
Abstract
Genome-wide association studies have linked polymorphisms in the gene G72 to schizophrenia risk in several human populations. Although controversial, biochemical experiments have suggested that the mechanistic link of G72 to schizophrenia is due to the G72 protein product, pLG72, exerting a regulatory effect on human D-amino acid oxidase (hDAAO) activity. In an effort to identify hDAAO inhibitors of novel mechanism of action, we designed a pLG72-directed hDAAO activity assay suitable for high-throughput screening (HTS). During assay development, we confirmed that pLG72 was an inhibitor of hDAAO. Thus, our assay employed an IC20 pLG72 concentration that was high enough to allow dynamic pLG72-hDAAO complexes to form but with sufficient remaining hDAAO activity to measure during an HTS. After conducting an approximately 150,000-compound HTS, we further characterized a class of compound hits that were less potent hDAAO inhibitors when pLG72 was present. Focusing primarily on compound
Introduction
Human D-amino acid oxidase (hDAAO) is a flavoenzyme expressed in liver, kidney, and brain. 1 Using flavin adenine dinucleotide (FAD) as a cofactor, hDAAO stereospecifically catalyzes the degradation of D-amino acids. 1 Several D-amino acids, most notably D-serine, are full agonists at the glycine site of the NMDA receptor (NMDAR), 2 a key excitatory receptor that is critical for several physiological processes such as cognition, learning, and brain structural plasticity. 3 Alterations in NMDAR activation states are proposed to underlie various pathological conditions, such as schizophrenia, neuropathic pain, and stroke. 3 By controlling the levels of the NMDAR coagonist D-serine, DAAO has been shown to be important for regulating cognition and learning. 4 Because DAAO inhibitors have activity in rodent assays relevant to disease states such as cognitive impairment associated with schizophrenia, 4 multiple industrial and academic groups have sought development of hDAAO inhibitors for potential utility in treating central nervous system (CNS) disorders.
One prominent hDAAO binding partner potentially relevant for schizophrenia is pLG72, the largest protein product of the G72 gene. 5 More than 10 years ago, an influential genome-wide association study (GWAS) identified G72 as an anthropoid-specific gene linked to schizophrenia; this study further presented yeast two-hybrid and biochemical data demonstrating that pLG72 bound to and regulated hDAAO activity. 5 Because of prior evidence linking hDAAO pathways to schizophrenia, the G72 genetic link to schizophrenia had an immediate mechanistic explanation. Following that initial publication, GWAS studies and meta-studies in several populations confirmed that G72 polymorphisms influence risk for schizophrenia and perhaps other psychiatric diseases. 6
Despite genetic evidence linking the G72 gene and the pLG72 protein to hDAAO function and psychiatric disease, many mysteries remain regarding the precise function of pLG72 and its relationship to hDAAO. The most relevant issue concerns the biochemical outcome of pLG72 association with hDAAO. While the original report provided evidence that pLG72 activated hDAAO, 5 subsequent publications presented careful data demonstrating hDAAO inhibition by pLG72.7,8 Although a potential model synthesizing these competing observations is that pLG72 activates hDAAO over short periods of time but then initiates a slow, minutes-long conversion of hDAAO to an inactive conformation, 7 this controversy has yet to be convincingly settled. Furthermore, pLG72 protein is expressed weakly in whole-brain homogenates, perhaps because it is expressed only in specific brain regions such as the cerebral cortex 7 and amygdala. 9 Although the original publication detected pLG72 messenger RNA (mRNA) in brain, spinal cord, and testes, 5 there are reports of negative results in which the pLG72 mRNA and protein have not been detected in brain. 10 Finally, experiments in cell lines and in transgenic mice suggest the pLG72 gene primarily affects mitochondrial function and has psychiatric effects independent of pLG72 association with hDAAO.9,11
We initiated an effort to (1) generally identify hDAAO inhibitors of novel mechanism of action, (2) develop tool compounds to potentially explore hypotheses surrounding the function of the pLG72-hDAAO interaction, and (3) design compounds with potential utility for treating schizophrenia by targeting the pool of pLG72-hDAAO in the cerebral cortex. To accomplish these ambitious goals, we designed a pLG72-directed hDAAO activity assay (hDAAO/pLG72 assay) to screen for compounds that inhibited hDAAO when pLG72 was present. In our hands, pLG72 was a dose-dependent inhibitor of hDAAO. Therefore, we designed our assay with an IC20 concentration of pLG72. Under these conditions, we hypothesized that pLG72 would be present at a high enough concentration to interact dynamically with hDAAO but not at a concentration high enough to fully inactivate the enzyme. In these balanced conditions, sufficient hDAAO activity would remain to be assayed in a high-throughput screen (HTS). During hit confirmation, we identified several classes of compounds that differentially inhibited hDAAO depending on the presence of pLG72. Because of their structural homogeneity, availability from commercial sources, and rapid opportunity for exploring structure-activity relationships, we describe here the characteristics of one class that were less active as hDAAO inhibitors when pLG72 was present. As we show here, these “class C compounds” were not active site hDAAO inhibitors but instead bound covalently to hDAAO cysteines to inhibit hDAAO activity. Although these compounds may not be effective as in vivo tool or therapeutic compounds (goals 2 and 3 above), our results confirm that our HTS strategy was capable of identifying hDAAO inhibitors of novel biochemical mechanism of action (goal 1 above).
Materials and Methods
Compound Synthesis and Protein Production
Compound
Summary of Enzymatic Inhibition Data.
