
Abstract
The everlasting pharmacological development is continuously producing new substances with potential doping abuse. Among these, secretagogues are very prone to misuse by athletes for their properties to release growth hormone (GH) and some limitations in the actual analytical methods to detect them. In this paper, an in-depth study on the key variables of the radio receptor method previously developed by our group is performed and a fit-for-purpose protocol is established. Thus, this sensitive and robust screening method is proposed as an intelligent and preventive antidoping method to detect new growth hormone secretagogues (GHSs) in exceptional suspicious urine samples obtained from athletes and will support the current detection methods based on liquid chromatography–mass spectrometry (LC-MS).
Keywords
Introduction
With the advancements in pharmacological and biotechnological developments, the arrival of new chemical and biological entities with the potential for use for doping in sports is continuously increasing. This fact is reflected in the World Anti-Doping Agency (WADA) List of prohibited substances, which evolves every year. 1 Especially relevant is the part of the list involving peptide hormones, growth factors, related substances, and mimetics where continuous additions have been specified in recent annual revisions. Of particular interest, in the Prohibited List for 2015, are the paragraphs involving growth hormone secretagogues (GHSs) and GH-releasing peptides (GHRPs).
In addition to being specifically mentioned in the list, the analysis for this type of growth factor has been made mandatory for all sports and federations from 2015 on, with the percentage of samples to be analyzed for them being variable, depending on the risk estimated among those different sports. According to the WADA “Technical Document for Sports Specific Analysis” (TDSSA), such a percentage ranges from 5% to 30% of total samples for most sports and disciplines. 2 Thus, all antidoping laboratories are to be prepared to detect the presence of compounds belonging to this pharmacological class of growth hormone secretagogues/growth hormone–releasing peptides (collectively addressed as GHSs).
Following the pharmaceutical evolution, antidoping targeting methods have been developed in recent years using liquid chromatography–mass spectrometry (LC-MS) to screen for those compounds of the GHS group known to be present underground or in Internet-distributed products and those already marketed (so far only GHRP-2 or pralmorelin for diagnostic purposes in Japan).3–10 Nevertheless, this field is a playground for many Pharma companies, and the number of drugs with a similar effect is not exactly known, but it can be estimated from the literature that at least 57 compounds are under development and could be potential targets for future use or abuse.11–15
Preventive antidoping research and “intelligence” applied to the suspected use or trafficking of new doping agents is gaining wide acceptance. Based on information gained from testing, whistle-blowing, police activities, suspicious exceptional sporting results, and several other sources, the testing authorities often accumulate much data on the use of new doping agents that have not yet been identified. In this regards, a methodology able to detect activity for any substance in a group of doping agents with similar biomolecular or pharmacological mechanisms would be of great advantage for confirming the abuse of new known or unknown chemical and biological entities. 16 For GHSs, a complex and tremendously heterogeneous family of molecules, this means that the prime target is the mechanism of interaction with the receptor GHS-R1a, for which the endogenous natural ligand is ghrelin, a 3-octanoylated 28–amino acid peptide. 17 We have developed a radio receptor displacement assay where compounds suspected to have GHS-like properties are studied by their capacity to in vitro displace labeled ghrelin from the GHS-R1a receptor expressed at the membrane of recombinantly engineered cells.18,19 Following up on the initial proposal addressing GHSs in urine, in the present article, a further and detailed study of several other variables that are relevant for robust test is presented, thus rendering a detailed method fit for detecting the presence of this family of compounds in urines from subjects where information gathered from intelligence sources may have flagged them as highly suspicious for the abuse of new potential growth factors for doping purposes. This approach will be of special value for all cases where the first battery of targeted tests, that is, those based on LC-MS analysis of well-known compounds, render a negative outcome.
Materials and Methods
Chemicals
Ghrelin was purchased from RayBiotech (Norcross, GA). Growth hormone-releasing peptide 2 (GHRP-2, pralmorelin) was kindly supplied by Dr. S. Kageyama (Tokyo Laboratory, Anti-Doping Center, Mitsubishi Chemical Medience, Tokyo, Japan). Testosterone glucuronide, erythropoietin, pituitary growth hormone, and recombinant somatropin were purchased from National Institute for Biological Standards and Control (NIBSC, Hertfordshire, UK). Radiolabeled ghrelin ([125I-His9]ghrelin) was purchased from PerkinElmer (Waltham, MA). All other chemicals were of the highest grade commercially available.
Cell Culture, Membrane Preparations, and Competition Binding Assay
Cell culture, membrane preparations, and competition binding assay parameters have previously been published by Pinyot et al. 18 Briefly, HEK293 cells stably expressing GHS-R1a were obtained from Dr. R. Smith (Baylor College of Medicine, Houston, TX) through Dr. F. Casanueva (University of Santiago de Compostela, Santiago de Compostela, Spain). Cells were cultured in 100 mm dishes in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 U/mL), 2 mM glutamine (Invitrogen, Paisley, UK), and G418 (Invitrogen, 800 μg/mL). Cells were grown under a humidified atmosphere of 95% air and 5% CO2 at 37 °C up to 70%–80% confluence. Cultured cells were trypsinized and pelleted by centrifugation at 1000 g for 5 min and frozen at −80 °C in DMEM with 10% DMSO (Sigma-Aldrich, St. Louis, MO) at a concentration of 5 × 106 cells/mL.
