
Abstract
Friedreich ataxia (FRDA) is an autosomal recessive neuro- and cardio-degenerative disorder for which there are no proven effective treatments. FRDA is caused by decreased expression and/or function of the protein frataxin. Frataxin chaperones iron in the mitochondrial matrix and regulates the iron–sulfur cluster (ISC) assembly complex. ISCs are prosthetic groups critical for the function of the Krebs cycle and the mitochondrial electron transport chain. Decreased expression of frataxin is associated with decreased ISC assembly, mitochondrial iron accumulation, and increased oxidative stress, all of which contribute to mitochondrial dysfunction. In media with beta-hydroxybutyrate (BHB) as carbon source, primary FRDA fibroblasts grow poorly and/or lose viability over several days. We screened a random, short-hairpin-RNA (shRNA)-expressing library in primary FRDA fibroblasts and identified two shRNAs that reverse the growth/viability defect in BHB media. One of these two clones increases frataxin expression in primary FRDA fibroblasts, either as a vector-expressed shRNA or as a transfected short-interfering RNA (siRNA).
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive neuro- and cardio-degenerative disorder, with a prevalence of approximately 1 in 40,000 in European populations. (Recent reviews include those by Koeppen and Mazurkiewicz, 1 Collins, 2 and Gomes and Santos. 3 ) FRDA is characterized by progressive ataxia of all four limbs, dysarthria, areflexia, sensory loss, and muscle fatiguability. Skeletal deformities and cardiomyopathy are found in most patients, impaired glucose tolerance and diabetes mellitus are found in ~30% of patients, and reduced visual acuity and hearing loss are occasionally seen. Onset of symptoms usually occurs around puberty, and most patients are confined to a wheelchair by their late 20s. Myocardial failure and/or arrhythmias are the most common causes of premature death.
FRDA is caused by mutations in the nuclear gene, FXN, which encodes the highly conserved protein frataxin. Most disease alleles harbor a GAA repeat expansion in the first intron, which results in decreased transcription. Frataxin localizes primarily to the mitochondrial matrix, where it chaperones iron and regulates the iron–sulfur cluster (ISC) assembly complex. ISCs are prosthetic groups that are important for the function of many proteins, both mitochondrial and cytosolic, including aconitase and mitochondrial respiratory complexes I, II, and III. Decreased frataxin function in yeast models, mouse models, and humans with FRDA is associated with mitochondrial iron accumulation, mitochondrial dysfunction, and increased oxidative stress.
We described previously the design, construction, and validation of the first short-hairpin-loop-RNA (shRNA)-expressing library that is completely random at the nucleotide level. 4 This library, which comprises approximately 300,000 random shRNA-expressing clones, allows unbiased identification of small-RNA therapeutics and biologic tools by screening for phenotype. Herein, we describe the results of screening our library in primary FRDA fibroblasts. We took advantage of the particular sensitivity of these cells to beta-hydroxybutyrate (BHB)-based media to identify shRNAs that reverse this sensitivity. One of the shRNA sequences we identified increases frataxin expression, either as a vector-expressed shRNA or as a transfected siRNA, thereby partially reversing the primary defect in FRDA.
Materials and Methods
Cells and Cell Culture
We obtained primary FRDA fibroblasts and age- and sex-matched control fibroblasts from Coriell (Coriell Institute for Medical Research, Camden, NJ). We previously confirmed that the FRDA fibroblasts have homozygous GAA repeat expansions in the first intron of the FRDA gene using long-range PCR: 5 GM3816 cells have moderate GAA repeat expansions of 223 and 490 repeats, GM4078 cells have moderate GAA repeat expansions of 357 and 553 repeats, and GM3665B cells have larger GAA repeat expansions of 790 and 1357 repeats. The Dulbecco’s modified Eagle’s medium (DMEM) used for cell culture was either from Sigma-Aldrich (D5030; Sigma-Aldrich, St. Louis, MO) supplemented with uridine (50 µg/ml), stabilized glutamine (G-MAX), and sodium bicarbonate (110 mg/L), or from Life Technologies (11966-025; Life Technologies, Carlsbad, CA). Both formulations are glucose free. To this base medium, either glucose or DL-b-hydroxy-butyrate (215011000; Acros Organic, Geel, Belgium) was added at 5 mM final concentration. During cell expansion, the medium used was low-glucose DMEM (Life Technologies 11885-076). All media were supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT) and 1% penicillin/streptomycin. The human myoblast cell line (NBT) 6 was grown in Ham’s F10 medium supplemented with 15% FBS, 1% penicillin/streptomycin, sodium pyruvate (100 µg/ml), uridine (50 µg/ml), and creatine monohydrate (1 mM). DL156 primary FRDA fibroblasts (repeat expansions of 500 and 570 repeats) were a kind gift of Dr. David Lynch of the Children’s Hospital of Philadelphia. Dr. Lynch obtained the cells from one of the patients in his FRDA study, patients who were recruited using the Friedreich Ataxia Research Alliance database, by listing on Clinicaltrials.gov (NCT01965327), and through the practice of Dr. Lynch. All subjects in his study provided written informed consent prior to participation, and they were provided with a modest stipend for each visit.