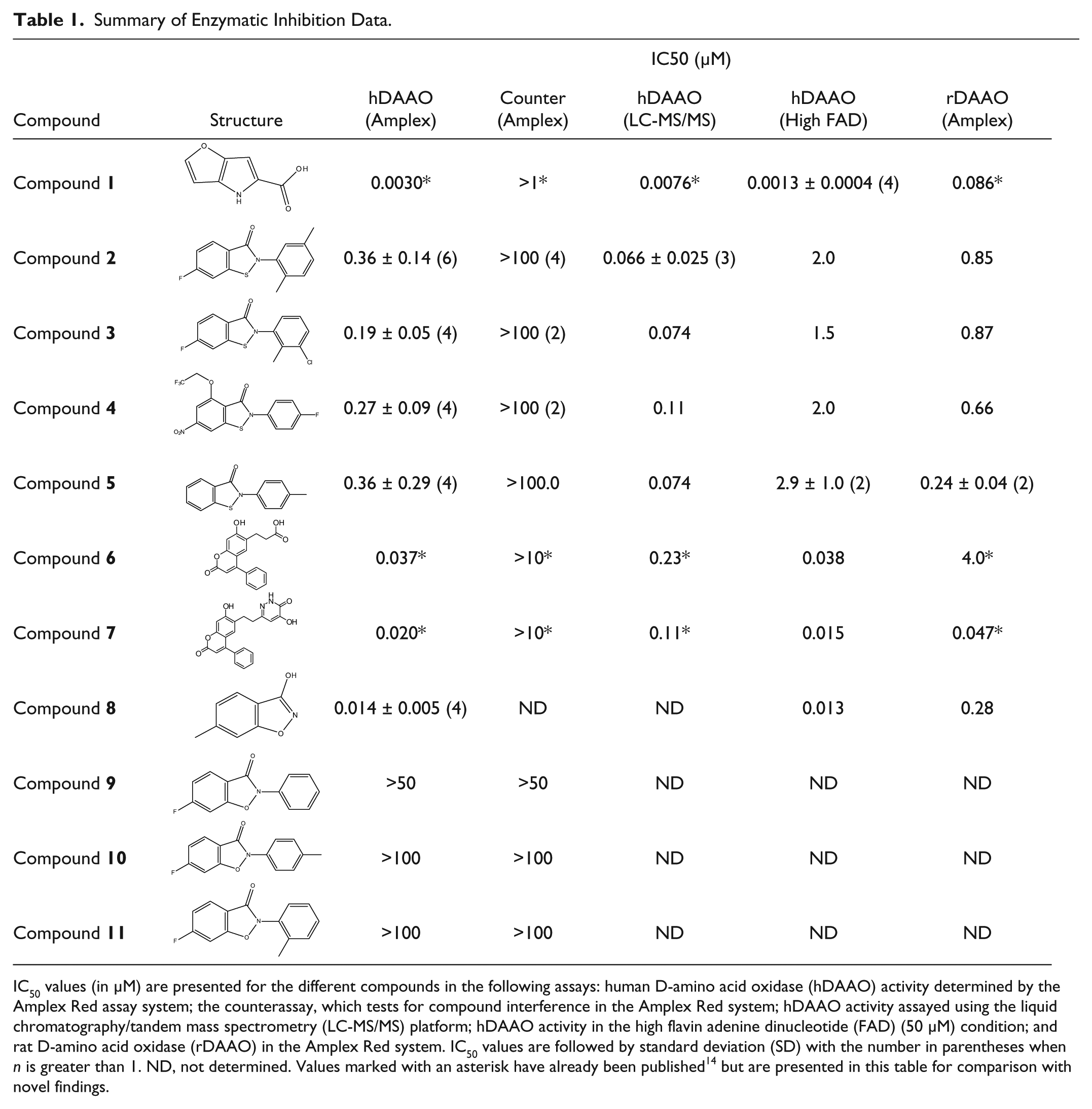
IC50 values (in µM) are presented for the different compounds in the following assays: human D-amino acid oxidase (hDAAO) activity determined by the Amplex Red assay system; the counterassay, which tests for compound interference in the Amplex Red system; hDAAO activity assayed using the liquid chromatography/tandem mass spectrometry (LC-MS/MS) platform; hDAAO activity in the high flavin adenine dinucleotide (FAD) (50 µM) condition; and rat D-amino acid oxidase (rDAAO) in the Amplex Red system. IC50 values are followed by standard deviation (SD) with the number in parentheses when n is greater than 1. ND, not determined. Values marked with an asterisk have already been published 14 but are presented in this table for comparison with novel findings.
hDAAO protein (NP_001908.3, 347 amino acids, 40 kDa) was expressed using an N-terminal 6-His-tag in BL21(DE3) Escherichia coli cells and purified by elution on a nickel column followed by size exclusion chromatography. 17 pLG72 was prepared as reported in Molla et al. 18 and was stored in 20 mM Tris-HCl (pH 8.5), 100 mM NaCl, 5% glycerol, 5 mM 2-mercaptoethanol (βME), and 0.1% N-lauroylsarcosine (NLS). Rat DAAO (rDAAO) was purified and stored as previously described. 19 Glucose oxidase protein was purchased from Life Technologies (Carlsbad, CA; Amplex Red Glucose/Glucose Oxidase Assay Kit). Bovine D-aspartate oxidase (DASPO) was purchased from Gabrielle Tedeschi (University of Milan, Milan, Italy).20,21
HTS
All enzymatic assays were conducted at room temperature (20–23 °C). For all concentration-response curves, data were analyzed by Prism (GraphPad Software, La Jolla, CA) using a standard, four-parameter equation:
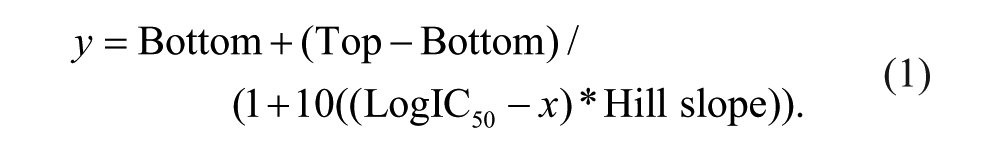
In the primary screen, approximately 150,000 compounds were screened in the hDAAO/pLG72 assay. To eliminate nonspecific compounds, all compounds in the primary HTS were counterscreened for inhibition of the flavoenzyme glucose oxidase (GO assay) in parallel. The GO assay was chosen for counterscreen because GO is a well-characterized flavoprotein for which an easy-to-use kit exists for enzyme assay (Life Technologies). The general assay format was a coupling assay in which hydrogen peroxide produced by hDAAO or GO enzymatic activities was used by horseradish peroxidase (HRP; Sigma-Aldrich, St. Louis, MO) to oxidize Amplex Red (Life Technologies) and convert it into resorufin. The quantity of resorufin was then determined by a fluorescence plate reader.