Membranes were prepared as follows: One aliquot of 5 × 106 cells was thawed and centrifuged at 4 °C for 10 min at 4000 g. The cell pellet was resuspended in 1 mL of homogenization buffer (50 mM Tris-HCl and 10% sucrose, pH 7.4) and sonicated for 5 min. Disrupted cells were centrifuged again at 4 °C for 10 min at 4000 g. Membrane pellet was resuspended in 5 mL of binding buffer (25 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, 2.5 mM ethylenediaminetetraacetic acid [EDTA], and 0.4% bovine serum albumin [BSA], pH 7.4) and used fresh or stored at 4, −20, or −80 °C until used to study storage conditions.
For competition binding assays, membranes obtained from 50,000 cells were incubated in triplicate with [125I-His9]ghrelin (at a concentration of 15 pM; specific radioactivity 2200 Ci/mmol) and different GHSs or urine samples spiked with doping substances in a final volume of 300 µL of fresh binding buffer or stored binding buffer in different conditions to study their stability (frozen, freeze-dried, and 4 °C). Prior to addition to the membranes, [125I-His9]ghrelin aliquots were counted in a 1470 Wizard gamma scintillation counter (PerkinElmer) to obtain the value of the total radioactivity initially present in each experiment. The samples were incubated from 30 to 70 min at 25 °C under continuous shaking. The reaction was stopped by rapid centrifugation at 16,000 g for 5 min at 4 °C. The cell pellet was rinsed from one to four times with ice-cold 50 mM Tris-HCl pH 4, 7.4, or 9 in order to wash the radiolabeled ligand present in the tube surface and centrifuged again at 16,000 g for 3 min at 4 °C each time to prevent the pellet from resuspending. Finally, the pellet was measured in the gamma counter and the results were analyzed with appropriate software (see below).
Reference Urine Samples
Two reference urine samples were included in each competition binding experiment, one as a blank or negative control (not spiked with GHSs) as the maximum possible binding (100% of relative specific binding [RSB]) and the other as a positive control, spiked with 7.5 μM GHRP-2, as the minimal possible specific binding (0% RSB). All sample binding values were calculated relative to these limits.
Experimental Urine Samples
Urine samples from a human recombinant growth hormone (rhGH) excretion study were available at the Barcelona antidoping laboratory.
The study had been approved by the local research ethics committee and subjects signed an informed consent before participation. Each subject had received a daily subcutaneous injection of 0.067 mg·kg−1 (1 mg of rhGH is equivalent to 3 IU) of rhGH (Genotonorm, Pfizer Laboratories, New York, NY) daily for 14 days. Twenty-four-hour urine samples were collected, aliquoted, frozen immediately, and kept at −20 °C until analysis. Two urine samples collected during and after treatment were used.
For the effect of the food intake study, urine samples were collected from 27 volunteers before and after a conventional lunch, aliquoted, frozen immediately, and kept at −20 °C until analysis.
To estimate the concentrations at the limit of decision, urine samples from the same volunteer spiked six times with decreasing amounts of GHRP-2 were studied. After the competition binding assay with membranes obtained from a total of 50,000 cells, concentrations were calculated and a limit of decision was established as that concentration involving an RSB of 76.3%, as described by Pinyot et al. 19
For analyses, urine samples were thawed and centrifuged for 15 min at 3500 g. After desalting and purified by receptor affinity as described by Pinyot et al., 19 samples were reconstituted in 150 μL of binding buffer and analyzed by competition binding assay.
Data Analysis
All results were analyzed with GraphPad Prism 5 software (San Diego, CA), and graphics were produced with the same software. Statistical analysis was performed using Excel and R programs. To contrast independent continuous variables, the Student t test was used. The null hypothesis was rejected for values of p ≤ 0.05.
Results
The GHS radio receptor assay verified with 10 different GHSs with Ki values between 1.1E-06 and 2.1E-10 M (supplementary data) 18 has been thoroughly interrogated in this paper to evaluate additional variables that are of relevance in the antidoping field and render an optimal test in those situations where established LC-MS methods do not provide an answer. The only GHS approved by health authorities so far, GHRP-2, was selected to evaluate important variables such as functional sensitivity, membrane stability, stability of binding buffer, influence of wash steps and pH, interference with other major doping substances, and the effect of food intake. Together with the parameters evaluated earlier (membranes vs intact cells; desalting; assay accuracy; gender, age, and exercise effects, threshold; excretion; and linearity),18,19 ultimately a robust and reliable protocol is suggested (vide infra) with high detection capability.
Storage Conditions of the Isolated Membranes and Binding Buffer
Different storage conditions of isolated membranes were analyzed in order to establish their long-term stability. For this, isolated membranes stored at 4, −20, and −80 °C were tested. Rapid decrease of activity was observed upon storage at 4 °C ( Fig. 1A , top). The binding assays showed values of 58.6% and 87.2% of RSB in membranes stored for 30 days at 4 and −20 °C, respectively, and 95.3% and 99.7% of RSB in membranes stored at −80 °C for 30 and 660 days, respectively ( Fig. 1A , bottom). While storage at 4 °C can only be permitted during processing of the analytical batch, the storage of −20 °C can be authorized for periods of less than 1 month (short-term storage). For long-term storage, only −80 °C presented statistically significant stability independent of storage time.