Library Preparation and Introduction
Our random, shRNA-expressing library comprises approximately 300,000 random 29-mer sequences, separated from the reverse complement of each sequence by a noncomplementary loop-encoding sequence, cloned in the green, fluorescent-protein (GFP)-expressing vector, pSIREN (Clontech, Mountain View, CA). The details of the library construction and validation have been described. 4 To infect human cells, the library, or single clones identified by the screen, were packaged using amphitropic retroviral packaging plasmids. 7 FRDA fibroblasts were infected in 6-well plates at 100,000 cells/well density. The retroviral supernatants were added, and the cells were spun at room temperature for 90 min at 1000 g in the presence of 5 mg/ml polybrene (Sigma-Aldrich). After 3 h, the cells were transferred to an appropriately sized flask. Infection efficiency was monitored by GFP expression on a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA); ideally, the GFP% was kept at ~30% or less such that, by Poisson distribution, the majority of the infected cells received only one construct. At the end of the screening process, genomic DNA of the remaining cells was extracted using the QIAamp Mini DNA kit (Qiagen, Venlo, the Netherlands). The shRNA-expressing cassette was amplified by PCR using appropriate primers, restriction-digested, and cloned back into the pSIREN vector; primer sequences and procedural details have been described previously. 4
siRNA Transfection
RNA oligonucleotides were from IDT DNA (IDT, Newark, NJ). As suggested by the manufacturer, the passenger (sense) strand was synthesized as a 25-mer ending with two deoxy-ribose bases (AT), whereas the guide strand (antisense) was synthesized as a 27-mer with two uridines at the 3′ end [Dicer substrate RNA interference (RNAi) design; IDT]; thus, the final double-stranded RNA (dsRNA) was designed to have one blunt end and one asymmetric 3′ overhang (the two uridines). The synthesized oligos were resuspended in water and mixed in annealing buffer (100 mM potassium acetate, 2 mM magnesium acetate, 30 mM Hepes pH 7.4). The solution was heated at 90 °C for 1 min and kept at 37 °C for 1 h to favor small interfering RNA (siRNA) double-strand annealing. Cy3-oligo derivatives were prepared by using the Silencer siRNA labeling kit (Ambion, Naugatuck, CT) and used to check for transfection efficiencies. Double-strand siRNAs were transfected by using RNAi-max reagent (Life Technologies) at a final concentration of 10 nM as per the manufacturer’s instructions. Cells were transfected every 3–4 days for up to 14 days.
Quantification of Cell Growth and Viability
Cell growth was quantified by counting cells using the Countess Counter (Life Technologies). Cell viability was assessed by chemiluminescence measurement of intracellular ATP using the CellTiter-Glo assay kit (Promega, Madison, WI) as per the manufacturer’s instructions. The effects of hit shRNAs and siRNAs were compared to those of a control selected at random from the library (after construction but prior to screening, which is equivalent to a randomly selected sequence from a pool of random sequences made in an oligonucleotide synthesizer).
Isolation and Deep Sequencing of Guide Strands
GM3816 cells were infected with shRNA clones as described above and sorted for GFP-positive cells. To co-immunoprecipitate the Argonaute protein and Argonaute-associated guide strands, we used the anti-Argonaute monoclonal antibody 2A8 (a kind gift of Dr. Zissimos Mourelatos, University of Pennsylvania). Briefly, cells were lysed in RBS buffer (20 mM Tris-HCl pH7.4, 200 mM sodium chloride, 2.5 mM magnesium chloride, 0.5% IGEPAL CA-630, 0.1% Triton X-100) supplemented with ethylenediaminetetraacetic acid (EDTA)-free protease inhibitors (Roche, Basel, Switzerland) and RNAsin (N211A; Promega, Madison, WI), and sonicated briefly a few times. After centrifugation, the clear lysate was incubated for 2 h at 4 °C in the presence of the antibody adsorbed on protein G (15920-010; Invitrogen, Carlsbad, CA). Beads were washed five times with RBS buffer, and the RNA was extracted using 500 µl of Trizol (Ambion) as per the manufacturer’s instructions. The co-precipitated RNAs were then ligated to adaptors, reverse transcribed, and amplified by PCR (FC-102-1009; Illumina, San Diego, CA). A band of the appropriate length (~93 bp) was isolated after electrophoresis on a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel, purified, and sent for deep sequencing at the sequencing core facility of the University of Pennsylvania. The sequence list was trimmed of the adaptors’ sequences and sorted alphabetically, and the sequence of the clone was searched for.