More specifically, test compounds (10-mM stock solution in DMSO; Sigma-Aldrich) were diluted in the assay buffer (50 mM phosphate buffer, 150 mM NaCl, and 0.06% human serum albumin [Sigma Aldrich], pH 7.4) and added to the assay plate (low-volume 384-well plate; Corning, Corning, NY) using a CyBi-Well pipettor (CyBio, Jena, Germany) at 6 µL/well for a final concentration of 10 µM. In the hDAAO/pLG72 assay, the 5× enzyme mixture containing hDAAO, pLG72, FAD (Sigma Aldrich), and NLS in assay buffer and the 5× start solution containing HRP, Amplex Red, and D-serine in assay buffer were each added at 2 µL/well using FlexDrop PLUS (PerkinElmer, Waltham, MA). The final concentrations of each reagent were as follows: 6.25 nM hDAAO, 0.7 µM pLG72, 4 µM FAD, 0.005% NLS, 0.07 U/mL HRP, 0.035 mM Amplex Red, and 7.5 mM D-serine. In the GO assay, a similar procedure was used in which enzyme and start solution were both added as 5× master mixes. The final reagent concentrations in the GO assay were as follows: 1.25 mU/mL GO, 0.07 U/mL HRP, 0.035 mM Amplex Red, and 40 mM D-(+)-glucose. The assay plates were incubated for 1 h, and the fluorescence of resorufin was measured using an EnVision plate reader (PerkinElmer) with 535 nm excitation and 590 nm emission. In all experiments, hDAAO and pLG72 were incubated for a minimum of 30 min prior to activity assessment.
Primary hits were defined as compounds with >50% inhibition of hDAAO in the presence of pLG72, <20% inhibition of GO, and ≥50% purity of compound (as assessed by standard liquid chromatography/mass spectrometry analysis). As control, the IC50 of compound
The reproducibility and concentration dependency of primary hits were evaluated in the hDAAO/pLG72 assay, the GO assay, and the assay of hDAAO activity in the absence of pLG72 (hDAAO solo). Confirmed hits were defined as compounds that inhibited hDAAO/pLG72 with an IC50 <10 µM and were negative for GO inhibition (IC50 >10 µM).
Additional Enzymatic Assays
Enzymatic assays other than the HTS and hit confirmation activities described above were performed in 96-well black-walled plates (Corning Costar, Corning, NY). DMSO was used as solvent for compound solutions, except where specifically indicated. In all experiments, DMSO vehicle concentration was carefully maintained such that all controls and experimental groups had precisely the same DMSO concentration. No [DMSO]f exceeded 1.5%.
The hDAAO and rDAAO activity assays in the Amplex Red platform were performed as described previously 14 using roughly the same coupled assay format as described above for the HTS. The hDAAO reaction was allowed to incubate for 1 h, and accumulated fluorescent resorufin reaction product was measured with a FlexStation II (Molecular Devices, Sunnyvale, CA). While the final FAD in this standard protocol was 450 nM, for the “high FAD” condition, 50 µM FAD was used. In the counterassay, hDAAO was omitted and replaced with 800 nM hydrogen peroxide (Sigma-Aldrich) to produce the fluorescent product. For assaying rDAAO, the hDAAO protocol was followed with the following alteration: rDAAO was used instead of hDAAO, and D-serine concentration was increased to 30 mM.
DASPO activity and inhibition by test compounds also was assayed using the Amplex Red format. The assay protocol was the same as described above with the following exceptions: 0.36 nM DASPO was used as enzyme, 1.6 mM D-aspartate (Sigma-Aldrich) was used as substrate, and the reaction was allowed to proceed for 30 min prior to measurement of fluorescent resorufin product. The positive control inhibitor was potassium sodium tartrate solution (Sigma-Aldrich), which was diluted in aqueous buffer solution.
For an assay of hDAAO activity using an alternative detection method, liquid chromatography/tandem mass spectrometry (LC-MS/MS) was used to directly measure the benzoylformic acid product of the hDAAO reaction, when D-phenylglycine was used as substrate. This assay was described in greater detail previously. 14
For determining hDAAO inhibition in the presence of reducing agents, the LC-MS/MS platform was used, as the Amplex Red platform was sensitive to high amounts of reducing agents. In these experiments, the standard LC-MS/MS–based activity assay was conducted, except that 5 mM glutathione (Sigma-Aldrich) or 5 mM βME (Pierce, Rockford, IL) was added to buffers as indicated in Results and figure legends.
As described previously, 14 saturation experiments were used to determine compound mechanism of action with respect to substrate (D-serine) and cofactor (FAD). In the Amplex Red system, D-serine was maintained at a constant concentration, while the cofactor was varied over seven different concentrations or vice versa. These saturation curves were repeated in the presence of varying amounts of inhibitor. After a 1-h reaction, product was measured in endpoint mode using the FlexStation II. To determine Vmax,obs and KM,obs values at each inhibitor concentration, data of enzymatic activity (y) at given substrate concentrations (x) were plotted and fit with a one-site, specific binding equation (GraphPad Prism):
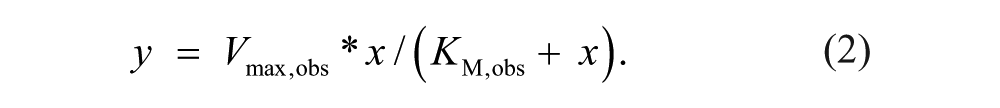
Several experimental designs were used to estimate rate of compound dissociation from hDAAO (off-rate). In each case, the assay used the Amplex Red assay platform described above. In the jump-dilution protocol, high concentrations of inhibitor were incubated with hDAAO and FAD for 30 to 90 min to form inhibited complexes. Reaction mixture (Amplex Red, D-serine, HRP) was then added, and fluorescent reaction product was measured kinetically using the FlexStation II. Examination of concentration-response curves was used such that at the “high” concentration, hDAAO was 80% to 100% inhibited, and at the “low” concentration, hDAAO was 0% to 20% inhibited. Utilization of this assay for hDAAO inhibitor quantitative off-rate determination has been described previously. 14 Accumulated product formed (P) at a given time (t) is fit with equation (3) to determine initial reaction velocity (vi), steady-state reaction velocity (vs), and off-rate (k):
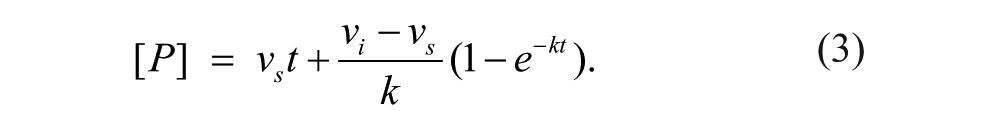
An alternative assay for off-rate determination with longer time scale was developed. Here, 200 nM hDAAO was incubated for 30 min with 4 µM inhibitor in 0.24 µM FAD. Samples then were diluted 200-fold, such that 1 nM hDAAO and 20 nM inhibitor were present. In this dilution, the FAD concentration was 61.5 µM. After a time of recovery that could last up to 24 h to allow for compound dissociation, 130 µL of diluted mixture was added to 70 µL of the Amplex Red reaction mix for hDAAO activity determination. The final [D-serine] was 50 mM, and the final [FAD] was 40 µΜ. Activity was assessed over 10 min kinetically on the FlexStation II, and activity was defined by the slope of the linear progress curve of product formation. For a given time point, all results were normalized to activity of DMSO-treated hDAAO. A 20-nM inhibitor condition was used to ensure that this concentration was not inhibitory.
