 is required for TGF-β/Smad signaling ( Fig. 1 ), we reasoned that a PDLIM5 inhibitor would likely inhibit the TGF-β/Smad signaling. To further validate the screening results, we tested whether paclitaxel inhibits TGF-β/Smad signaling. We treated A549 cells with 50 nM paclitaxel for 2 d and measured the protein levels of PDLIM5, Smad2/3, and pSmad2/3 in the cell lysates. We found that treatment with paclitaxel (Taxol) inhibited the total Smad2 and pSmad2 and decreased the ratio of pSmad3 over total Smad3 while inducing total Smad3 ( Fig. 3 ). However, the levels of PDLIM5 and pSmad3 remained unchanged ( Fig. 3 ). These results demonstrated that paclitaxel indeed inhibits TGF-β/Smad signaling, as we predicted, validating our HTS platform as a suitable approach to identify PDLIM5 inhibitors as anticancer drug candidates.
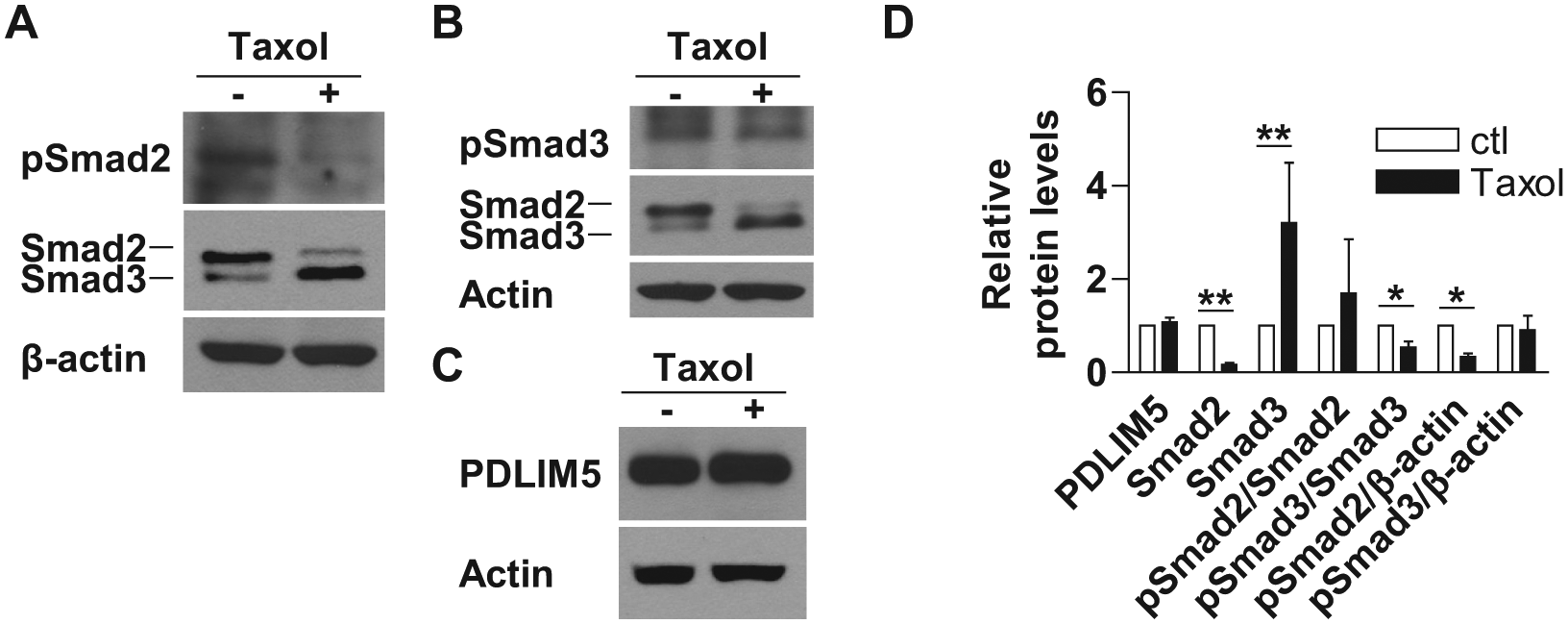
Treatment of paclitaxel inhibits TGF-β/Smad signaling. A549 cells were treated with 50 nM paclitaxel (Taxol) for 2 d followed by Western blot analysis for PDLIM5, Smad2, Smad3, pSmad2, and pSmad3. The quantification shown in
Paclitaxel-Mediated Inhibition of TGF-β/Smad3 Signaling Depends on PDLIM5 in NSCLC
To address whether paclitaxel inhibits TGF-β/Smad signaling via PDLIM5 in NSCLC cells, we treated A549-shCTL and A549-shPDLIM5 with paclitaxel or vehicle and incubated for 48 h followed with measurement of total and phosphorylated Smad2/3. We found that suppression of PDLIM5 had little effect on total Smad2, pSmad2, or the ratio of pSmad2/Smad2 ( Fig. 4 ). Although suppression of PDLIM5 had little effect on the ratio of pSmad3/Smad3, it decreased the amount of pSmad3 ( Fig. 4 ). Thus, paclitaxel inhibits TGF-β/Smad3 via PDLIM5.