Quantification of Frataxin Expression
Total RNA was extracted using the RNAeasy kit (Qiagen), including an on-column DNAase treatment. 0.5 µg of total RNA was retro-transcribed using the QuantiTect Reverse Transcription kit (Qiagen). Levels of frataxin were assessed by quantitative reverse transcriptase PCR (qRT-PCR) using a Taqman assay and b-actin or GAPDH as the reference gene. Primers and reagents were from Applied Biosystems (Waltham, MA). Samples were run in triplicate and analyzed using the Delta Delta C(T) method (2-ΔΔCt).
Statistical Analyses
Unless otherwise indicated, two-sided Student’s t-tests were used to compare means.
Results
Sensitivity of FRDA Cells to BHB
Given the known sensitivity of cells with mitochondrial dysfunction to media with BHB in place of glucose, 8 we compared the growth of primary FRDA fibroblasts (GM3816) to age- and sex-matched normal control fibroblasts (GM8400) in both glucose- and BHB-based medium, keeping the cells sub-confluent. Whereas the growth of normal control fibroblasts in BHB-based medium was slower than in glucose-based medium ( Fig. 1A ), the growth of FRDA fibroblasts in BHB-based medium slowed nearly to a halt after approximately 7 days ( Fig. 1B ). Similar results were obtained with additional primary FRDA fibroblasts (GM4078) and age- and sex-matched normal control fibroblasts (GM8402; data not shown). When the cells were plated in BHB-based medium at a density that allowed growth to confluence, the normal control fibroblasts grew faster and reached confluence earlier, after which they underwent confluence-associated growth arrest and began to lose viability; despite the fact that the FRDA fibroblasts reached