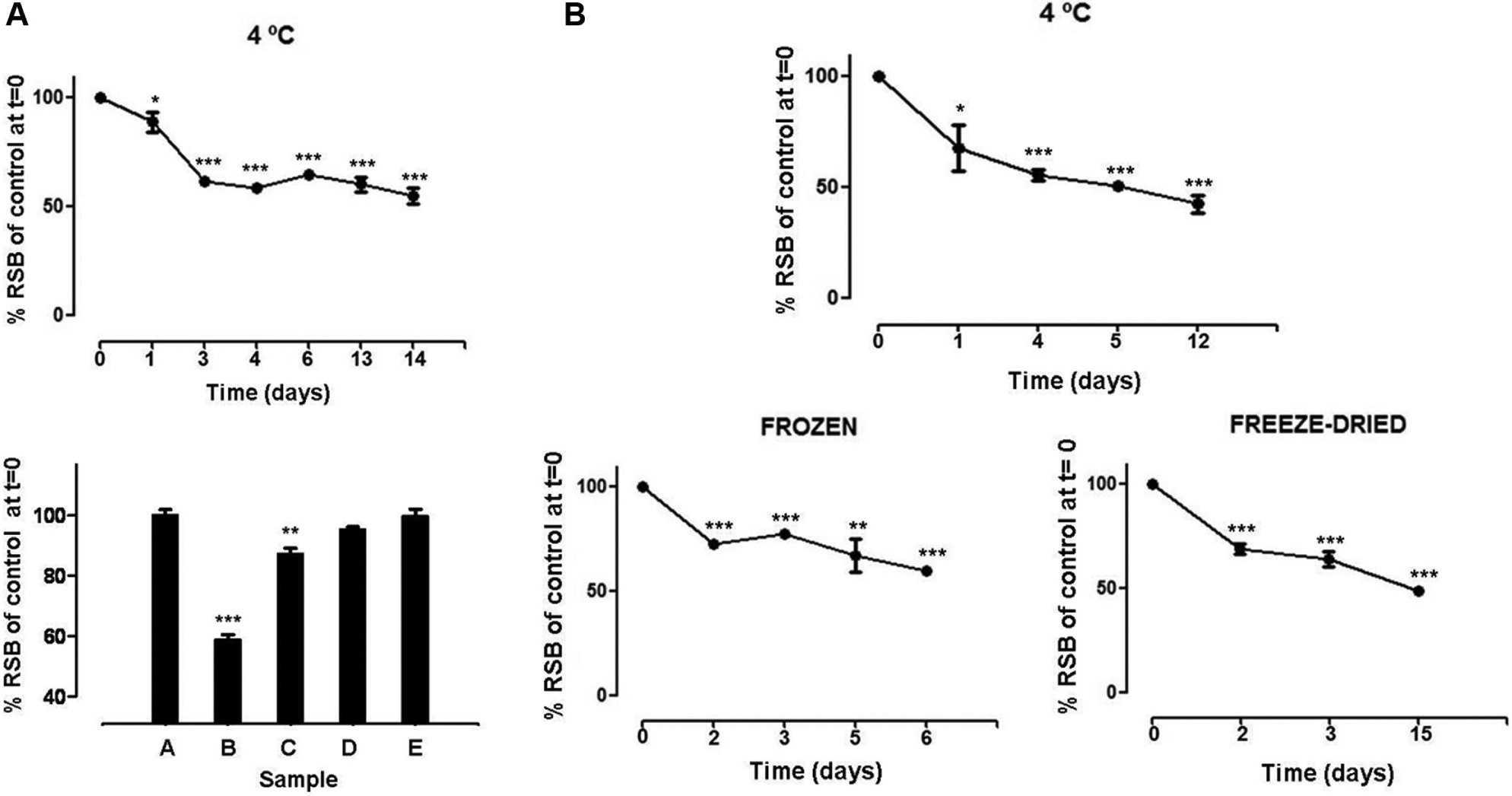
Stability of membranes and binding buffer stored under different conditions. (
Given that buffers are usually prepared in larger volumes, binding assays were performed with the following variables for the storage of binding buffer: 4 °C, frozen, and freeze-dried. Figure 1B shows an RSB of 67.6% in binding assays performed 24 h after preparing binding buffer and stored at 4 °C. Likewise, both frozen and freeze-dried binding buffers (resuspended in water at the time of use) showed RSBs of 69.1% and 72.5%, respectively, after 48 h storage. As such, statistically significant different values when binding buffer was prepared in advance were compared with freshly prepared binding buffer and stressed the absolute requirement for fresh preparation.
Incubation Time Conditions, Number of Washes, and pH Values after Incubation of Competition Binding Assay
To evaluate the relevance of the incubation time on the binding between the GHS-R1a receptor and GHS, samples spiked with 1 nM of ghrelin were assessed by competition binding assay at different incubation times. The RSB value was measured every 5–10 min for 70 min (starting 30 min after incubation). The results, displayed in Figure 2A , showed mean values of approximately 49% RSB at 35 and 40 min of incubation and 29% RSB at samples incubated for 70 min.
