
Abstract
Genomic DNA is constantly exposed to endogenous and exogenous damaging agents. To overcome these damaging effects and maintain genomic stability, cells have robust coping mechanisms in place, including repair of the damaged DNA. There are a number of DNA repair pathways available to cells dependent on the type of damage induced. The removal of damaged DNA is essential to allow successful repair. Removal of DNA strands is achieved by nucleases. Exonucleases are those that progressively cut from DNA ends, and endonucleases make single incisions within strands of DNA. This review focuses on the group of endonucleases involved in DNA repair pathways, their mechanistic functions, roles in cancer development, and how targeting these enzymes is proving to be an exciting new strategy for personalized therapy in cancer.
Introduction
Genomic DNA is constantly at risk of damage from numerous sources, including both endogenous products of metabolism such as reactive oxygen species and also exogenous sources, for example, UV light, radiation, and chemical agents. 1 Therefore, to maintain genomic stability, cells have developed effective surveillance mechanisms to detect DNA damage and induce either DNA repair or, if repair is not possible, cell death. Due to the range of different adducts and lesions induced in DNA, cells have developed multiple DNA repair pathways consisting of many proteins with varying functions crucial for effective repair. Germline mutations to DNA repair genes are often associated with cancer susceptibility. Failure to initiate a cellular response to DNA damage results in the accumulation of mutagenic lesions that are associated with aging, neurological disorders, and cancer.
Loss of effective DNA repair correlates with carcinogenesis; conversely, an upregulation of DNA repair is often seen in cancer cells in response to DNA-damaging agents such as platinum-based chemotherapy and radiation. The capacity of cancer cells to identify and repair the damage caused by therapeutic assault is a major driver of acquired resistance and therefore limits the effectiveness of these conventional treatment approaches. 2 Therefore, pharmacological targeting of DNA repair proteins may increase the efficacy of current treatments and overcome the risk of therapeutic resistance.3,4 Furthermore, the concept of synthetic lethality, whereby loss of function of either one of two interrelated genes is not lethal but loss of both genes results in cell death, provides a promising platform for pharmacological targeting of DNA repair genes.5,6