For determination of compound reversibility in the presence of βME, 2 µM hDAAO was incubated with 20 µM inhibitor for 30 min. hDAAO inhibitor mixes were diluted 2-fold into 5 mM βME. After variable time periods, a small aliquot was diluted 1000-fold into Amplex Red reaction volume. With this dilution to 5 µM, βME did not interfere with the Amplex Red fluorescent detection system. [D-serine]f was 50 mM, [FAD]f was 40 µΜ, [compound]f was 10 nM, and [hDAAO]f was 1 nM. Activity was assessed over 10 min kinetically on the FlexStation II. Activity was defined by the slope of the linear progress curve of product formation, and results were normalized to hDAAO plus DMSO controls.
LC-MS/MS Analytical Methods
Detection of degradation products from the reaction mixtures of compounds
LC-MS/MS Proteomic Methods
Covalent binding of compound
Product ion data were searched against the combined forward and reverse custom protein database comprising the hDAAO sequence using a locally stored copy of the Mascot search engine v2.3 (Matrix Science, London, UK) via Mascot Daemon v2.3. Peak lists were generated using Proteome Discoverer v1.4 (ThermoFisher). The database was appended with common background proteins. Search parameters were precursor mass tolerance 10 ppm, product ion mass tolerance 0.6 Da, two missed cleavages allowed (trypsin digest only), no fixed modifications, variable modifications of oxidized methionine, and deamidation on asparagine and glutamine and compound
Results
pLG72-Directed hDAAO Activity Assay HTS
The principle on which the proposed assay is based is that pLG72 binds to hDAAO,5,7,8 alters hDAAO structure in a time-dependent manner,7,22 and regulates hDAAO activity. This regulation has been reported as activation5,23 by some researchers and inhibition by others.7,8 In our hands, following a 30-min preincubation protocol and assaying hDAAO reaction product over the course of 60 min, pLG72 inhibited hDAAO with an IC50 of approximately 0.9 µM (
As the subset of hDAAO protein relevant for schizophrenia could potentially be found in complex with pLG72,
7
we sought to screen compounds capable of inhibiting hDAAO when pLG72 was present. We screened for hDAAO inhibition in the presence of 700 nM pLG72, which was an approximate IC20 pLG72 concentration (
In this hDAAO/pLG72 assay, we screened approximately 150,000 compounds from the Sumitomo Dainippon Pharma small-molecule corporate library. In the primary screen, compounds were screened in the hDAAO/pLG72 assay and counterscreened for inhibition of the flavoenzyme GO. Compound hits (>50% inhibition of hDAAO/pLG72 with <20% inhibition of GO) were subjected to purity analysis. Compounds with >50% purity were tested in concentration-response experiments in the two