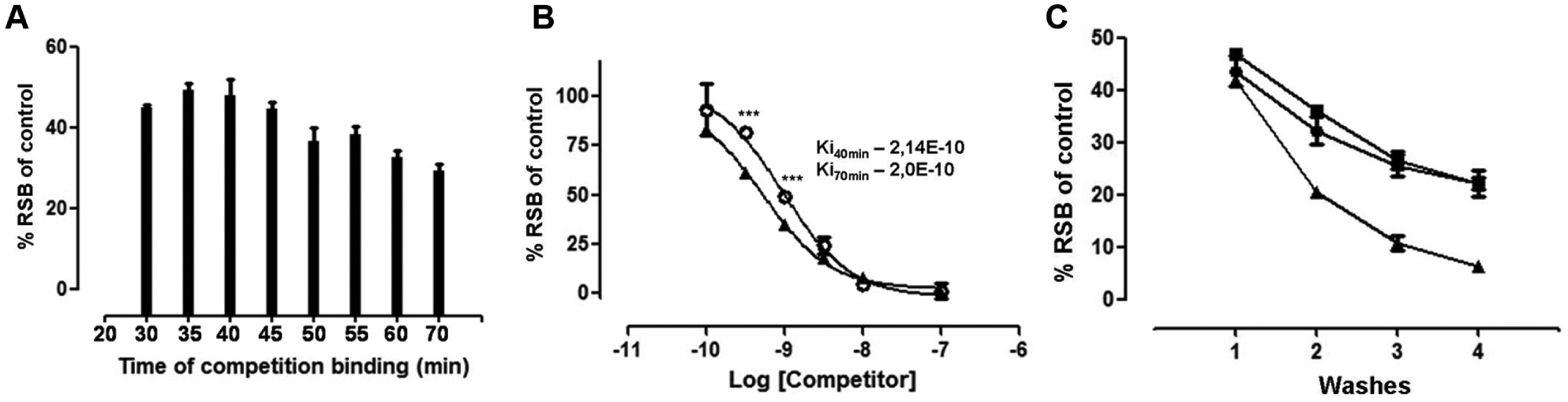
Effects of incubation time on the GHS competition binding assay and evaluation of the number of washes after competition. (
Therefore, 40 and 70 min were selected as incubation times to perform competition curves with ghrelin and to corroborate results obtained in the above experiment. These curves showed very similar Ki values for samples incubated in both incubation times in spite of statistical differences in RSB at some concentrations (1 nM and 500 pM ghrelin) ( Fig. 2B ).
The percent of binding of radioactive ghrelin to membranes was measured over four wash steps at pH 4, 7.4, or 9. As can be seen in Figure 2C , the results showed a decrease to about 50% RSB in the case of samples that were washed four times in buffer with pH 4 or 7.4, and 85% RSB at pH 9 with respect to samples that were washed once. When the samples were washed once at pH 7.4 and pH 4, the results were 93% and 89.5% RSB.
Food Intake Effect
Plasma ghrelin levels are naturally increased in humans before meals, while they are reduced after food ingestion. For these, in order to determine the effect on the competition binding assay caused by potential changes in the endogenous ghrelin levels due to food intake, urine samples from 27 volunteers (14 men and 13 women) before and after a conventional meal were evaluated. Preprandial urine samples yielded a mean value of 94.1% RSB with a coefficient of variation (CV) of 6.5%, while postprandial samples yielded 97% RSB with a CV of 6.4% ( Fig. 3 ). No significant difference was found among these samples (p = 0.103).
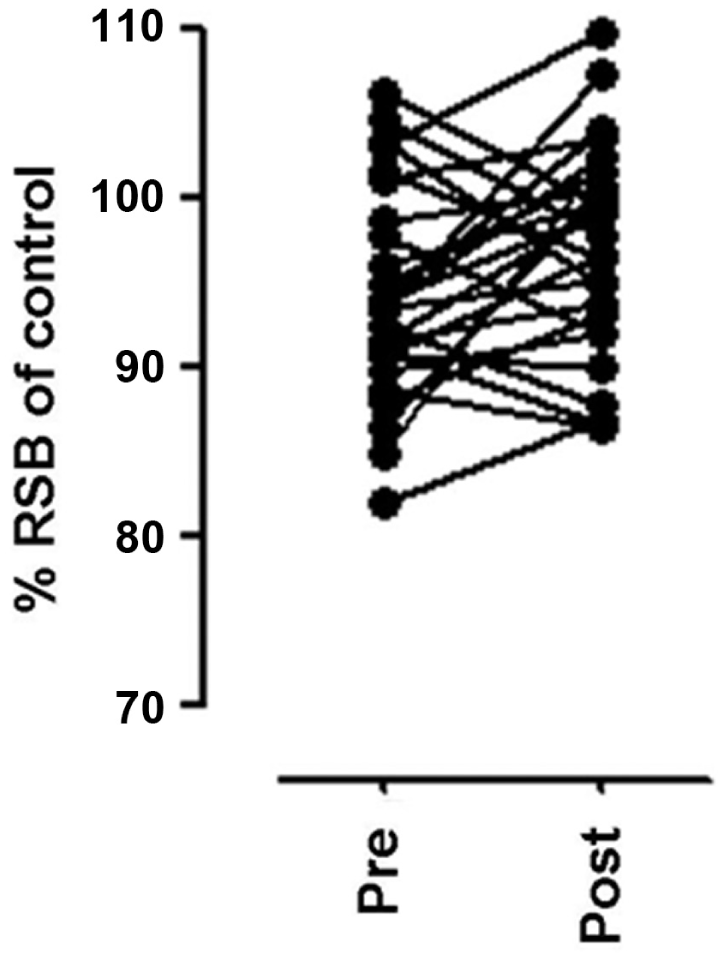
Evaluation of urine samples before and after conventional meal in 27 volunteers.
