
Abstract
Acid-sensing ion channel 1a (ASIC1a) is involved in several pathologies, including neurodegenerative and neuroinflammatory disorders, stroke, epilepsy, and inflammatory pain. ASIC1a has been the subject of intense drug discovery programs devoted to the development of new pharmacological tools for its modulation. However, these efforts to generate new compounds have faced the lack of an efficient screening procedure. In the past decades, improvements in screening technologies and fluorescent sensors for the study of ion channels have provided new opportunities in this field. Unfortunately, ASIC1a is mainly a Na+ permeable channel and undergoes desensitization after its activation, two features that make the use of the available screening procedures problematic. We propose here a novel screening approach for the study of ASIC1a activity in full automation. Our method is based on the stimulation of ASIC1a-expressing cells by protons and the use of electrochromic fluorescent voltage sensors as a readout of ion channel activation. This method will prove to be useful for drug screening programs aimed at ASIC1a modulation.
Introduction
Ion channels are important targets in drug discovery due to their implication in a large number of diseases. 1 Beside genetic disorders, ion channels are also implicated in neuroinflammatory and neurodegenerative diseases, such as multiple sclerosis, stroke, epilepsy, and Parkinson disease.2,3 In these pathologies, injurious conditions in the brain cause the extracellular pH to drop below physiological levels. This reduction of pH levels, which goes from 6.6 in multiple sclerosis to 6.0 in the ischemic brain,4,5 can modulate several ion channels in both neurons and glial cells. Among the pH-sensitive channels, acid-sensing ion channels (ASICs) are rapidly activated by protons and operate in the above-mentioned range of pH levels. 6 Indeed, a large body of experimental evidence suggests that the modulation of ASICs in animal models of neurodegenerative pathologies positively influences clinical outcomes and mitigates neurodegeneration.2,4,6 ASICs belong to the NaC/DEG channel superfamily that includes epithelial Na+ channels (ENaC) and degenerins (DEG). 6 They are responsible for proton-dependent Na+ and, to a less extent, Ca2+ inward currents in neurons. 7 Four ASIC genes (ASIC1–4) and two specific splice variants (a and b) for ASIC1 and 2 have been described in mammals. 8 In the mouse brain, neurons express both ASIC1a and ASIC2a at high levels, while in the human central nervous system (CNS), neurons preferentially express ASIC1a. 9 Many efforts have been made to develop new specific drugs for ASICs. However, the discovery of new molecules has been hampered by the inadequacy of the currently available screening procedures that, although compliant with high-throughput screening (HTS) requirements, are not ideal for the study of these channels that are mainly Na+ permeable and inactivate after the acidic treatment. 10 Manual and automated patch-clamp technologies are the gold standard for the investigation of ion channels. However, despite patch-clamp techniques rapidly moving toward automation, the application of these methods remains laborious, time-consuming, and expensive.
Improvements in ion channel screening procedures over the past decades have provided new opportunities in this field, developing numerous methods and technologies for HTS.11,12 Fluorescence assays using ion-specific or membrane potential sensors still remain the most commonly used methods in HTS campaigns due to the favorable combination of high automation, low costs, and information content. Several fluorescence-based optical sensors have been developed for many ionic species, but the study of Na+-permeable channels still poses problems in HTS. 13 In addition, the application of many of these sensors to HTS technologies makes the system not fast enough to be used for the study of channels undergoing inactivation. Our optical electrophysiological approach is based on the use of electrochromic voltage-sensitive dyes (Fast VSDs) for the study of ion channels and in particular ligand-gated channels. Fast VSDs are voltage-dependent fluorochromes that respond to membrane potential variations with sub-millisecond changes of their spectral properties. Despite having been extensively used in physiological studies (from cultured neurons to in vivo approaches),13,14 Fast VSDs have been rarely employed to monitor membrane potential variations as a readout of ionic fluxes in drug screening procedures (see, however, “Method for Optical Measuring Variations of Cell Membrane Conductance,” EP2457088, WO 2011009825, US 13/386225).
Here, we present an approach based on the use of a Fast VSD (di-4-ANEPPS) for the investigation of the ASIC1a channel activity that will be beneficial in speeding up the quest for novel small molecules acting on these channels.