screening assays (hDAAO/pLG72 and GO assays) plus assay of hDAAO activity in the absence of pLG72 (hDAAO solo assay). This workflow is described in Figure 1A . Confirmed hits were defined as compounds that inhibited hDAAO in the presence of pLG72 with an IC50 <10 µM that were negative for GO inhibition (>10 µM IC50). Among the 102 confirmed hits from this screening workflow, we separated hDAAO inhibitors into three categories based on their pLG72 dependency: class A compounds were more potent in the presence of pLG72, class B compounds (the largest class) were equipotent inhibitors of hDAAO in the absence or presence of pLG72, and class C compounds were less potent inhibitors of hDAAO when pLG72 was present ( Fig. 1B ).
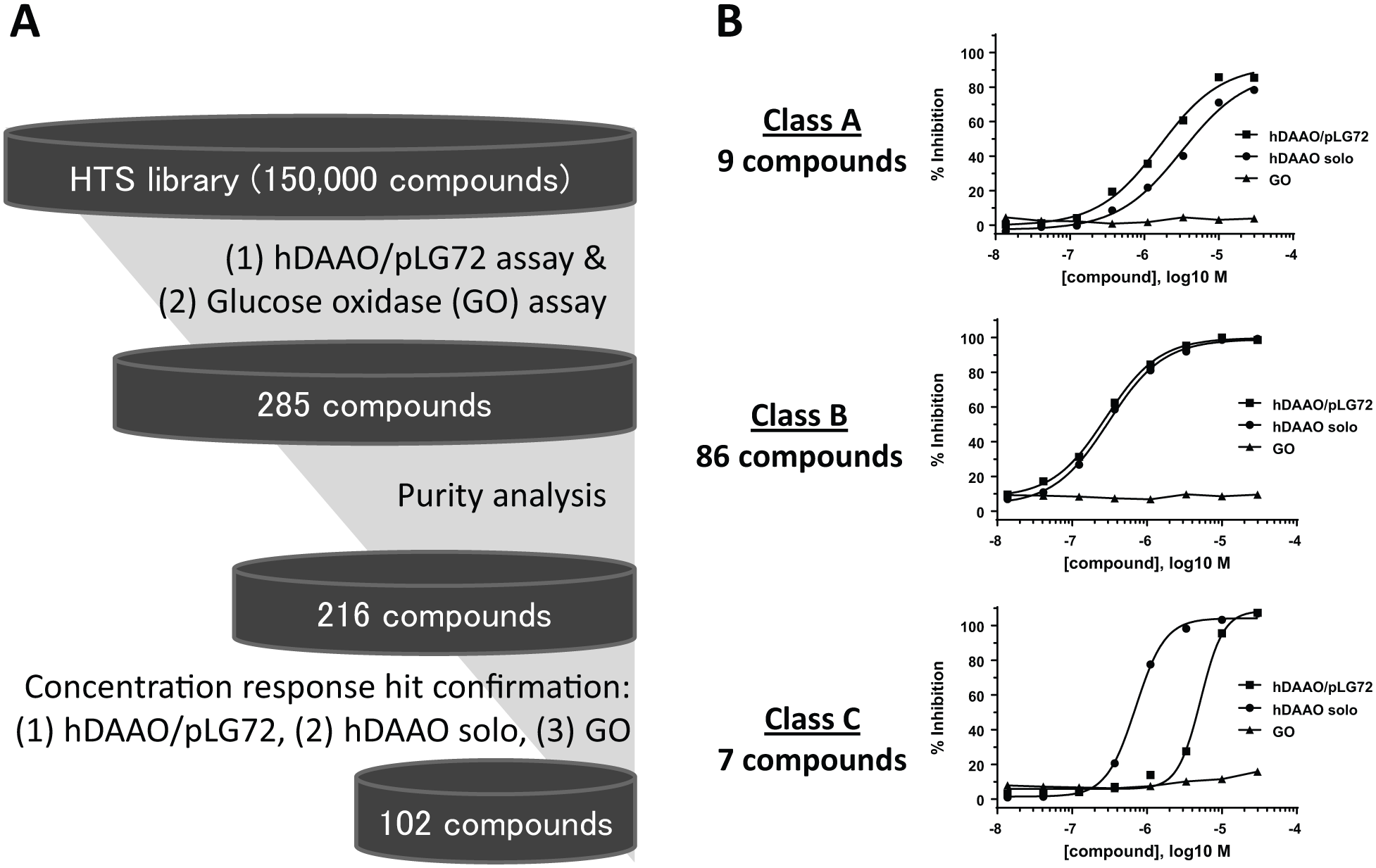
Results from the high-throughput screen (HTS). (
Biochemical Characterization of Class C Compound hDAAO inhibitors
Because the active-site hDAAO inhibitor compound
Using compound
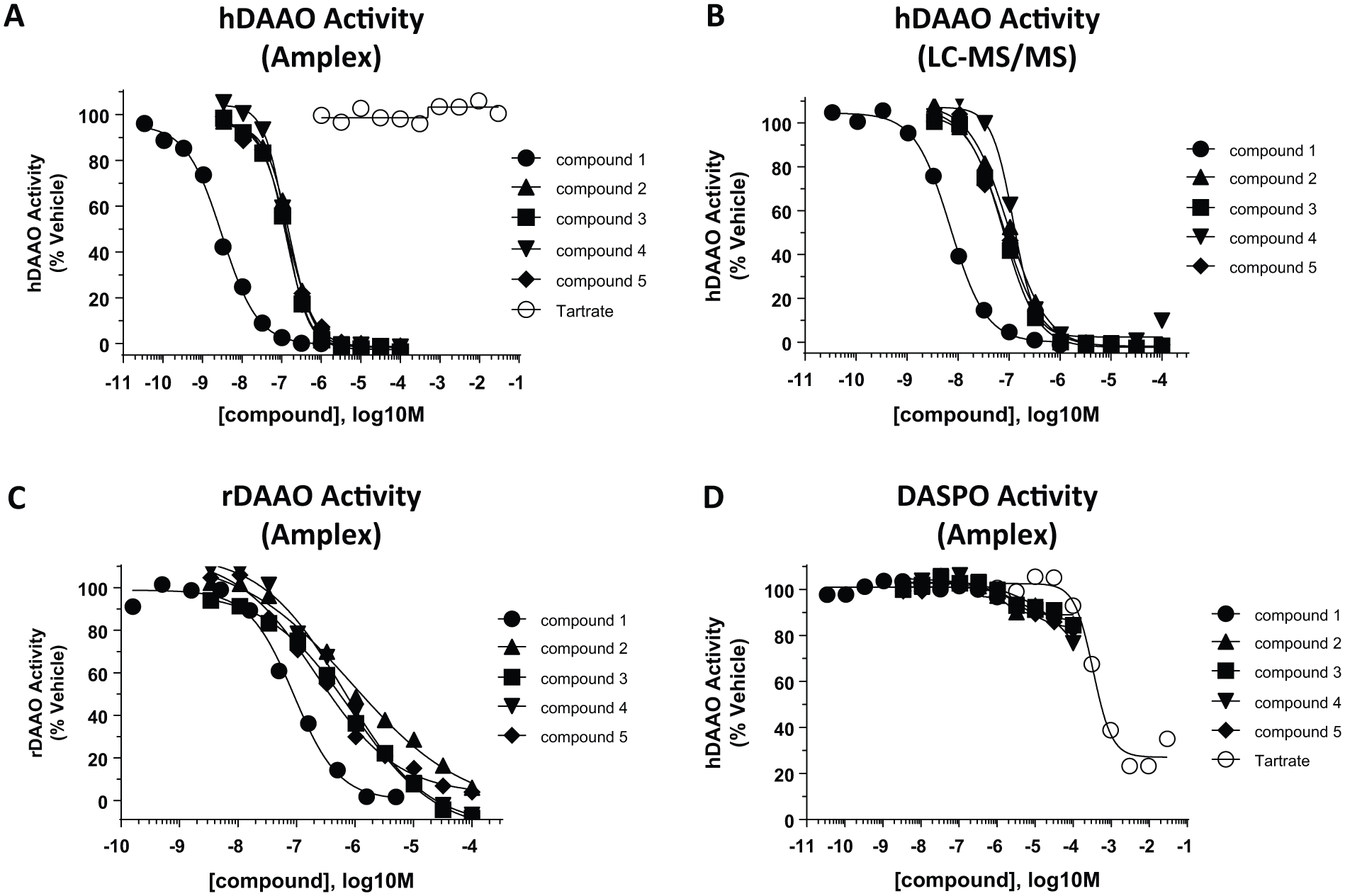
Class C compounds were specific D-amino acid oxidase (DAAO) inhibitors. (
To determine if class C compounds inhibited hDAAO via a novel mechanism, we tested the effect of FAD on inhibition. In these experiments, we changed FAD concentration in the Amplex Red assay from the standard condition (450 nM FAD, near the KM value) to a “high FAD” condition (50 µM FAD). In this assay, several compounds were marginally more potent in the latter, high FAD, conditions (
Fig. 3A
, left): compound
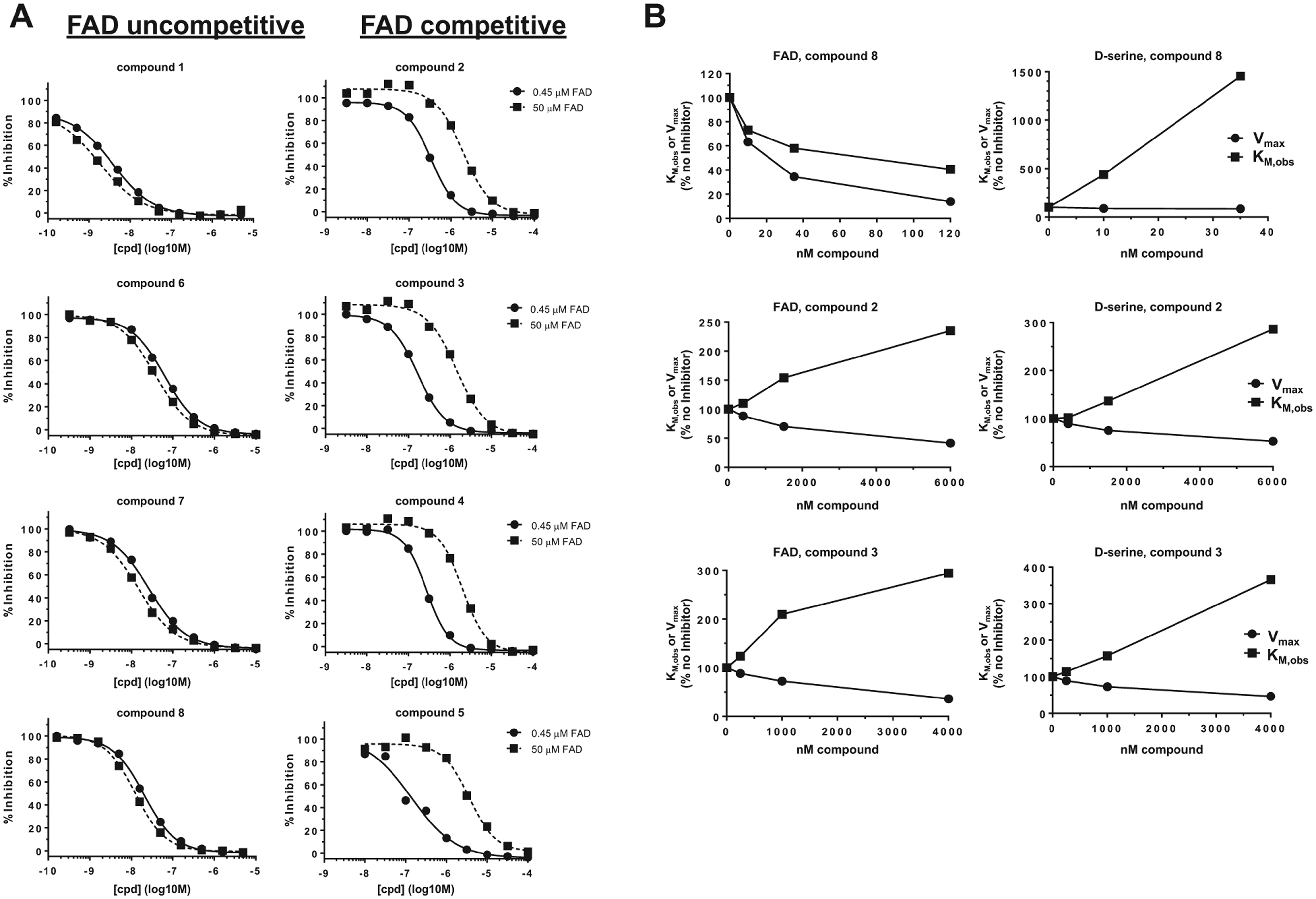
Class C compounds were flavin adenine dinucleotide (FAD) and D-serine mixed human D-amino acid oxidase (hDAAO) inhibitors. (
To test this hypothesis more thoroughly, we performed substrate and cofactor saturation experiments in which hDAAO relative ligand affinity (KM,obs) and maximum velocity (Vmax,obs) were measured in the presence and absence of various concentrations of inhibitor. In FAD saturation experiments, compound
Class C Compounds Inhibit hDAAO Covalently and Reversibly
To further define the mechanism of action of the class C hDAAO inhibitors, we used jump-dilution assays to determine compound off-rate. In these experiments, a high, inhibitory dose of test compound was incubated with hDAAO and then rapidly diluted, such that the test compound was then at a noninhibitory concentration. By observing the kinetic recovery of enzymatic activity from an initial velocity (vi) to a steady-state velocity (vs), the compound off-rate can be determined, as we have done previously for compound
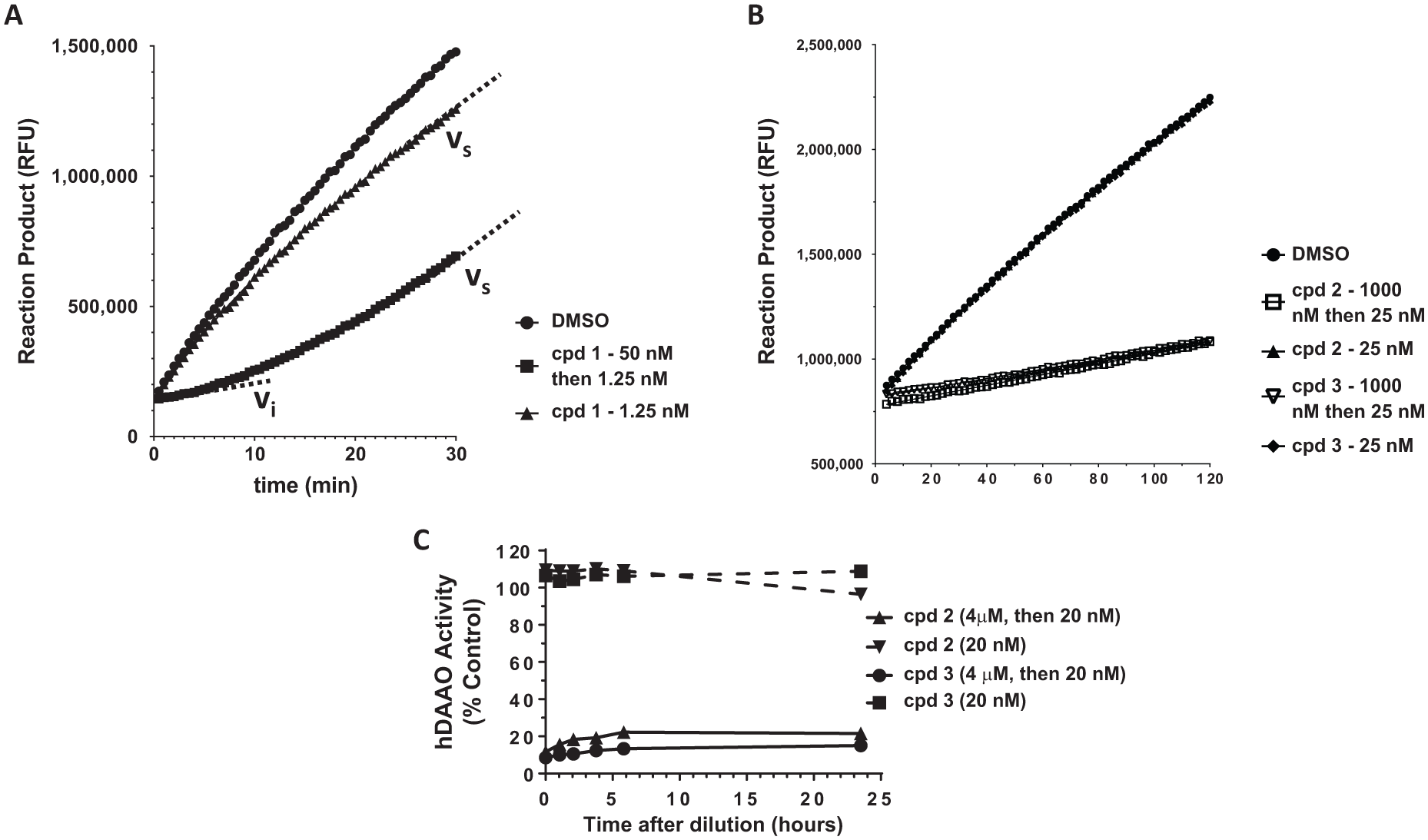
Compounds
Due to the lack of detectible off-rate (
Fig. 4
), we hypothesized that the class C compounds were inhibiting hDAAO via a covalent modification. This hypothesis was informed by literature reports that other isothiazolone compounds, via S-S thiol crosslinking,