Interference with Concomitant Doping Substances Potentially Present in Real Samples
Urines spiked with some of the more relevant doping substances (human recombinant erythropoietin [EPO], testosterone glucuronide [TES], human recombinant growth hormone [rhGH], and pituitary growth hormone [PIT]) were evaluated before and after adding a known concentration (1 nM) of GHRP-2 to evaluate the potential interfering effect on the GHS detection method. These substances were selected as representatives of frequently encountered doping substances to account for interference by both peptide and nonpeptide substances on the assay. As shown in Table 1A , urine without GHRP-2 had a mean value of RSB that was similar to that of the negative control urines, and samples containing GHRP-2 had values similar to those of the positive control urines for all substances tested. These results showed no interference in this assay by those selected doping substances.
Effect of Concomitant Presence of Some Doping Agents Potentially Present in Real Samples.
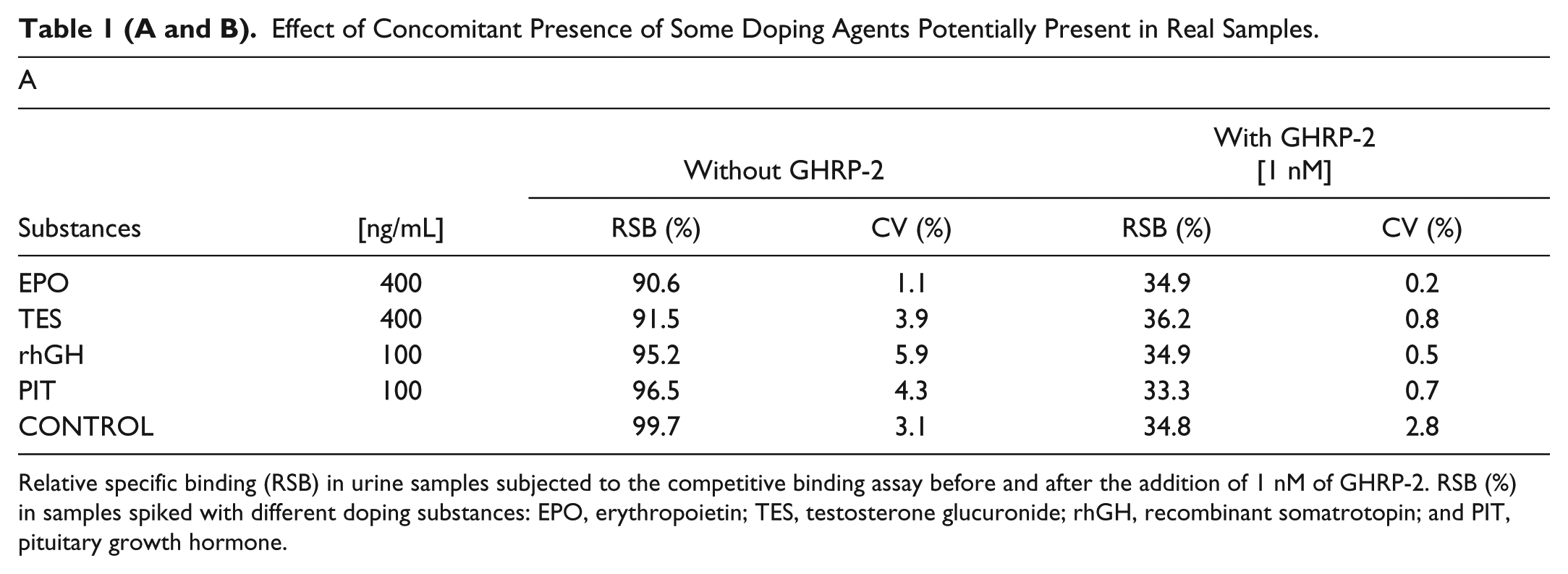
Relative specific binding (RSB) in urine samples subjected to the competitive binding assay before and after the addition of 1 nM of GHRP-2. RSB (%) in samples spiked with different doping substances: EPO, erythropoietin; TES, testosterone glucuronide; rhGH, recombinant somatrotopin; and PIT, pituitary growth hormone.