confluence later, their confluence-associated loss of viability was so rapid that the decline in viable cells overtook that of the normal control fibroblasts ( Fig. 1C ). Primary FRDA fibroblasts with large GAA repeat expansions (GM3665B) grew very slowly even in glucose-based medium; in BHB-based medium, these cells stopped growing after 18 days and then lost viability, even when kept sub-confluent ( Fig. 1D ).
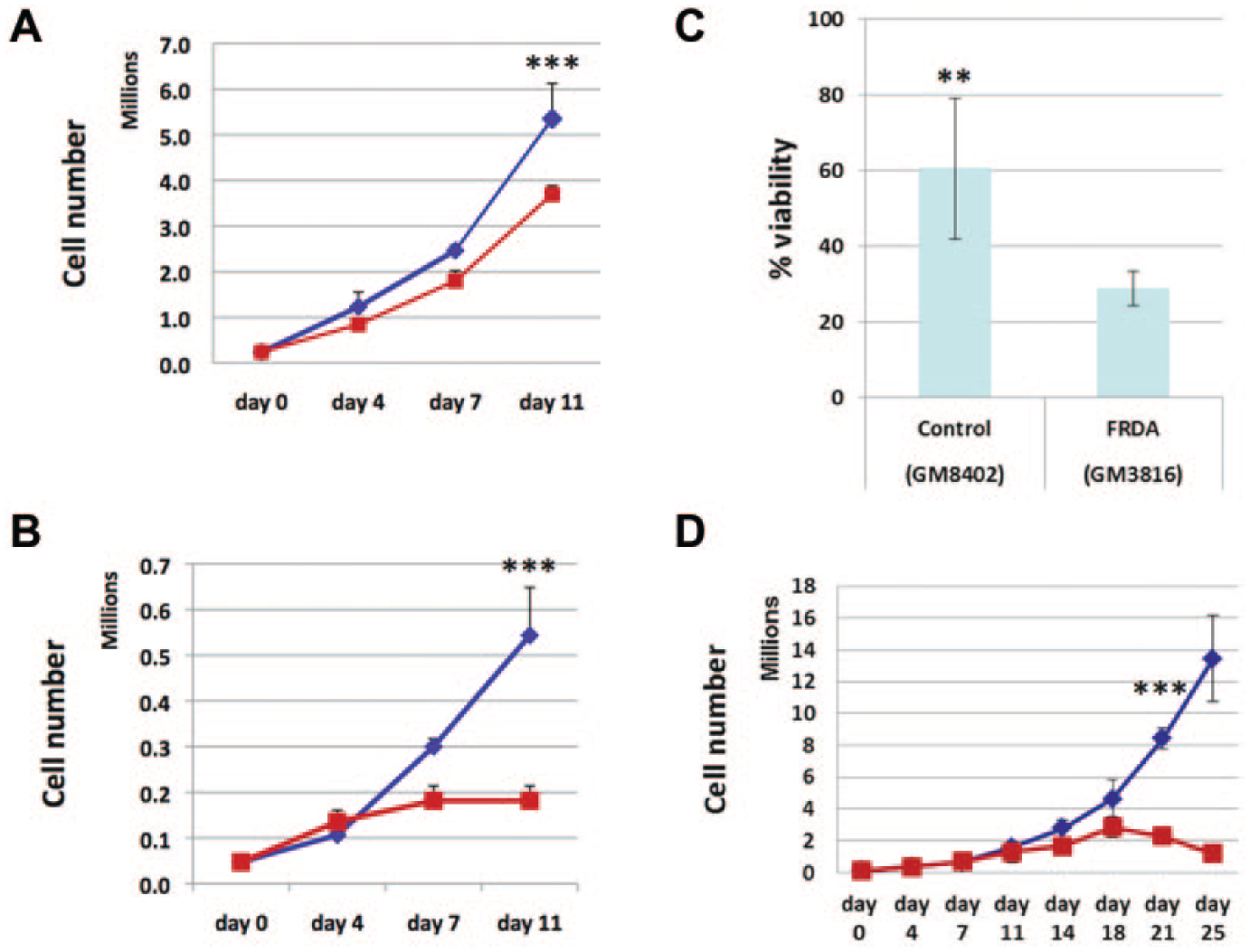
Increased sensitivity of Friedreich ataxia (FRDA) cells to beta-hydroxybutyrate (BHB). (
Library Screening
Taking advantage of the growth/viability defect of FRDA cells in BHB-based medium, and the specificity of this phenotype for mitochondrial dysfunction, we screened our 300,000-clone, random shRNA-expressing library for shRNAs that reverse this phenotype in primary FRDA fibroblasts in BHB-based medium. Our strategy is shown schematically in Figure 2 . Briefly, after packaging the library as retroviruses (Materials and Methods), we infected approximately 3 million GM3816 primary FRDA fibroblasts, aiming for 30% infection so that the majority of the cells received only one construct. We allowed the cells to recover from infection for 4 days, during which time they expanded to approximately 5 million cells. We then shifted the cells to BHB-based medium for 2 weeks, during which time ~99% of the cells lost viability. To enrich for true positives, we re-expanded the cells for 2 weeks in glucose-based medium (to ~2.4 million cells) and again shifted the cells to BHB-based medium for 2 weeks, during which time ~80–85% of the cells lost viability.
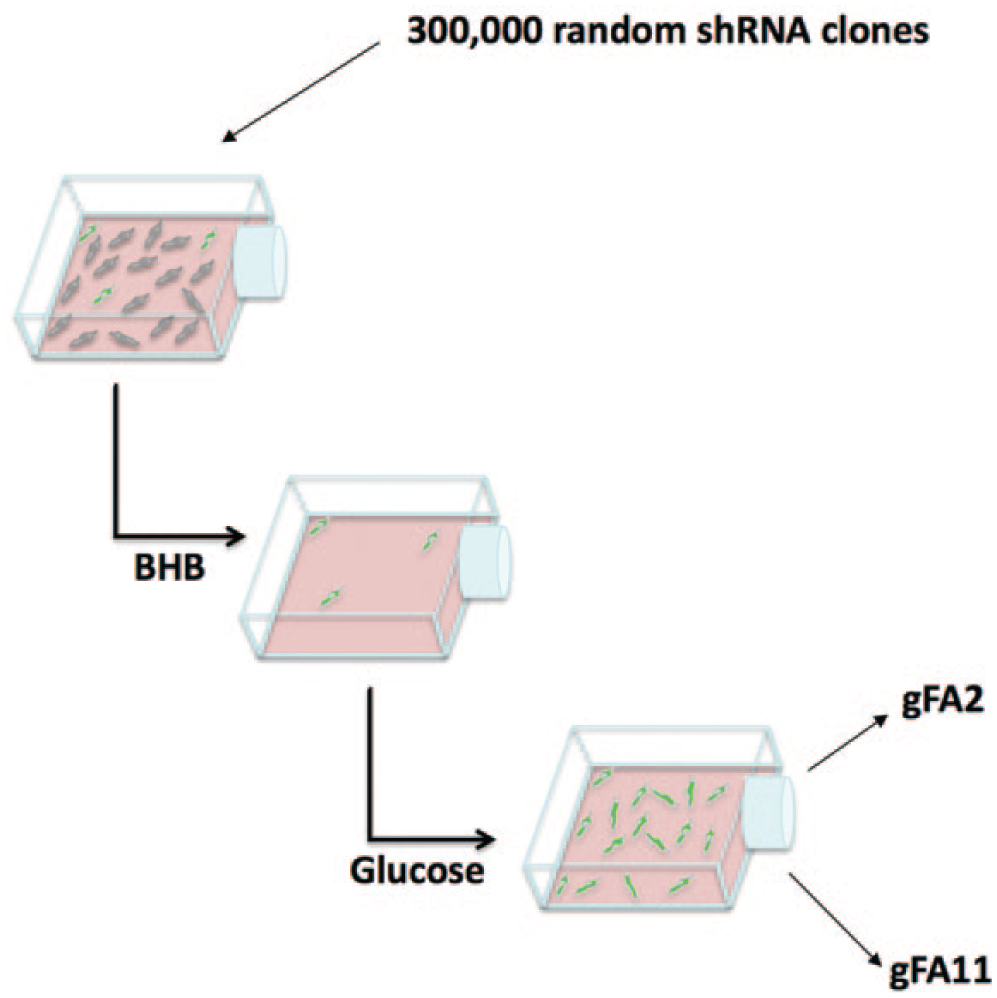