Materials and Methods
Generation of a Stable CHO-K1 Cell Line Expressing Mouse ASIC1a
Stable CHO-K1 cells 15 expressing the mouse ASIC1a were generated by transfecting cells with 10 µg pCDNA3-ASIC1a plasmid, encoding the full-length complementary DNA (cDNA) for the murine ASIC1a gene, using Lipofectamine 2000 reagent (Thermo Fisher Scientific, Waltham, MA). Upon transfection, cells were selected by 400 µg/mL G418 Disulfate Salt Solution (Sigma-Aldrich, St. Louis, MO) and further cloned by limiting dilution. Candidate cell clones were evaluated for ASIC1a expression by real-time polymerase chain reaction (PCR). Briefly, total RNA was extracted from each clone using the RNeasy Mini Kit (Qiagen N.V., Venlo, the Netherlands) according to the manufacturer’s recommendations, including DNase (Promega Corporation, Madison, WI) digestion. cDNA synthesis was performed by using the ThermoScript RT-PCR System (Thermo Fisher Scientific) and Random Hexamer (Thermo Fisher Scientific) according to the manufacturer’s instructions in a final volume of 20 µL. The LightCycler 480 System (Roche, Basel, Switzerland) and LightCycler 480 SYBR Green I Master (Roche) were used for the detection of ASIC1a with the following primers: ASIC1 F: 5′-ATGCTGGAGTTCTACGA CAGAGC-3′, ASIC1 R: 5′-CACAGGCAAGTATTCATC TTGCTG-3′. Samples were normalized using the following Histone H3 primers: H3 F: GGTGAAGAAACCTCATCGTT ACAGGCCTGGTAC; H3 R: CTGCAAAGCACCAATA GCTGCACTCTGGAAGC. Each sample was run in triplicate, and the mean cycle threshold (Ct) of ASIC1a was normalized to the mean Ct of the housekeeping gene (Histone H3). ASIC1a-expressing cells were further assayed by Western blot analysis at different time points. Cells were scraped into homogenization buffer (10 mM Tris-HCl [pH 7.4], 260 mM Sucrose, Protease Inhibitor Cocktail, including DNase; Sigma-Aldrich) with a rubber. Protein content was quantified using the Pierce BCA Protein Assay (Thermo Fisher Scientific). Total protein extracts (20 µg) were resolved on 10% acrylamide gels and transferred onto nitrocellulose membranes before receiving the blockage with 5% milk in tris-buffered saline solution with tween 20 (TBST; 50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.05% Tween-20; Sigma-Aldrich) for 1 h at room temperature (RT). The following primary antibodies (anti-ASIC1a, 1:100 [Santa Cruz, Dallas, TX] and mouse anti–β actin 1:30,000 [Sigma-Aldrich]) were incubated overnight at 4 °C. Membranes were then rinsed in 5% milk in TBST and further incubated for 1 h with peroxidase-conjugated antibodies: goat anti-mouse (1:20,000; Bio-Rad, Hercules, CA) and goat anti-rabbit (1:5000; Bio-Rad). Signals were revealed by ECL Plus (General Electric, Fairfield, CT).
Cell Culture
CHO-K1 cells were cultured in DMEM-F12 1:1 Mix with Ultraglutamine I (Lonza Group Ltd., Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS; EuroClone S.p.A., Pero, MI, Italy), 13 mM HEPES buffer (Lonza Group Ltd.), 0.375% Sodium Bicarbonate Solution (Lonza Group Ltd.), 1 mM Sodium Pyruvate Solution (Lonza Group Ltd.), and 1% Pen/Strep Solution (Lonza Group Ltd.) at 37 °C in a humidified atmosphere with 5% CO2. ASIC1a-expressing cells were maintained in complete medium supplemented with 400 µg/mL G418. Cells were seeded at the density of 1300 cells/cm2.
Primary Neuronal Culture
Neurons were established from the cerebral cortex of C57/BL6 (Charles Rivers Laboratories International Inc., Wilmington, MA, USA) embryos at the developing embryonic age of 17.5. Brains were dissected in cold HBSS (Thermo Fisher Scientific) supplemented with 0.6% glucose (Sigma-Aldrich) and 5 mM HEPES (pH 7.4) (Sigma-Aldrich). Cerebral cortices were mechanically dissociated in single cells and resuspended in culture medium containing 50% D-MEM (Lonza Group Ltd.); 50% Ham’s-F12 (Thermo Fisher Scientific); 5 mM HEPES (pH 7.4), 0.6% glucose, 0.5% glutamine (Thermo Fisher Scientific); 30 nM Na-Selenite (Sigma-Aldrich); 20 nM progesterone (Sigma-Aldrich); 60 nM putrescine (Sigma-Aldrich); 100 µg/mL apo-transferrin (Sigma-Aldrich); 0.025 mg/mL bovine insulin (tebu-bio, Le-Perray-en-Yvelines, France); and 5% FBS (EuroClone S.p.A.) in the absence of antimitotic and antibiotic drugs. Cells were collected for total RNA extraction 13 days after plating.