Here we focus on a group of enzymes termed endonucleases, which are involved in several DNA repair pathways and act to cleave either damaged DNA or nucleotide by-products of repair, thus facilitating progression to DNA synthesis and ligation. We provide an overview of the DNA repair pathways within which these enzymes are involved and how they function ( Figs. 1 – 3 ). We then go on to discuss their role in cancer and their prognostic value. Finally, we describe their potential as drug targets for personalized therapy in cancer ( Table 1 ).
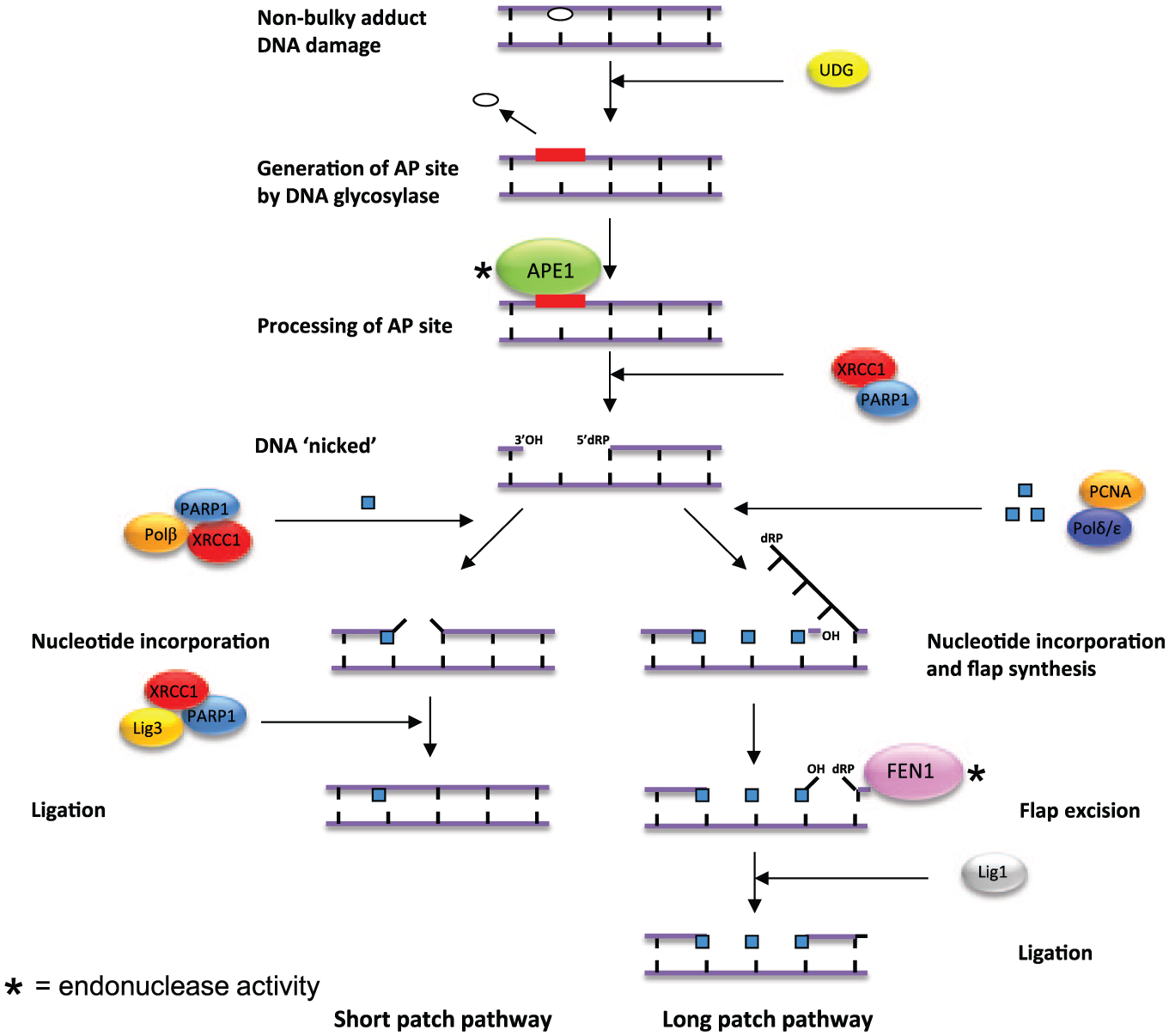
The role of endonucleases in short-patch and long-patch base excision repair following damage from nonbulky adducts. AP endonuclease 1 (APE1) is essential for cleavage of cytotoxic apurinic/apyrimidinic (AP) sites. Flap endonuclease 1 (FEN1) acts only in long-pathway base excision repair (BER) and cleaves the nucleotide flap in preparation for ligation.
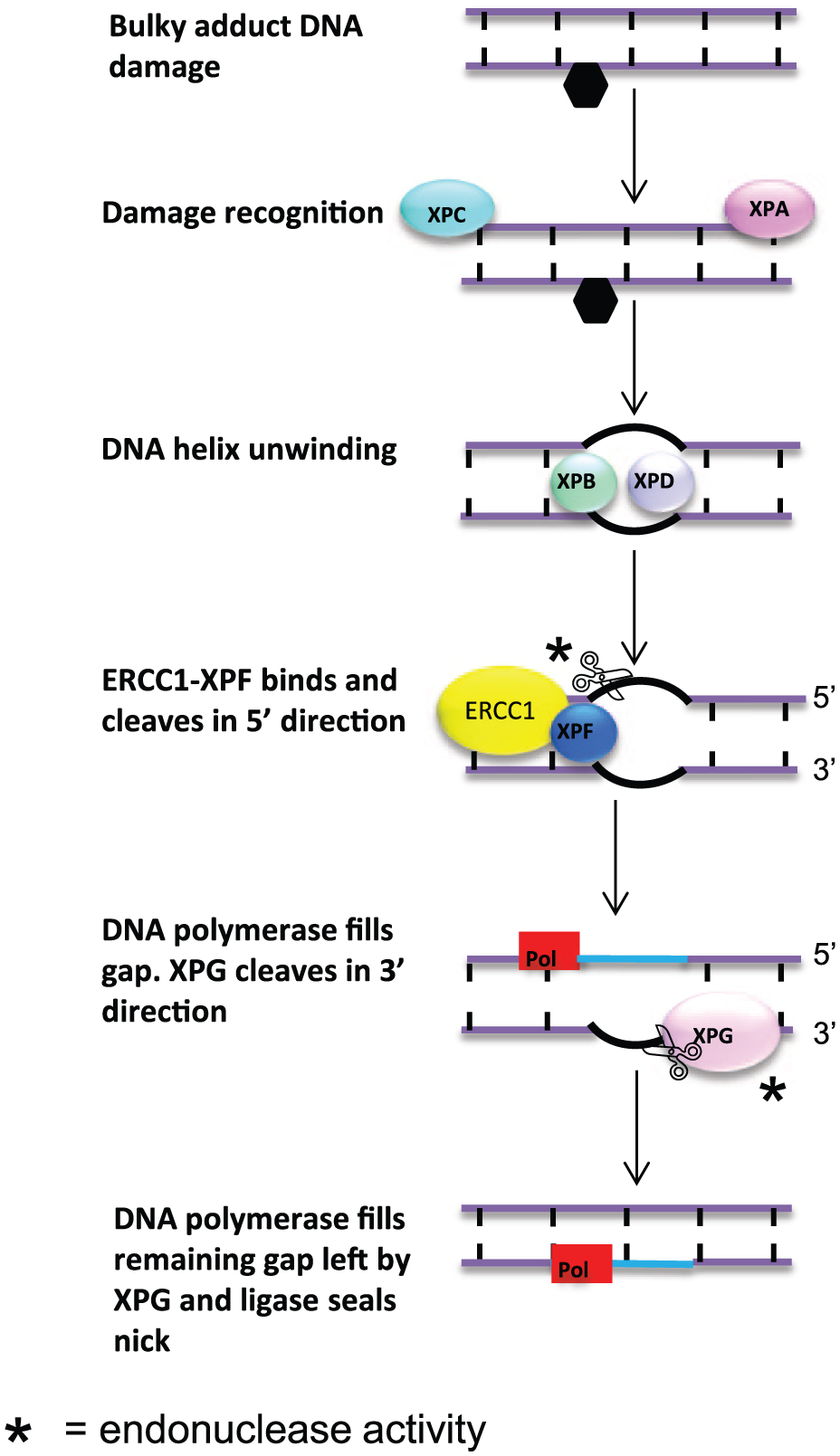
Nucleotide excision repair of bulky adducts. Following recognition of damage by XP group C (XPC) and XP group A (XPA), the DNA helix is unwound by XP group B (XPB) and XP group D (XPD). ERCC1-XPF then preferentially cleaves DNA in the 5′ direction. Following cleavage, DNA polymerase partially fills in the gap. XP group G (XPG) then cleaves at the 3′ end, and DNA polymerase completes repair.