28
are capable of forming covalent bonds with cysteines in target proteins.
29
Consistent with this model, we synthesized and tested for hDAAO inhibition compounds
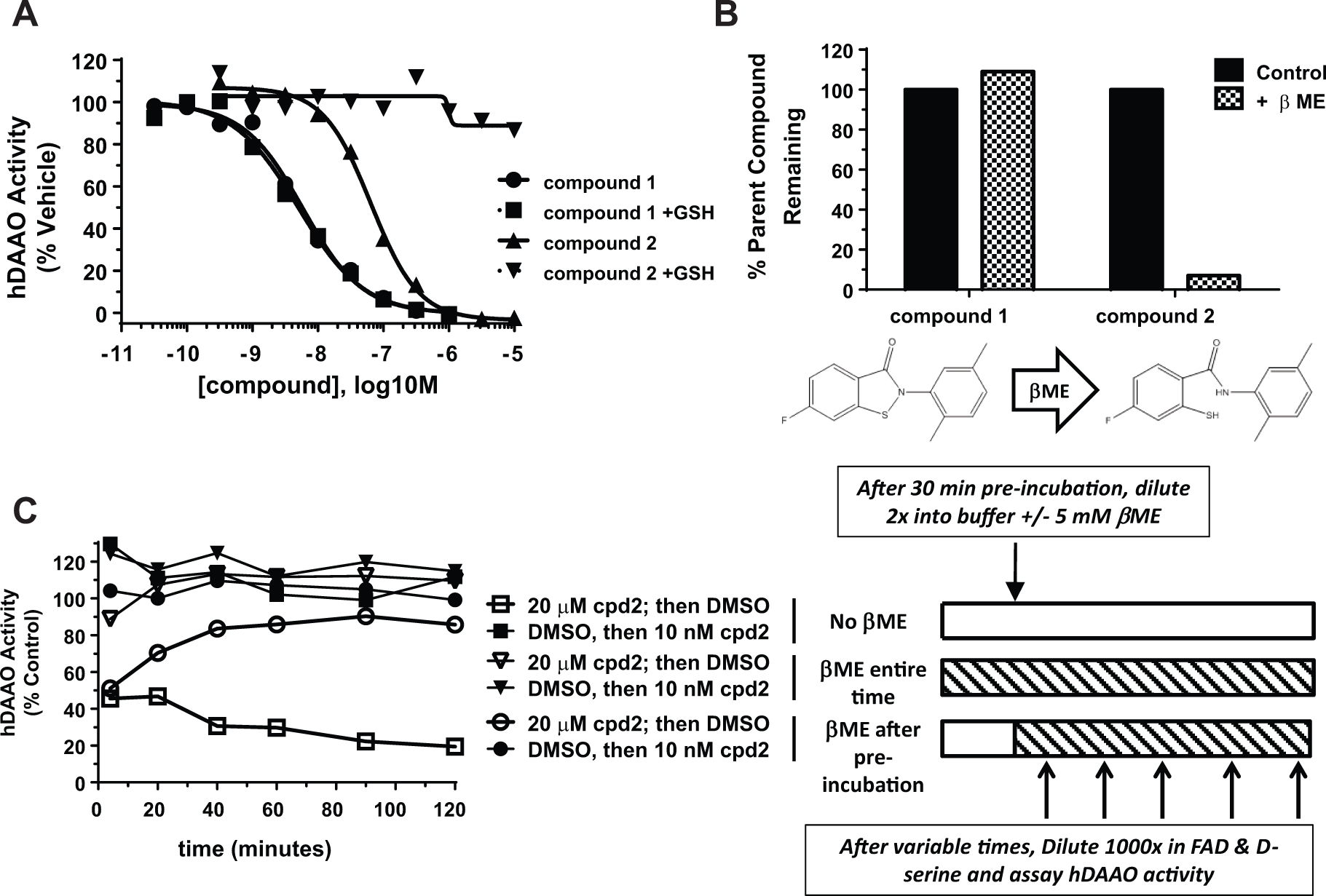
Compound
Because compound
Compound 2 Covalently Binds to All Five Cysteines of hDAAO
For a direct method to determine if compound
Discussion
In this study, we designed an HTS to identify hDAAO inhibitors sensitive to the presence of the hDAAO binding partner, pLG72. Approximately 85% of confirmed hDAAO inhibitors emerging from the screen were insensitive to pLG72 (class B compounds;
Fig. 1B
). However, the remaining inhibitors were either more potent (class A) or less potent (class C) hDAAO inhibitors when pLG72 was present. Because of their structural homogeneity, availability from commercial sources, interest in the wider scientific community (see below), and rapid opportunity for exploring structure-activity relationships, we focused our efforts on characterizing the class C compounds. These compounds were both FAD and D-serine competitive hDAAO inhibitors, diverging from the known mechanism of previously described, active-site hDAAO inhibitors. Our biochemical characterization of the prototypic class C compound
Although the role of pLG72 remains enigmatic, our study provides further data that may help elucidate pLG72 function in relation to hDAAO. Our prior reports demonstrating that pLG72 was an hDAAO inhibitor used relatively high concentrations of hDAAO and pLG72 in near stoichiometric concentrations. 7 In this study, to facilitate an assay suitable for HTS, our experimental design was different: we employed a classical pharmacological assay design (treating hDAAO as receptor and pLG72 as ligand) in which hDAAO was held at a relatively low (6.25 nM) concentration, and pLG72 concentration was increased until a functional outcome on hDAAO oxidative enzymatic activity could be measured. As a further difference, the newly designed assays were carried out over longer periods of time, with a 30-min hDAAO and pLG72 preincubation, followed by 1-h incubation after addition of substrates; reaction product was then measured in endpoint mode. Past assay formats from our laboratory included the 30-min preincubation but measured initial reaction rate 7 rather than accumulation of product over time. Despite these differences, we confirmed that the primary effect of pLG72 was hDAAO inhibition. Furthermore, the general affinity of pLG72, based on our IC50 determination of approximately 0.9 µM ( Fig. 1A ), was consistent with previous measurements of pLG72-hDAAO binding affinity using surface plasmon resonance. 