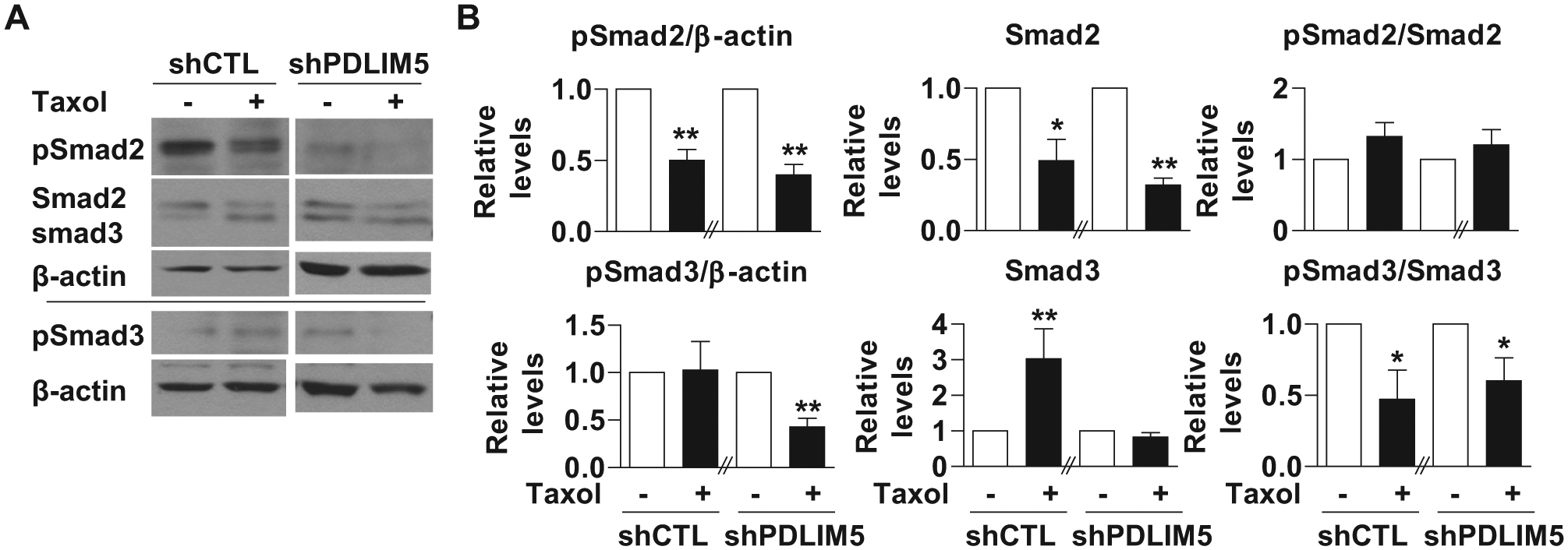
Paclitaxel-mediated inhibition of TGF-β/Smad3 signaling depends on PDLIM5 in non–small-cell lung carcinoma. A549-shCTL and A549-shPDLIM5 cells were treated with 50 nM paclitaxel (Taxol) for 2 d followed by Western blot analysis for PDLIM5, Smad2, Smad3, pSmad2, and pSmad3. Actin was used as control for equal loading. The quantification shown in panel
Discussion
In this pilot study of PDLIM5-targeted drug discovery, (1) we have discovered that PDLIM5 differentially regulates TGF-β1/Smad signaling in PASMC and alveolar epithelial cells, (2) we have established an HTS platform to screen for PDLIM5 inhibitors and performed a pilot HTS with the 1200-compound Prestwick library, and (3) we have identified and confirmed paclitaxel as a PDLIM5 inhibitor. Thus, our results indicate that PDLIM5 is a novel therapeutic target for PH, and this assay is suitable for HTS for PDLIM5 inhibitors.