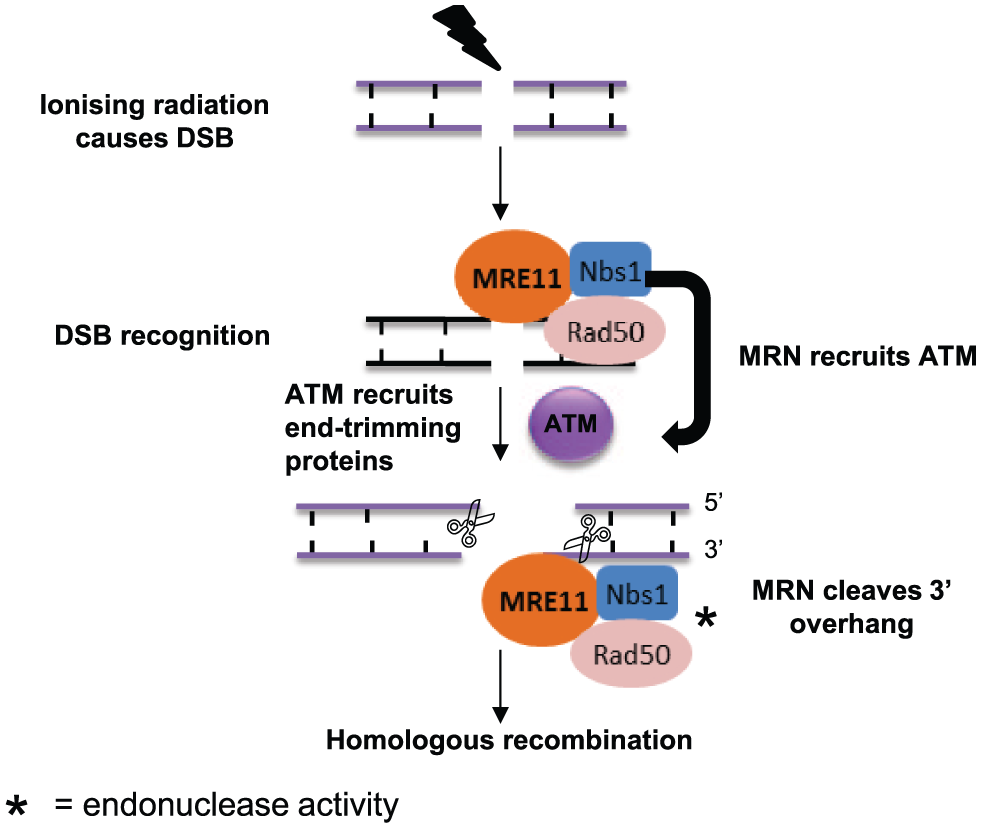
Initiation of homologous recombination by MRE11-RAD50-NBS1 (MRN) complex following a double-strand break (DSB). The MRN complex first acts to recognize the DSB and recruit ataxia-telangiectasia mutated (ATM). MRN goes on to act as an endonuclease and preferentially cleaves the 3′ overhang generated during DSB processing, after which homologous recombination proceeds.
Overview of the Role of Endonucleases in Cancer.
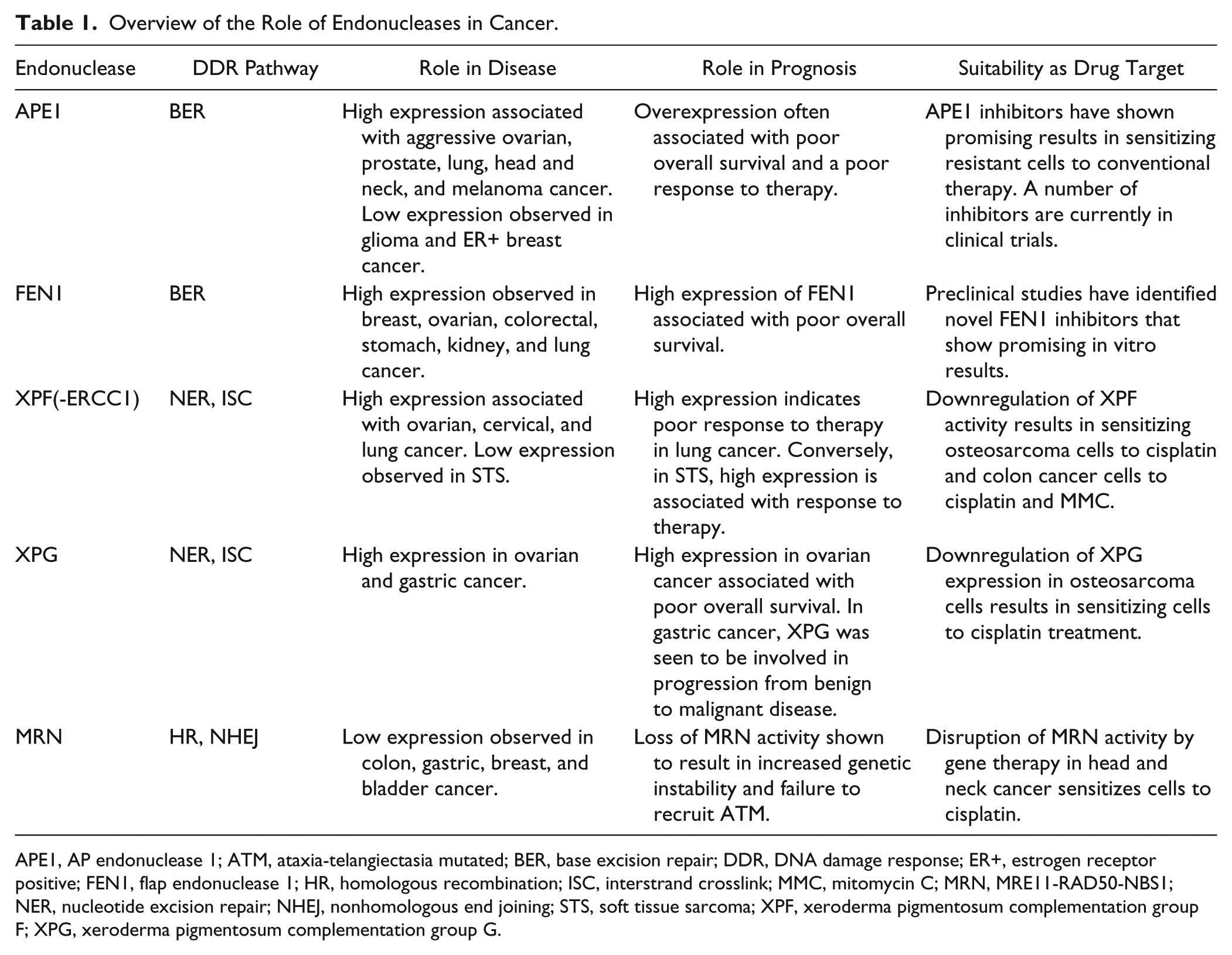
APE1, AP endonuclease 1; ATM, ataxia-telangiectasia mutated; BER, base excision repair; DDR, DNA damage response; ER+, estrogen receptor positive; FEN1, flap endonuclease 1; HR, homologous recombination; ISC, interstrand crosslink; MMC, mitomycin C; MRN, MRE11-RAD50-NBS1; NER, nucleotide excision repair; NHEJ, nonhomologous end joining; STS, soft tissue sarcoma; XPF, xeroderma pigmentosum complementation group F; XPG, xeroderma pigmentosum complementation group G.
Endonucleases in DNA Repair
Base Excision Repair Endonucleases
Base excision repair (BER) is responsible for the repair of damage to DNA bases caused by alkylation and oxidation in addition to deamination of bases and also single-strand breaks (SSBs). 