Patch-Clamp Whole-Cell Recordings
For patch-clamp recordings, cells plated onto glass dishes (diameter: 8 mm) coated with poly-L-lysine (Sigma-Aldrich) were transferred to a submerged-type recording chamber perfused with pH 7.4 Krebs-Ringers HEPES (KRH; 2 mL/min) at RT. Cells were visualized using a PatchPro2000 upright microscope (Scientifica, Uckfield, UK) equipped with a 40× objective. Whole-cell patch-clamp recordings from ASIC1a-expressing CHO-K1 cells were obtained either in a voltage clamp at a holding potential of −60 mV or in a current clamp at −60 mV using a MultiClamp 700B amplifier interfaced with a PC through a Digidata 1550 (Molecular Devices, Sunnyvale, CA). The recording pipettes were filled with 145 mM K-gluconate, 6 mM NaCl, 0.2 mM EGTA, 10 mM HEPES, 1 mM MgCl2, 0.4 mM GTP-Na2, and 4 mM ATP-Mg, adjusted at pH 7.35 with KOH, and had a resistance of 4 to 6 MΩ. Signals were low-pass filtered at 10 KHz and acquired at 50 KHz. Access resistance (Ra) was in the range of 15 to 25 MΩ, constantly monitored during recordings, and compensated by 70%. To activate the ASIC1a currents, a glass pipette (tip diameter: 2–3 µm) containing pH 6.0 KRH was positioned in the vicinity (10–20 µm) of the recorded cell using a micromanipulator (PatchStars; Scientifica). Local puffs of low-pH solution (10-s duration) were delivered at minimal pressure by a Picospritzer device (Parker Hannifin, Hollis, NH) driven by TTL step commands generated by pClamp digital output signals.
Drugs and Solutions
The composition of KRH solution was 130 mM NaCl, 5 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2 mM CaCl2, and 0.1% and 0.6% glucose for HTS and patch-clamp experiments, respectively. For patch clamp, 25 mM HEPES was used. For HTS experiments, KRH with 5 mM or 40 mM HEPES was used to obtain pH 7.4 KRH and acidic KRHs, respectively. All reagents were purchased from Sigma-Aldrich. Drugs: psalmotoxin 1 (PcTx1; Peptide Institute, Osaka, Japan), diminazene aceturate (DA; Sigma-Aldrich). Dyes: di-4-ANEPPS (Sigma-Aldrich); 4-(Dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran (DCM; Exciton, Dayton, OH).
VSD Fluorescence Recordings
Parental (CHO-K1) and ASIC1a-expressing cells were seeded in 96-well black microplates (Greiner Bio-One GmbH, Kremsmünster, Austria) at 125,000 cells/cm2 density 48 h before the experiment. Confluent cells were washed three times with pH 7.4 KRH, incubated with 10 µM VSD for 10 min at RT, and then washed twice with pH 7.4 KRH. These steps were performed in automation by using a Multiwash III microplate washer (TriContinent Scientific, Grass Valley, CA). For concentration-response curves, drugs were added to pH 7.4 KRH into the wells at the end of washes to obtain a preincubation of at least 10 min before starting the experiment. Microplates were processed in full automation by using a microscope equipped with a fluidic system for the delivery of the acidic stimulus (IN Cell Analyzer 1000; General Electric). The system was equipped with a CoolSNAP HQ monochrome camera (Roper Scientific, Tucson, AZ), and an objective 20×/0.45 ELWD Plan Fluor (Nikon, Chiyoda, Tokyo, Japan). For each well, VSD fluorescence was detected using the following Chroma filter set (excitation wavelength: 535 ± 25 nm, dichroic long-wave pass: 565 nm, emission wavelength: 620 ± 30 nm), and 30 frames were acquired at 1 Hz with a 100-ms exposure time. Acidic KRH was dispensed at 50 µL/s 10 s after the start of acquisition to activate ASIC1a channels. The volume of the injected solution was previously determined to achieve the desired final pH.
Image Processing and Data Analysis
The pipeline to analyze the time-lapse movies generated by the automated microscope was developed in MATLAB (MathWorks, Natick, MA). Briefly, the movie was corrected for stage drift by applying a rigid transformation. Cell membranes were then identified by thresholding, and their average fluorescence intensity over time was computed. For each condition tested, the fluorescence intensity curves from different fields of view (FOVs) were normalized to their respective initial value and then averaged together (sample average,
Statistical Analysis
Data are expressed as the mean ± standard error of the mean (SEM) or ± standard deviation (SD) as indicated. Statistical tests were carried out using GraphPad Prism 5.01 (GraphPad Software, La Jolla, CA). Nonlinear regression curve fitting was used for concentration-response experiments. The Z factor was calculated according to Zhang et al. 16
Results
Generation and Validation of ASIC1a-Expressing Cells
We generated stable CHO-K1 cell clones expressing the murine ASIC1a gene. Real-time PCR was used to identify clones overexpressing ASIC1a, and among them, we selected the C9 clone showing an expression level that is close to the physiological levels observed in neurons ( Fig. 1A ). We further assayed ASIC1a protein expression levels in C9 cells by Western blot analysis at different time points. The level of ASIC1a protein expression was detectable up to 80 days in cultures ( Fig. 1B ).