Library screening. We infected primary Friedreich ataxia (FRDA) fibroblasts (Coriell GM3816) with a 300,000-clone, random shRNA library. Four days post infection, the cells were switched to Dulbecco’s modified Eagle’s medium (DMEM) containing 5 mM BHB. After 2 weeks, only 1% of the cells survived. The cells were then moved into DMEM with glucose for 2 weeks. The entire cycle was repeated two more times. Finally, we extracted genomic DNA from the selected cells, amplified the shRNA-expressing cassette by PCR, and cloned the amplicons back into the parental vector. Two clones were highly represented and chosen for further analysis: gFA2 and gFA11.
To further enrich for true positives, we re-expanded the cells in glucose-based medium (to ~1 million cells), which took 5 weeks, and again shifted the cells to BHB-based medium, this time for 4 weeks (to increase the stringency of selection), during which time ~99% of the cells lost viability. We then expanded the cells in glucose-based medium for 5 weeks, at which point the cells were nearing the end of their lifespan. The cells were then collected, genomic DNA was extracted, and shRNA-encoding sequences were retrieved and cloned back into the parent vector (Materials and Methods). Sixteen clones were sequenced, and two were chosen for further study because they were repeated (suggesting enrichment): clone gFA2 (repeated four times) and clone gFA11 (repeated three times).
Hit shRNA Confirmation and Characterization
To confirm that clones gFA2 and gFA11 encode true-positive hit shRNAs, we packaged each as a retrovirus, infected primary FRDA fibroblasts with large GAA repeat expansions (GM3665B), flow-sorted for GFP-positive cells, and followed growth in BHB-based medium. Both clone gFA2 and clone gFA11 enhanced growth significantly compared to a control random clone in BHB-based medium (