7 Although we cannot conclusively resolve the discrepancy between our experiments demonstrating hDAAO inhibition with prior experiments demonstrating hDAAO activation by pLG72, the time dependency of the interaction is critical for understanding the complex interaction between pLG72 and hDAAO. Under short incubation periods, pLG72 has no effect or even an activating effect on hDAAO. However, over longer periods of incubation (many minutes to hours), pLG72 acts as a type of “negative chaperone,” placing hDAAO in a conformation that renders the enzyme inactive and promotes hDAAO degradation in cellular environments. 30
Although it is unclear if class C compounds will be useful for clarifying the biological function of pLG72-hDAAO complexes, by seeking hDAAO inhibitors with differential sensitivity to the presence of pLG72, our approach revealed novel, nonactive site-directed hDAAO inhibitors. Unlike canonical active site hDAAO inhibitors, class C compounds displayed a mixed-inhibition mechanism of action with respect to both D-serine and FAD (
Fig. 3
). With their immeasurably long off-rates (
Fig. 4
), we hypothesized that the class C compounds were binding to hDAAO covalently. Indeed, compound
Conceivably, flavoenzymes bound to FAD have cysteines buried within the protein structure inaccessible to solvent. Consistent with this model, crystal structures of hDAAO (e.g., protein data bank ID: 4QFC 14 ) reveal that four of the five cysteines have <10% solvent-accessible surface (SAS; determined with Discovery Studio software [Accelrys, Burlington, MA]), while the remaining cysteine (C264) has <15% SAS. Under conditions of FAD association, these cysteines would have limited access to a modifying compound in solution. However, hDAAO, rather unique among flavoenzymes, samples the non-FAD-bound, apoprotein structure at physiological FAD concentrations, and previous work demonstrated that the apoprotein structure is an open conformation with reduced tertiary structure. 31 Because FAD diminished the inhibitory potency of class C compounds ( Fig. 3 ), class C compounds may have better, or perhaps exclusive, access to hDAAO cysteines when hDAAO is in the apoprotein (FAD-unbound) conformation. Consistent with this model, stripping DASPO of FAD is very difficult, 20 and DASPO was fully active without adding FAD to the reaction. These observations suggest that DASPO rarely adopts the open, apoprotein conformation, perhaps forming the basis of its resistance to class C compounds. Finally, the defining feature of class C compounds, reduced inhibitory hDAAO potency in the presence of pLG72, is consistent with a model in which the pLG72-hDAAO interface shields hDAAO cysteines from class C compound access, either directly or via altered protein conformation. Further experiments would be necessary to test these biophysical hypotheses and determine the structure of the hDAAO-pLG72 complex.