Interestingly, our findings suggest that PDLIM5 has opposing effects on TGF-β/Smad signaling, depending on the cell type: in PASMC, PDLIM5 inhibits TGF-β/Smad signaling, whereas in alveolar epithelial cells, PDLIM5 promotes TGF-β/Smad signaling ( Fig. 5 ). In the context of TGF-β/Smad in human lung diseases, we postulate that PDLIM5 may be a factor that is antihypertensive yet profibrosis (TGF-β/Smad induces epithelial-mesenchymal transition [EMT] and contributes to pulmonary fibrosis). Therefore, a PDLIM5 activator is required for the treatment of PH, whereas a PDLIM5 inhibitor is required for pulmonary fibrosis. Because PDLIM5 has opposing effects on TGF-β/Smad signaling in PASMC and alveolar epithelial cells, a PDLIM5 inhibitor in alveolar epithelial cells will likely act as a PDLIM5 activator in PASMC. Thus, a PDLIM5 inhibitor indentified from this system can be used as PDLIM5 activator for the treatment of PH. Consistently, paclitaxel was reported to prevent and reverse experimental PH. 20
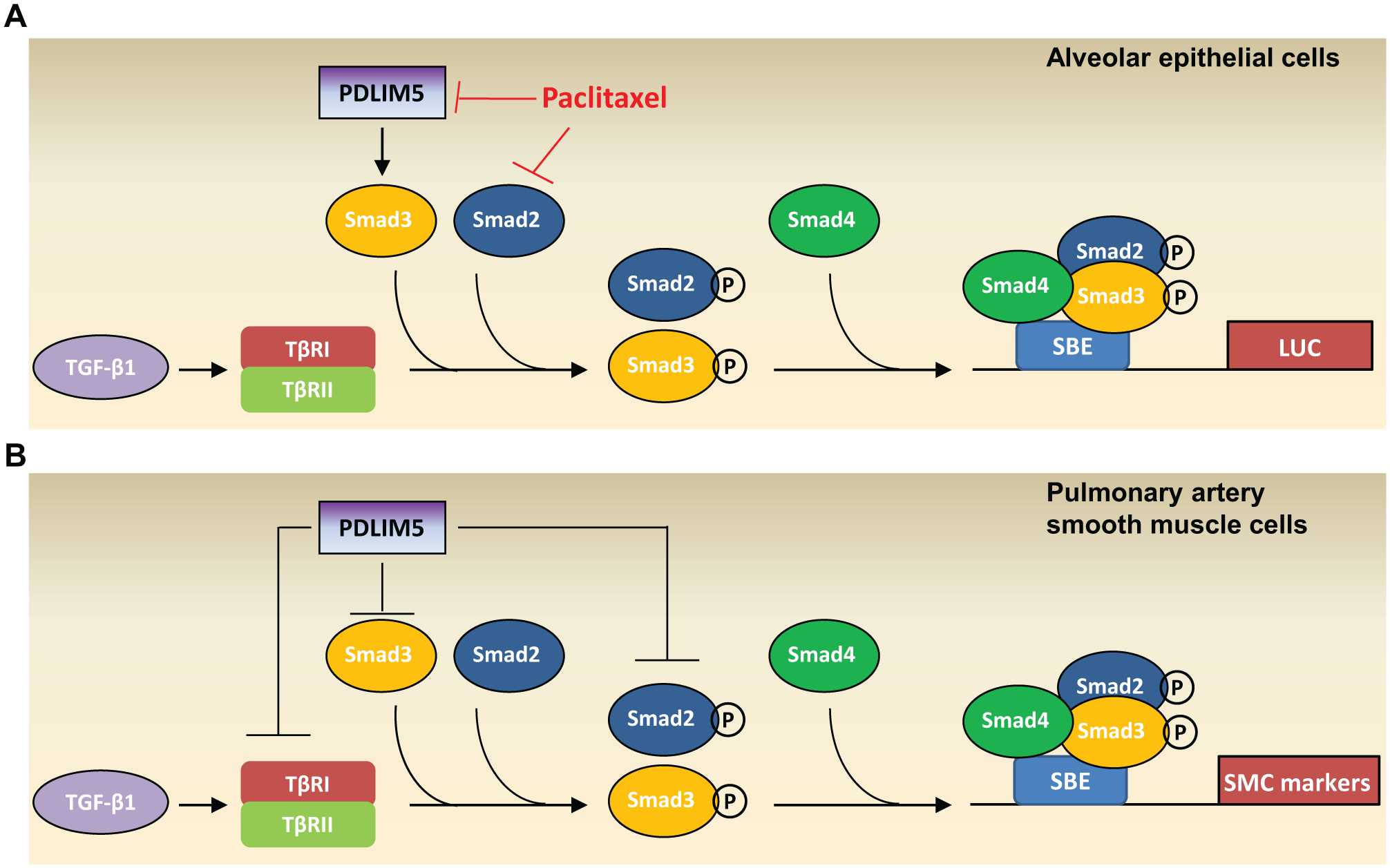
Schematic diagrams depicting differentiated PDLIM5-mediated regulation of the TGF-β/Smad signaling in different cell types. (
TGF-β is a potent and pleiotropic cytokine that participates in the pathogenesis of numerous human diseases.21,22 The pleiotropy of TGF-β has been demonstrated by its capability to produce multiple effects: canonical Smad signaling, direct signaling by type II receptor kinases to regulate tight junctions, stabilization of actin filaments, and regulation of MAPK and PI3K outputs.21,23 In cancer, TGF-β has paradoxical dual functions: tumor suppression at the early stage and tumor promotion at the later stage.21,22 In the early stage of cancer, TGF-β inhibits cell proliferation and induces apoptosis to inhibit tumorigenesis, whereas in the late stage, TGF-β promotes tumor spreading in part by inducing EMT.24–26 The dual function of TGF-β suggests that the TGF-β signaling action is highly context dependent and is influenced by spatiotemporal environments, therefore making TGF-β–targeting drug development particularly challenging.
24
Thus, the role of PDLIM5 inhibitors in lung cancer may have undesired effects. The key to this issue may lie in the fact that PDLIM5 has differential roles on Smad2 and Smad3. Consistently, we have shown that paclitaxel inhibits Smad2 expression and Smad3 phosphorylation (
Figs. 3