Fig. 3A
), as well as in nonstringent, glucose-based medium (
Fig. 3B
). Because the primary defect in most FRDA cells is decreased transcription of the FXN gene, we used qRT-PCR to determine whether either clone gFA2 or clone gFA11 increases FXN messenger RNA (mRNA), packaging each as a retrovirus, infecting primary FRDA fibroblasts with moderate GAA repeat expansions (GM3816), and growing the cells in BHB-based medium. Clone gFA2 increased FXN mRNA approximately 1.7-fold as a vector-expressed shRNA compared to a control random clone (
Fig. 4A
), whereas clone gFA11 did not increase FXN mRNA significantly (data not shown). The increase in FXN mRNA associated with clone gFA2 was independent of the endogenous control used (
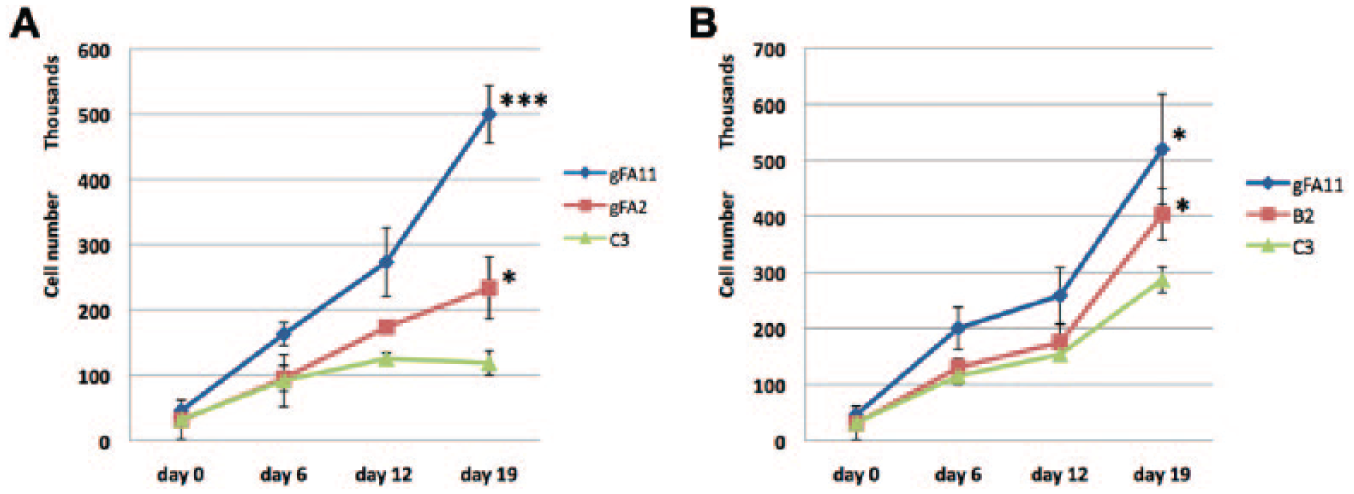
Hit short hairpin RNAs (shRNAs) enhance growth of Friedreich ataxia (FRDA) cells in beta-hydroxybutyrate (BHB)-based medium. (
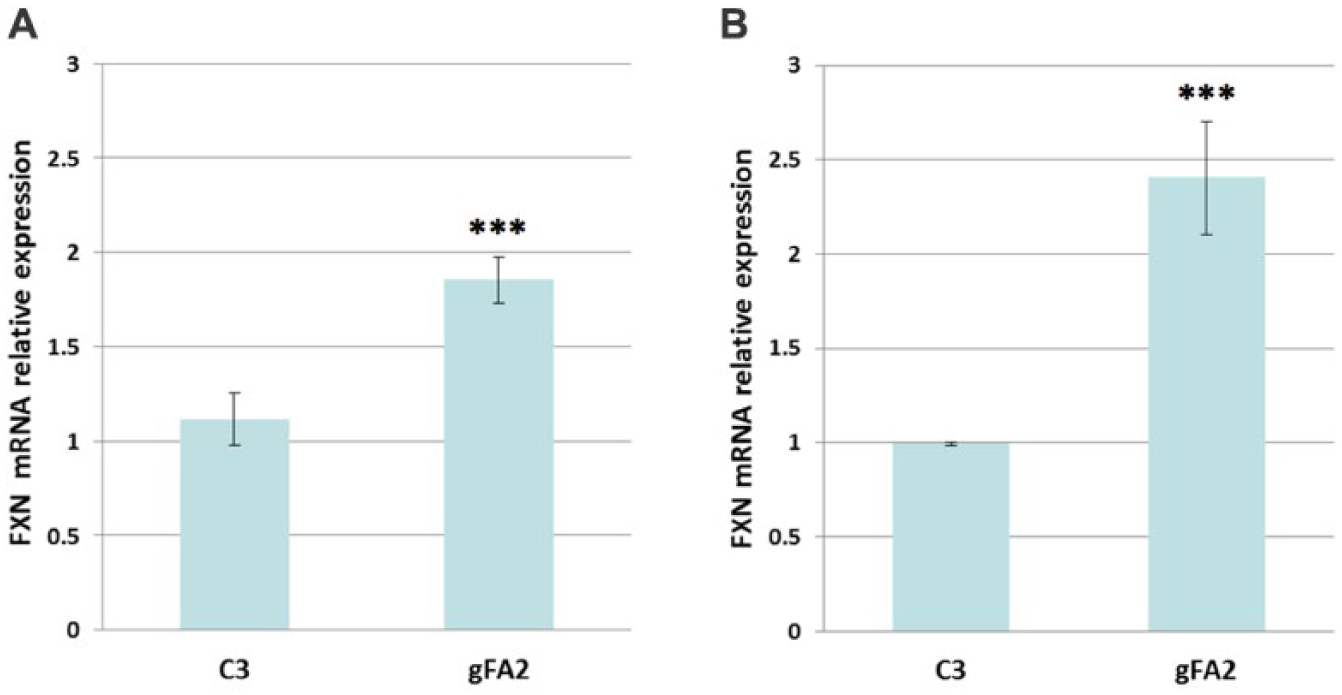
Clone gFA2 increases FXN mRNA. (
Small-RNA therapeutics are generally based on siRNAs rather than shRNAs: siRNAs are processed by the same cellular machinery as mature shRNAs and can be introduced into cells by transfection. To test the efficacy of the gFA2 sequence as a transfected siRNA (and also to confirm that gFA2 is processed through the canonical RNAi pathway), we first determined the intracellular guide-strand usage of gFA2 shRNA. As described in Materials and Methods, we used an antibody that recognizes all four human and mouse Argonaute proteins, and co-immunoprecipitates microRNAs (miRNAs).