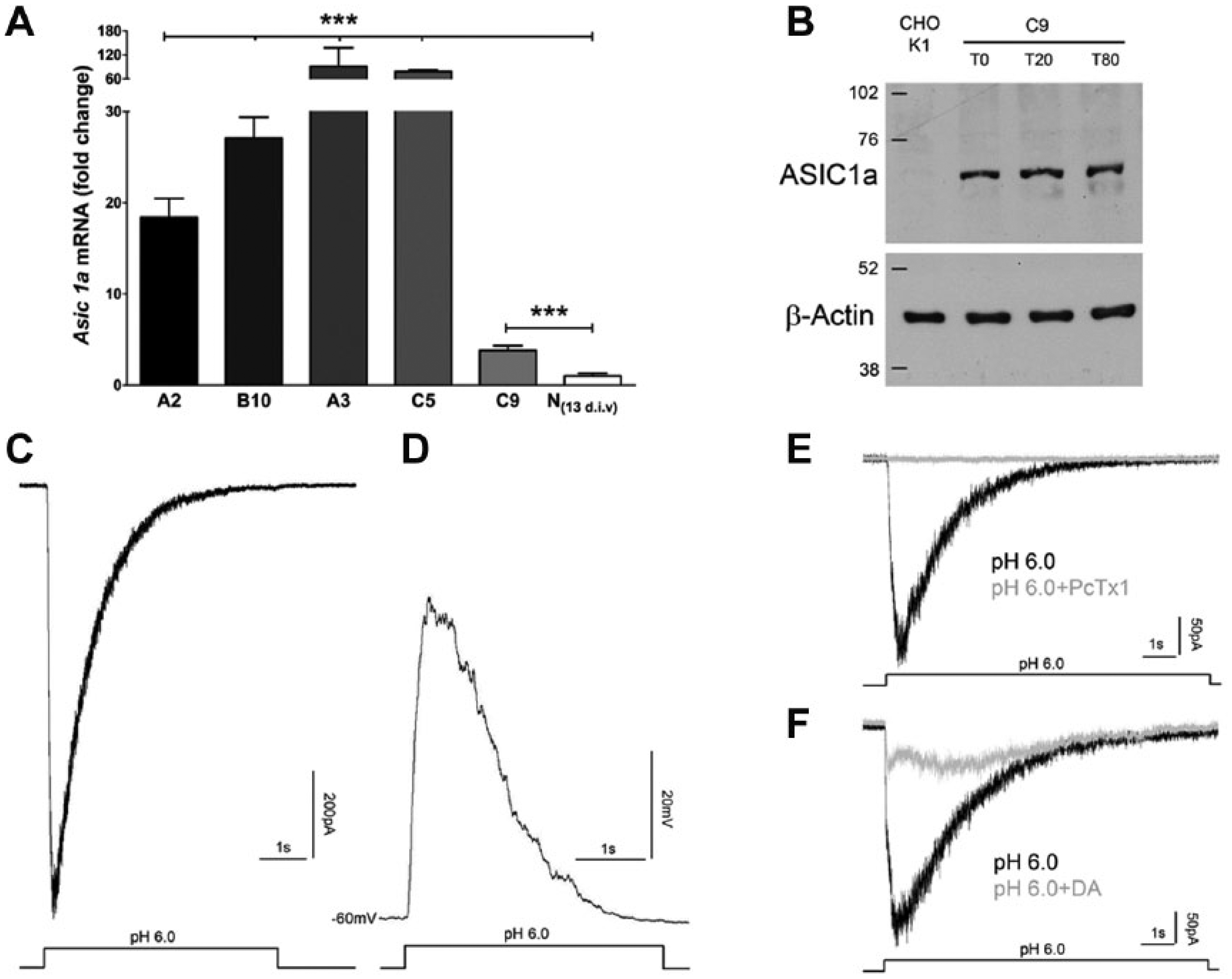
Generation and validation of the acid-sensing ion channel 1a (ASIC1a) clone. (
Beside the gene expression analysis, we used patch-clamp electrophysiology to study whole-cell currents elicited in the C9 clone by a low pH treatment to confirm their origin from ASIC1a channel opening. In these experiments, pressure ejection of an acidic extracellular saline (pH 6.0 KRH) evoked inward currents activating in a few hundred milliseconds and desensitizing in a few seconds ( Fig. 1C ). In the current-clamp configuration, the same acidic treatment resulted in a transient cell membrane depolarization (peak voltage change: 54.05 ± 15 mV, n = 5; Fig. 1D ). In contrast, the acidic puff did not evoke inward currents in parental CHO-K1 cells (data not shown). When repeated channel activation was elicited, acidic stimulation was applied at 2-min intervals to avoid rundown. 17 Previous studies showed that the tarantula venom peptide PcTx1 inhibits specifically the ASIC1a homotrimeric channels both in vitro and in vivo.6,18–20 In our electrophysiological experiments, 100 nM PcTx1 completely abolished acidic-evoked current in C9 cells 5 min after application ( Fig. 1E ), thus providing a functional confirmation of ASIC1a-mediated currents. Among small molecules able to inhibit ASICs, DA, belonging to the diarylamidines family, was reported to reduce ASIC1a currents at low micromolar concentration. 21 In accordance, a 5-min bath preincubation with 10 µM DA reduced the acid-evoked currents in C9 cells by 65.8% ± 4.8% (n = 5, p < 0.05, t test; Fig. 1F ).