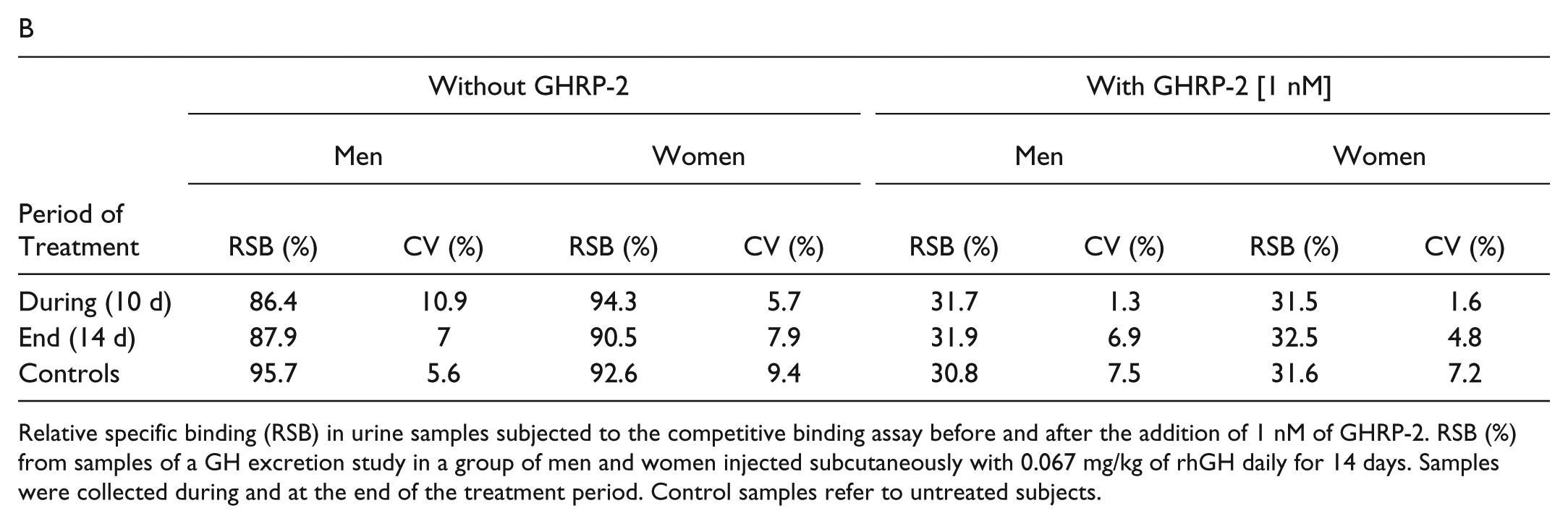
Relative specific binding (RSB) in urine samples subjected to the competitive binding assay before and after the addition of 1 nM of GHRP-2. RSB (%) from samples of a GH excretion study in a group of men and women injected subcutaneously with 0.067 mg/kg of rhGH daily for 14 days. Samples were collected during and at the end of the treatment period. Control samples refer to untreated subjects.
On the other hand, analyses on urines from rhGH excretion study samples were performed. Urine samples were collected from a group of four men and four women, Caucasian volunteers, dosed subcutaneously with 0.067 mg/kg/day of rhGH for 14 days. At 10 and 14 days after administration, urine samples from volunteers were analyzed before and after spiking a known concentration of GHRP-2. Both samples groups, displayed in Table 1B , showed results similar to those of the blank specimen.
Limit of Decision
After optimizing the method, an experiment to set the limit of decision for GHRP-2 was performed. To establish this limit, six urine samples corresponding to different concentrations of GHRP-2 were measured in separate binding experiments to calculate the mean and CV. RSB results displayed in Table 2 showed that the limit of decision can be estimated as 400 pM in compliance with the decision limit at 76.3% RSB established by Pinyot et al. 19 based on interindividual variability. This concentration corresponds to approximately 0.4 ng/mL of GHSs in urine, even though this may vary as a function of the particular substance.
Determination of Limit of Decision for GHRP-2.
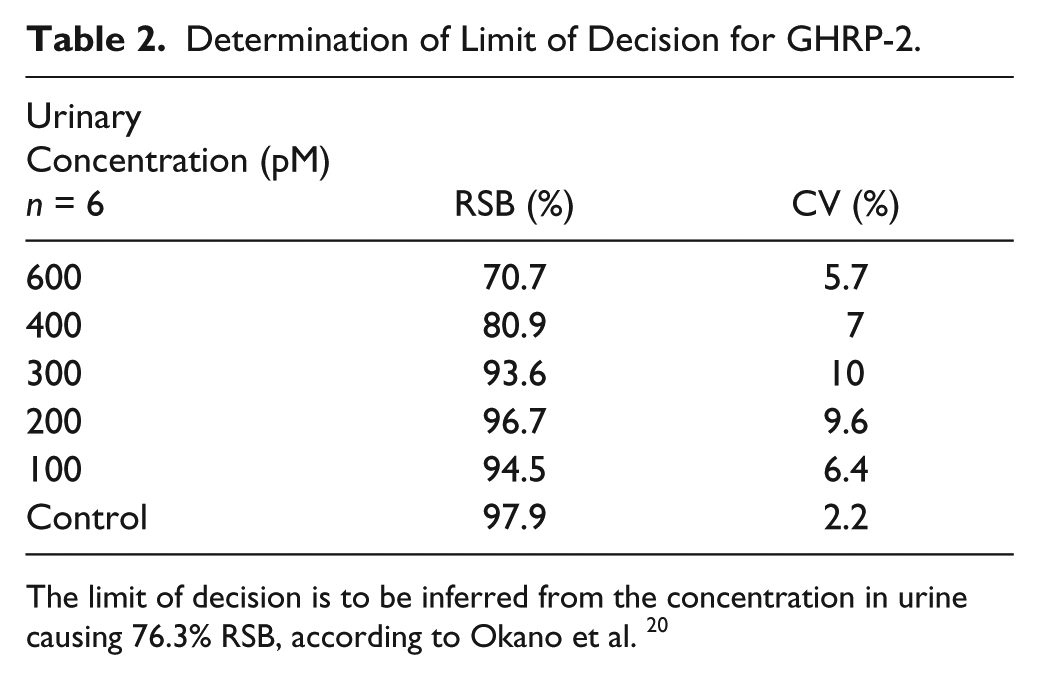
The limit of decision is to be inferred from the concentration in urine causing 76.3% RSB, according to Okano et al. 20
Discussion
Anticipating doping abuse of emerging compounds produced by pharmaceutical companies demands the development of detection methods that are reliable and efficient and ideally precede illicit use. In this scope, GHSs are attractive to athletes for several reasons: the great variety in the chemical structure of the distinct GHS, the availability of these substances through the Internet, and the limitation of detection methods to address all potential emerging GHSs. GHSs have the ability to release GH in a way that circumvents current GH-doping tests as mostly used today by the antidoping laboratories (based on the differential GH isoform approach). Administration of GHSs leads to elevated hGH levels and can also be combined with rhGH as a stimulation-masking agent. 20 Thus, screening methods based on competitive binding by the receptor site with radioactive and nonradioactive labeled ligands to support other detection methods based on LC-MS have been developed since 2010.6,10,18,21–23 Whereas LC-MS is the method of choice for known substances, the technique falls short for nontargetted antidoping analyses. The competitive binding assay established by Pinyot et al. may complement this gap because the nonradioactive methods (e.g., based on time-resolved fluorescence techniques 22 ) involve a complex methodology and a sensitivity that is 10-fold lower than that of the radioactive method. The original publication assessed variables such as membranes versus intact cells; desalting; assay accuracy; and gender, age, and exercise effects; others such as threshold, stability, excretion, and linearity have now been optimized to be fit for the purpose of preventive doping screening in sports.