9
The Argonaute-associated guide strands isolated from the GM3816 cells previously infected with gFA2 were sequenced in their entirety; approximately 56% of gFA2-derived sequences were from one strand of gFA2, and 44% from the other. The association of these sequences with the Argonaute protein suggested that they were responsible for the biological activity of clone gFA2. Accordingly, we designed the gFA2 siRNA shown in
Figure 5
(which also included commonly used strategies to increase potency and stability). We used this siRNA to transfect primary FRDA fibroblasts with large GAA repeat expansions (GM3665B) in BHB-based medium and used qRT-PCR to measure the effect on FXN mRNA; clone gFA2 increased FXN mRNA significantly as a transfected siRNA compared to a control random siRNA, with FXN mRNA levels increasing approximately 2.4-fold (
Fig. 4B
). In primary FRDA fibroblasts grown in glucose-based medium, transfected gFA2 increased FXN mRNA more modestly (1.2-fold;
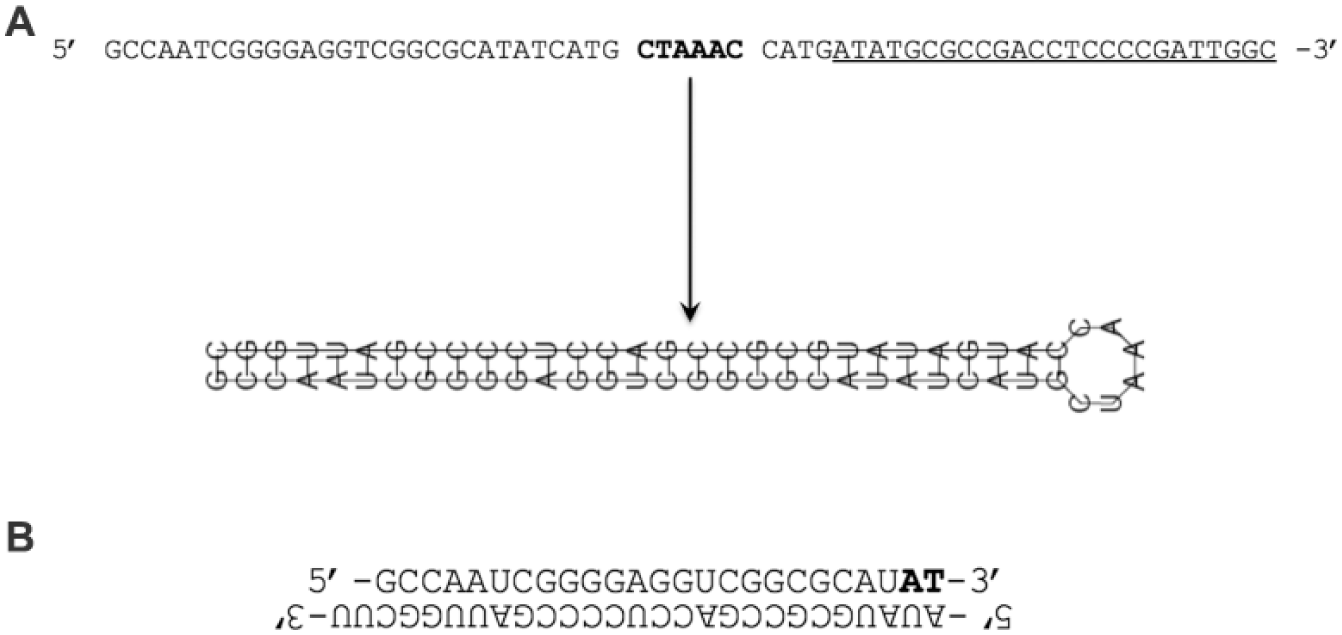
Schematic showing the gFA2 short hairpin RNA (shRNA) sequence and the gFA2 small interfering (siRNA) construct used for the gFA2 experiments depicted in
Discussion
The existing paradigm for the use of RNAi in the development of small-RNA therapeutics is to interfere with the expression of a single gene using a shRNA or siRNA. However, the most potent endogenous miRNAs, on which shRNAs and siRNAs are modeled, target hundreds of mRNAs simultaneously through ~7-nucleotide “seed-sequence” matches, which are sufficient to interfere, at least partially, with expression. A given 7-nucleotide sequence should occur, on average, every 16,384 nucleotides (four to the seventh power), which is ~366,000 times in the human genome (of ~3×109 base pairs). Unsurprisingly, therefore, small-RNA therapeutic initiatives based on targeting single mRNAs are complicated by off-target effects, which diminish therapeutic indices. In addition, the ability to target sets of genes is precisely what makes miRNAs so bioactive, and single-gene targeting fails to take advantage of this.