Fluorescence-Based Method for the Study of ASIC1a Channels
Inward currents generated by ASIC1a activation induce changes in membrane potential ( Fig. 1D ) that can be detected by using Fast VSDs (hereafter defined as VSDs). VSDs intercalate the lipophilic components of biological membranes and shift their fluorescence spectra in response to changes of membrane potential. 14 Recording VSD fluorescence at a fixed wavelength results in a variation of intensity that is sufficiently fast to decode transient potential changes in the sub-millisecond time scale. 22
CHO-K1 parental cells (hereafter, ASIC1a–) and ASIC1a-expressing C9 cells (hereafter, ASIC1a+) were loaded with 10 µM di-4-ANEPPS (
Fig. 2A
and
2D
, respectively). We treated ASIC1a– and ASIC1a+ with an acidic solution (pH 5.0 KRH;
Fig. 2B,E
) that was injected in cells kept at pH 7.4. This procedure rapidly changed the pH level that reached pH 6.7 in approximately 1 s (
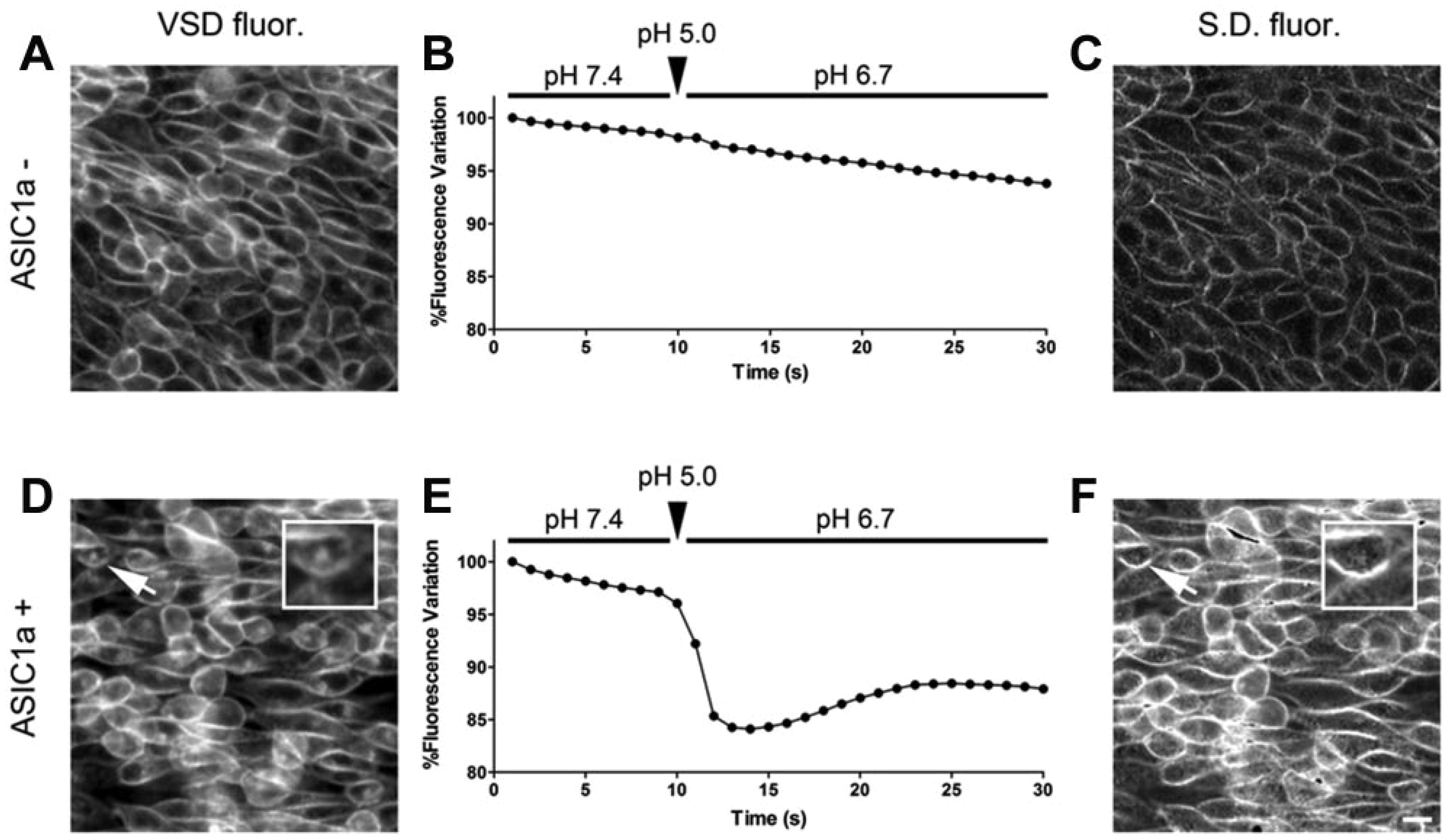
Voltage-sensitive dye (VSD) fluorescence-based method for the study of acid-sensing ion channel 1a (ASIC1a). (
Setup of the Fluorescence-Based Method in Full Automation
Having established the feasibility of the approach, we next set up the conditions for the exploitation of the assay by using an automated microscope equipped with a fluid dispensing system. Cells were seeded in 96-well plates at 125,000 cell/cm2 and kept in culture for 48 h before the experiment. By this procedure, cells could reach full confluence and recover from trypsinization, which might cleave the extracellular portion of ASIC1a channels, thereby affecting channel gating properties. Finally, since peripheral wells may have alterations in the response to test compounds (phenomenon known as the edge effect 23 ), they were not considered in the analysis to avoid misinterpretations.