To this end, several variables were studied to improve the overall runtime, the sensitivity, and the robustness of the method to establish a definitive optimal test protocol. Variables studied to improve the runtime of the methods were storage conditions of the isolated membranes and binding buffer. Results have shown a good stability for the membranes under appropriate conditions but a poor overall stability of the binding buffer used in the method. Membranes prepared from cells and stored at −80 °C for up to 2 years would have an efficiency equal to that of membranes prepared fresh, which avoids the need of membranes to be freshly prepared and allows, if suitable, obtaining them from an external source, establishing sufficient resources for special competition events, and guaranteeing the equivalence of reagents throughout the different laboratories. In this respect, it is important to indicate that membranes stored at 4 and −20 °C showed a significant degradation. Potential degradation of the receptor by proteases18,19 in membranes stored at these temperatures cannot be discarded. Regarding binding buffer, after testing several conditions, neither of them showed the sensitivity achieved with a fresh one. Thus, it is mandatory that these two critical reagents are always addressed as indicated here.
After optimizing the runtime protocol of the screening method, five additional variables were studied to assess the sensitivity and robustness of the method developed by Pinyot et al.18,19: incubation time conditions of binding assay (40 vs 70 min), number of washes at different pH values, food intake effect, interference with concomitant doping substances potentially present in real samples, and limit of decision.
In biomolecular interactions seeking optimal binding between the ligand and receptor, allowing for the equilibrium to take place may affect the sensitivity of the screening method. The time of incubation was evaluated in a competition binding assay with incubations of 70 and 40 min, following the initial screening of a full time course, and in spite of some differences in RSB found between the two times tested, the Ki values obtained from competition experiments were practically identical in both assays. This means that after 40 min, equilibrium has settled and prolonged incubations do not impact negatively on the binding. This allows for flexibility, with no impact on the final result, in the analytical protocol of at least 30 min following the initial receptor-mediated binding.
Background levels found in the samples due to nonspecific interactions during the binding assay can affect the sensitivity and robustness of the method. For example, it is well known that improper washing of the samples could result in a high background or modify the interaction between the ligand and receptor. 24 This very important step in ligand binding assays was assessed by testing the number of wash steps as well as the pH of the wash buffer. It could be established that in this particular assay, the lowest background, in combination with the highest value of ligand bound to the receptor, was obtained when we used a wash buffer with pH 4 or 7.4 and samples were only washed once. The lower binding results at pH 9 were attributed to the partial loss of the octanoyl group in serine 3 of labeled ghrelin, which is required for the binding by the receptor.24,25 As such, a limited number of wash steps and an acid or neutral pH in this method are critical to obtain a good signal from samples, and our recommendation is to use pH 4 and a single wash cycle ( Fig. 4 ).
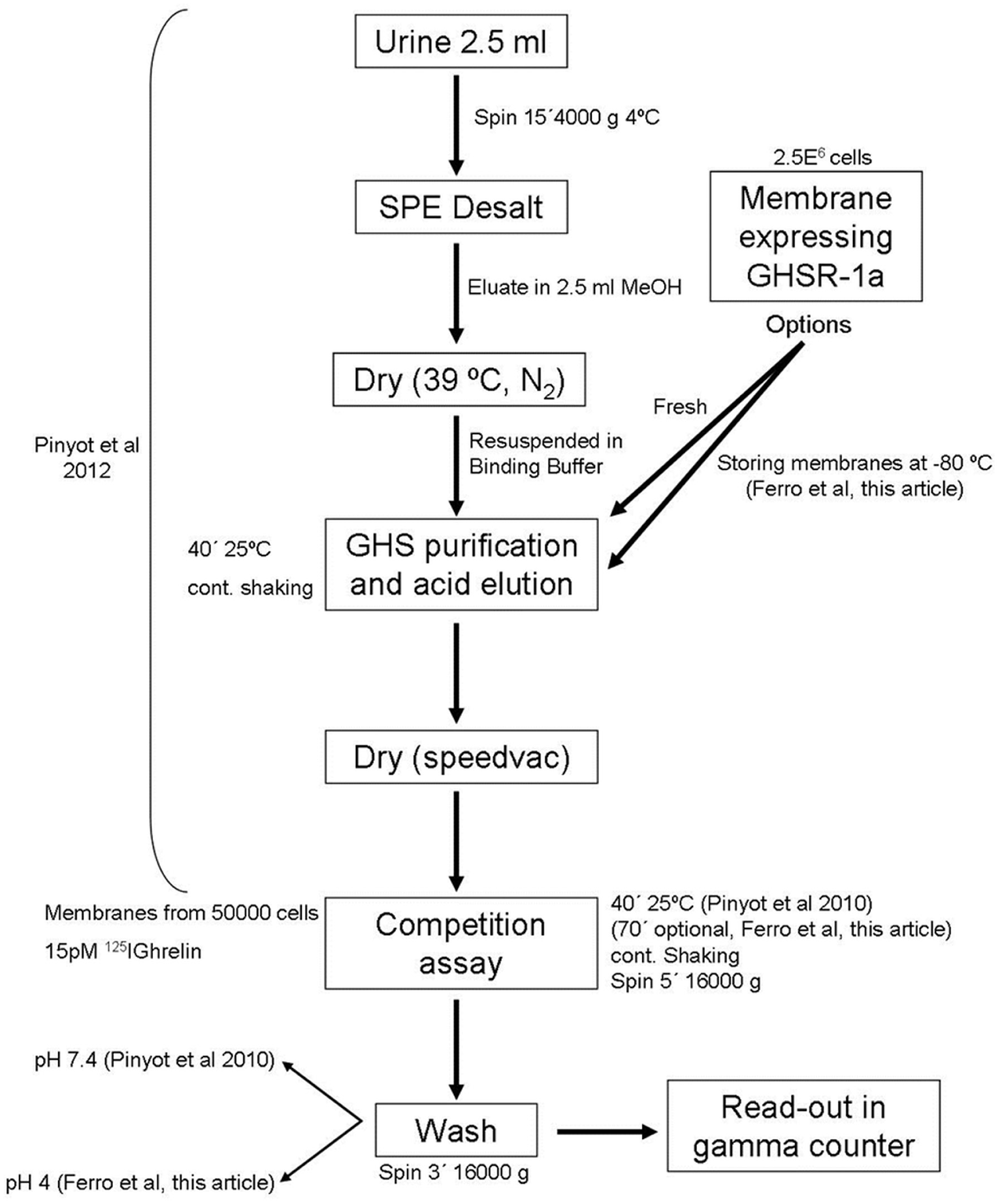
Optimal protocol to detect GHS abuse in urine.