The core substructure of the class C compounds, 2-phenyl-2,3-dihydro-1,2-benzothiazol-3-one, has been termed ebsulfur. 26 Biological studies of various analogues of these compounds have revealed general features consistent with our results. In experiments performed more than two decades ago, benzisothiazolone was shown to inhibit growth of various bacterial strains 28 and, consistent with our results in Figure 5 , glutathione reduced the biological potency of these compounds. 28 In more recent experiments, ebsulfur has been explored as a potential treatment for African trypanosomiasis. Ebsulfur inhibited the trypanosome enzyme trypanothione reductase via covalent inhibition, and trypanosome strains higher in glutathione levels were more resistant to ebsulfur treatment. 26 In unrelated studies, Trevillyan et al. 29 demonstrated that isothiazolone compounds, similar in structure to class C compounds, inhibited kinase targets via S-N bond reduction and covalent binding to cysteines in the kinase active site; these effects were blocked by the reducing agents βME and dithiothreitol. Taken together, our results are consistent with this general class of redox-sensitive compounds inhibiting protein targets via covalent binding to cysteine residues.
The identification of a class of compounds inhibiting hDAAO via a novel mechanism was initially quite exciting, since most hDAAO inhibitors occupy the enzyme’s active site. However, although covalent inhibition can be advantageous therapeutically, chemical reactivity generally is considered a negative attribute for a compound undergoing drug development. Because of their propensity to bind cysteines, isothiazolone compounds recently have been placed on lists of pan-assay interference compounds (PAINS), with advice to remove them from screening libraries.
32
Despite these concerns, ebsulfur and other class C-like compounds have been pursued as potential therapies for a variety of indications. As described above, various studies have proposed ebsulfur as a potential antimicrobial agent. A recent study suggested that ebsulfur and the related compound ebselen act as lithium mimetics and, therefore, could potentially treat bipolar disorder.
33
A report from the Sanford-Burnham Institute described the development of class C compounds as inhibitors of the enzyme phosphomannose isomerase (PMI), which could have utility for the treatment of the congenital disorder of glycosylation type Ia.
34
In a subsequent report, the authors characterized the compound we termed compound
Footnotes
Acknowledgements
For preparing compounds
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.