Before starting the experiment, the following procedure was repeated three times: the multiwell plate was gently shaken to facilitate the removal of detached cells, and then two-thirds of the solution was substituted with pH 7.4 KRH dispensed at 150 µL/s by the 12-port manifolds of a cell-washing machine. Afterward, cells were incubated with VSD for 10 min at RT. Three additional rounds of washes were performed to eliminate the excess of staining. Finally, 100 µL pH 7.4 KRH was left in the each well before starting the experimental procedure. To properly set the pH treatment, we established a calibration curve in each experiment mixing increasing volumes of the acidic KRH (pH 5.0) to a fixed volume of pH 7.4 KRH (
A proper volume of pH 5.0 KRH (determined by a calibration curve, as described above) was automatically administered 10 s after starting the acquisition of images; pH value reached 6.7 in approximately 1 s (see
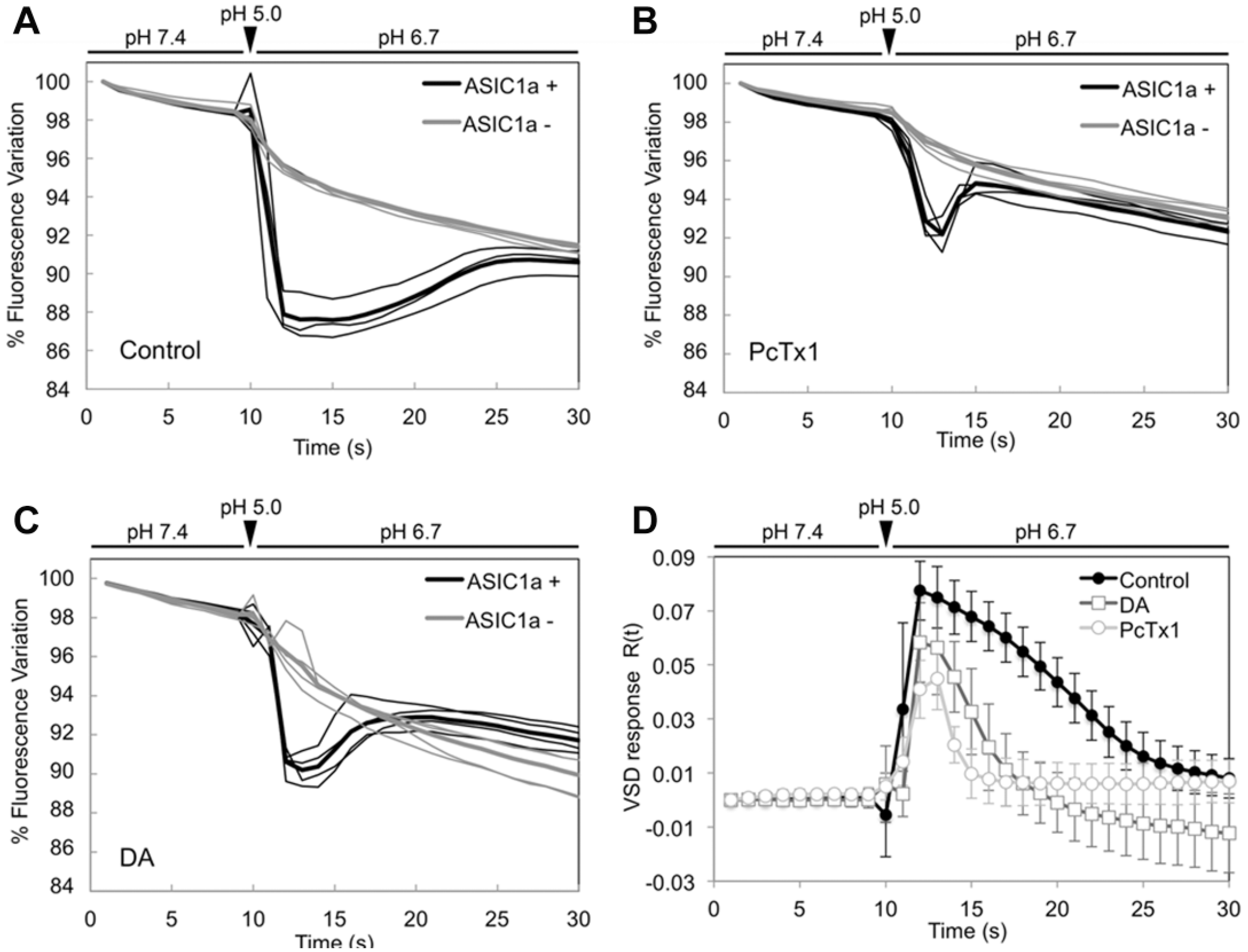
Voltage-sensitive dye (VSD) fluorescence curves for acidic stimulation in control cells and cells treated with known acid-sensing ion channel 1a (ASIC1a) inhibitors. (
The same protocol was employed in experiments in which cells had been previously treated with ASIC inhibitors. More specifically, we estimated the response of ASIC1a in the presence of 100 nM PcTx1 and 100 µM DA added 10 min before starting the experiment. As expected, the pharmacological inhibition of ASIC1a mediated by PcTx1 strongly reduced the normalized VSD fluorescence, although a residual signal was detected ( Fig. 3B,D ). Similarly, 100 µM DA substantially reduced the fluorescence intensity in ASIC1a+ cells ( Fig. 3C,D ).