After several reports about the increase of circulating ghrelin levels in plasma before meals, while they are reduced after food ingestion,26,27 an evaluation of this screening method in samples before and after a conventional meal was necessary. In fact, patients with anorexia had shown markedly elevated plasma levels (1.05 ng/mL vs 0.5 ng/mL for the control group), 28 but these elevated ghrelin concentrations returned to normality after feeding. These studies demonstrate circadian rhythms in ghrelin levels throughout the day. If indeed so, and considering that the concentration in urine for endogenous ghrelin is about 60% of that circulating in blood, 29 these changes could theoretically affect the competition assay. Our assessment of pre- and postprandrial samples showed a marginal 3% lower binding in samples evaluated before food ingestion, but no significant difference was found between these samples and the results obtained after food ingestion. These differences are much below the threshold at 76.3% RSB established by Pinyot et al. 19 based on interindividual differences and should not give any misleading result. Nevertheless, one should not exclude the possibility of a delayed effect in urine samples after food intake. However, this would be difficult to demonstrate without a dedicated clinical study, as other events (e.g., some drugs30,31) may have a faster impact on urinary or plasma ghrelin levels.
Doping is often performed using various substances at the same time, with variable influence on the endocrine system, so it is important to verify the robustness of the screening method described18,19 in such circumstances. Results obtained from urine samples spiked with GHSs and other doping substances (TES, EPO, rhGH, PIT) showed no interference of the latter with the method described, regardless of their different physical-chemical characteristics or mechanism of action.
At last, the sensitivity and precision of the assay were assessed. The lower limit of quantification tested for GHRP-2 in urine samples ( Table 2 ), with the coefficient of variation well within the accepted limit of 20% for ligand binding assays, was similar to the effective limit of decision that corresponds to 400 pM (value around 76.3% RSB), which represents the threshold for calling a sample suspicious for any growth hormone secretagogue taking into account interindividual variability. 19
In summary, a definitive protocol, displayed in Figure 4 , to detect GHSs in urine samples for antidoping purposes or other pharmaceutical interests has been established. This protocol can also detect any metabolites from GHSs with a capacity of interaction with the GHS-R1a receptor present in urine. The variables studied in this work complete the initial protocol by Pinyot et al. and determine the bases of a sensitive and robust screening method. This method could be employed to support the actual detection method by GHSs based on LC-MS, 6 which is very useful for well-known compounds but may render a negative outcome for emerging compounds, and will allow preliminary studies on those samples suspected of GHS abuse.
Footnotes
Acknowledgements
The authors thank F. Casanueva and R. Smith for HEK293 cells expressing GHS-R1a, S. Kageyama for GHRP-2, and clinical research unit staff (Esther Menoyo, Esther Papaseit, Clara Pérez-Mañá, Cristina Llop, Clara Gibert, Soraya Martin, and Marta Pérez) for their assistance. This work was carried out with the financial support of the Ministerio de Ciencia e Innovación (DEP2009-09717) and the Catalan government (DIUE 2009SGR492). The authors also express their gratitude to the U.S. Anti-Doping Agency (USADA) for the initial support to the approach.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was carried out with the financial support of the Ministerio de Ciencia e Innovación (DEP2009-09717) and the Catalan government (DIUE 2009SGR492).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.