To complement the single-gene-targeting paradigm, we designed and synthesized the first shRNA-expressing libraries that are completely random at the nucleotide level.4,10 Using a pooled approach, we can screen hundreds of thousands, 4 or millions, 10 of shRNAs in a single tissue-culture dish, selecting for a beneficial phenotype and retrieving “hit” shRNA-encoding sequences by PCR. In effect, we have miniaturized high-throughput screening such that each “drug” is tested in a single cell. Because our libraries are completely random, our screens are unbiased: we let the cells tell us which shRNAs are most effective and least toxic. Because of the size of our libraries, all possible seed sequences are covered many times over, all but guaranteeing hit sequences. Our approach bypasses the problem of off-target effects and harnesses the power of multigene targeting. Many cell-culture disease models unsuitable for microtiter-plate formats are amenable to our approach.
Herein, we describe the successful application of our approach in a disease model. Specifically, we screened our 300,000-clone, random, shRNA-expressing library in a disease model of the inherited neuro- and cardio-degenerative disorder Friedreich ataxia. Most disease alleles in FRDA harbor a GAA repeat expansion in the first intron of the FXN gene, thereby decreasing expression of the protein frataxin, a protein critical in the assembly of iron–sulfur clusters in the mitochondrial matrix. We took advantage of the sensitivity of primary FRDA fibroblasts to beta-hydroxybutyrate (BHB)-based media, and the specificity of this sensitivity for mitochondrial dysfunction, to identify shRNAs that reverse this sensitivity. To confirm enrichment for true positives, we generally monitor the increasing percentage of cells with the library marker (GFP);4,10 autofluorescence of primary FRDA fibroblasts precluded the use of GFP for precise quantification of enrichment, so we inferred enrichment by the stringency, and the number of rounds, of selection. One of the shRNA sequences that we identified increases frataxin expression, either as a vector-expressed shRNA or as a transfected siRNA. The increase in frataxin expression was seen in glucose-based, as well as BHB-based, medium and did not depend on the presence of a GAA repeat expansion, as the increase was seen in normal control cells. This partial reversal of the primary defect in FRDA validates our phenotypic approach.
A limitation of our approach is that, because the targets of our hit shRNAs are gene sets, parsing the mechanisms underlying beneficial phenotypes is unlikely to be straightforward. We are currently using bioinformatic analyses to try to understand better how clone gFA2 increases frataxin expression, and how clone gFA11, which does not increase frataxin expression, reverses the growth defect of FRDA cells in BHB-based media. Clones that work as both transfected siRNAs and vector-expressed shRNAs should more amenable to such analyses because they more likely act through canonical RNAi. Another limitation is that, although siRNAs have been delivered in vivo successfully to hepatocytes and endothelial cells, delivery to most cell types in vivo remains problematic. Nevertheless, because our screens last days to weeks, shRNAs that are even slightly toxic are culled out (i.e., we screen simultaneously for efficacy and a lack of toxicity). As evidenced by the results described herein, our approach has the potential to identify shRNAs and siRNAs with favorable therapeutic indices in vivo as delivery problems to target tissues are increasingly solved.
Footnotes
Acknowledgements
We thank Dr. Zissimos Mourelatos for helpful discussions and for his generous gift of 2A8 antibodies. We thank Dr. David Lynch for the primary, DL156 FRDA fibroblasts.
Abbreviations
BHB beta-hydroxybutyrate
FRDA Friedreich ataxia
GAA guanine adenine adenine
GAPDH glyceraldehyde 3-phosphate dehydrogenase
GFP green fluorescence protein
ISC iron–sulfur cluster
qRT-PCR quantitative reverse transcriptase polymerase chain reaction
RNAi RNA interference
shRNA short-hairpin-loop-RNA
siRNA short-interfering-RNA
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Friedreich’s Ataxia Research Alliance, the Hamilton and Finneran families, the National Institute of General Medical Sciences (R01 GM090304), and NetScientific.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.