Assay Validation
We validated our assay by performing concentration-response curves for both protons and the ASIC1a inhibitors PcTx1 and DA. As expected, ASIC1a activation depended on pH, showing a pH50 around 7.0, which is consistent with the data reported in literature ( Fig. 4A ).24–27
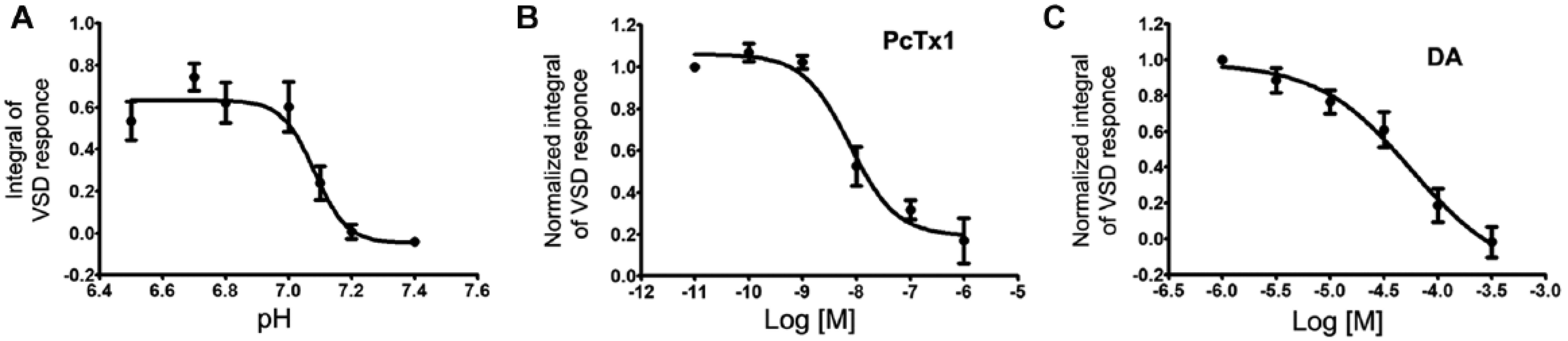
Assay validation. (
We next evaluated the effect of PcTx1 by drawing a concentration-response curve in which ASIC1a+ cells received the pH 6.7 stimulus in the presence of increasing concentrations of PcTx1. We obtained an IC50 = 7 nM, a value comparable to that reported in the literature ( Fig. 4B ).18,24 Also, the application of DA resulted in a substantial inhibition of ASIC1a activity, although the IC50 value (57 µM) was somewhat higher than previously published ( Fig. 4C ). 21
Finally, the Z factors of our approach applied to pH levels (pH 7.4 vs. pH 6.7) and PcTx1 levels (0 µM vs. 1 µM) were 0.62 and 0.47, respectively.
Altogether, these results confirm the suitability of our automated assay for the screening of candidate drugs acting on proton-evoked ASIC1a currents. The performances of our assay are the following: the experimental procedure takes 1 h per plate, implying that at least six plates can be processed and analyzed in a working day. Typically, 10 molecules at a single dose or two concentration-response curves can be extrapolated from each plate, resulting in four dose-response curves or 20 molecules at a single dose per day in triplicate.