We found that paclitaxel does not alter expression levels of PDLIM5; rather, it inhibits the PDLIM5-mediated cell signaling ( Fig. 3 ). Therefore, we anticipate that PDLIM5 inhibitors will have limited effects on PDLIM5 protein levels but inhibit molecular signaling underlining cell proliferation, survival, and spreading. PDLIM5 is known to sequester ID2 in the cytoplasm to exert its inhibitory effects on transcription.28,29 PDLIM5 can also recruit activated PKC and its substrates to promote phosphorylation.6,8,30 This approach may minimize the adverse effect of loss of PDLIM5 on cell phenotype or function during screening.
Although suppression of PDLIM5 has little effect on cell proliferation of PASMC
5
or alveolar epithelial cells (
Fig. 1
), we have discovered that paclitaxel, a potent antiproliferative drug, is a PDLIM5 inhibitor. This may be a concern that antiproliferative hits may dominate a screen like ours. However, this seemingly undesired outcome may be explained by the difference between the IC50 and the dose we have used for our screen: the median IC50 values of paclitaxel for the NSCLC cell lines is typically higher than 23 µM or 0.38 µM for 1 or 5 d of treatment, respectively
31
; in our screening, we used 12.5 µM at 2 d of treatment (
Fig. 2
;
From our pilot screening, the majority of the compounds may not exert any effect on either TGF-β1 activity or PDLIM5 function; however, we have identified 50 compounds in the Prestwick Library that enhance PDLIM5 activity by at least twofold (data not shown). These compounds can also be valuable because PDLIM5 regulates TGF-β/Smad signaling in a tissue- or disease-specific manner and will be further investigated.
In summary, with the ultimate goal of identifying novel small molecules to treat PH, we have established a PDLIM5-targeting drug discovery platform for HTS. Future studies to screen larger libraries to expand our knowledge of druglike chemicals that target PDLIM5 signaling are warranted. Given the limited knowledge of PDLIM5 function, this system and identified probes will be very useful tools to elucidate the molecular mechanisms underlying PDLIM5 function.
Footnotes
Acknowledgements
We thank Dr. Gregory R. Thatcher, PhD, director of the UICentre, for suggestions and Miranda Sun for proofreading.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work is partly supported by an American Lung Association biomedical research grant, a Pulmonary Hypertension Association/Pfizer proof-of-concept award (in which the American Thoracic Society provides administrative support), an Inception Grant Arm 1 from UIC CCTS and UICentre, a Gilead Sciences Research Scholars Program in Pulmonary Arterial Hypertension award (to G. Zhou), and National Institutes of Health R01HL123804 (to G. Zhou).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.