Discussion
Brain pathological events, such as neuroinflammation or stoke, are featured by a substantial drop of the extracellular pH that can activate ASICs channels.2,4,6,20 Sustained acidosis can negatively influence large networks of neurons. 2 The importance of this phenomenon is highlighted by several studies in animal models of neurodegenerative disorders as well as in a recently published clinical trial in patients affected by progressive multiple sclerosis. 28 Thus, there is consensus on the fact that ASICs represent an interesting category of druggable channels particularly relevant in the drug discovery campaigns for several pathologies of the CNS. 1 Drug discovery for ASICs, and in general for ion channels, is still suffering for an unsatisfactory balance between throughput performances and information content, making the discovery process long and expensive. This is due to the intrinsic complexity of the ion channels physiology. Ion channel activation induces ion fluxes through the plasma membrane that transiently change the membrane potential. However, this phenomenon is intrinsically difficult to be investigated due to its small effect on the general cell physiology and its fast dynamics.
Patch clamp represents the most efficient and direct way to study ionic fluxes, but it is laborious, expensive, and time-consuming and thus not ideal for screening purposes, even with very small libraries of compounds. Automated patch clamp represents a valid solution for increasing screening performances, although it remains laborious and expensive. Alternatively, fluorescence-based platforms for ion channel screening have been developed and used for drug screening programs. Fluorescence ion detectors are efficient for calcium ions but more complex to be adopted for other ionic species (e.g., K+, Cl–, and Na+). In addition, the intracellular concentration of membrane-permeant ions is a relatively indirect reporter of ionic fluxes through ion channels.
Ionic fluxes and membrane potential variations are directly linked with a good correspondence in terms of both intensity and kinetics. 11 Fast electrochromic VSDs are able to encode ionic fluxes in a subsecond time scale, making them adequate for the study of fast operating channels. This strategy has been proficiently used for neurophysiological studies both in vitro and in vivo13,14 but never, at least to our knowledge, explored for the study of ionic fluxes for drug screening purposes. We have explored this possibility by comparing the electrophysiological and optical responses to an acidic treatment of the ASIC1a channels.
The kinetics of activation and inactivation of ASIC1a can be followed by using both classical electrophysiology and our optical method, showing similar results and thus confirming the possibility of using Fast VSDs for ion channel studies in drug screening processes. We validated our assay with selected ASIC1a specific inhibitors, and our results are in good agreement with the literature, demonstrating the specificity of our method. Moreover, the analysis of pH concentration-response curves showed a pH50 of 7.0 for ASIC1a, which is comparable, although not identical, to data reported in literature ranging from pH 6.3 and pH 6.8.24–27 We can speculate about the difference in pH sensitivity considering that our experiments are conducted in intact cells, while measures reported in literature are obtained with standard patch-clamp technology, in which the cytosol washout is usually performed. ASIC channels are intracellularly modulated by several protein kinases that play a relevant role in their modulation, including PKA and CaMKII. 29 Indeed, levels of second messengers may influence the activity of these kinases, thus changing the pH sensitivity of the channel. Similarly, our method estimates an IC50 value of 57 µM for DA while published experiments show an IC50 of 3 µM in CHO cells expressing ASIC1a. 21 However, published data, generated by standard patch clamp, may suffer from the above-mentioned cytosol washout and are usually obtained using pH 6.0 as acidic treatment instead of 6.7 as in our experiments. Moreover, it was shown recently that the efficacy and the kinetics of ASIC inhibitors are markedly dependent on pH, likely accounting, at least in part, for these differences. 30 Accordingly, also in our patch-clamp experiments, ASIC1a-expressing cells, stimulated with pH 6.0 and treated with 10 µM DA, showed a strong reduction of current peak-reproducing published data. An additional possibility accounting for the differences observed in drug effects could be found in the suboptimal speed of activation of ASIC1a induced by low pH. In fact, we reached our maximal activation in 1 s, when some channels may have already undergone inactivation. We can also mention that injecting an acidic stimulus to cells preincubated with the drugs may induce a transient displacement of the drug from the cellular microenvironment. These aspects may explain, at least in part, the small differences among the two techniques used in this study and in the literature.
An advantage of our assay is related to the method used to activate ASIC1a. The channel activation is carried out using the proton as a physiological agonist, thus avoiding the use of synthetic or natural molecules able to artificially open the channel or shifting its pH sensitivity. We have set up our acidic treatment using a final pH of 6.7, which is close to pH levels found in several pathological conditions such as multiple sclerosis or during ischemia under normoglycemic conditions.4,5
Our method has the following performances: the Z factor is 0.62 for the pH stimulation and 0.47 for PcTx1 inhibition, revealing a high quality of the assay; it enables the screening of either 20 molecules in a single dose or 4 molecules in concentration-response curves in triplicate per day.
In conclusion, we have set up an innovative method for the study of ASIC1a based on the use of protons as the physiological agonist and VSDs for the detection of channel activity. This method will prove to be useful for drug screening programs aimed at developing new ASIC1a modulators.
Footnotes
Acknowledgements
The authors deeply acknowledge Barbara Rossetti for her help in manuscript preparation and people from the Alembic Facility for technical assistance. CHO-K1 cells were a generous gift of AXXAM S.p.A.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grant FISM (grant number 2014/R18). Dr. Nausicaa Mazzocchi was supported by a fellowship from FONDAZIONE IBSA for Scientific Research (Call 2014).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.