7 There are two major BER subpathways; short patch (SP-BER) and long patch (LP-BER) ( Fig. 1 ). These pathways are complex and differ on the length of the DNA patch and which endonucleases are involved. Although LP-BER and SP-BER involve numerous different proteins, both pathways are initiated in the same manner in response to damaged bases ( Fig. 1 ). First, human AP endonuclease 1 (APE1) is activated following the removal of a damaged base and the subsequent creation of an apurinic/apyrimidinic (AP) site by DNA glycosylase. AP sites also arise spontaneously or as a result of damaging irradiation.8–10 APE1 is vital for the identification and processing of AP sites. If left unattended, these intermediate products of BER are cytotoxic, most likely due to interfering with replication forks. 11 APE1 acts to cleave the DNA phosphodiester backbone of AP sites, resulting in a nucleotide gap flanked by a 3′-hydroxyl group and a 5′-deoxyribose phosphate group. APE1 is a globular α/β protein, which is essential in the initiation of repair in both long-patch and short-patch BER; in addition, APE1 also has regulatory effects on redox activity. 12 Once APE1 has acted to cleave cytotoxic AP sites, repair continues via either the short-patch or long-patch pathway ( Fig. 1 ).
During LP-BER DNA, polymerases process the gap via lagging strand DNA synthesis. Once DNA has been primed and extended, the segments of DNA left on the lagging strand result in the generation of a “flap” structure of approximately 2 to 12 nucleotides. Removal of this flap by the endonuclease, FEN1, is therefore necessary. FEN1 acts to cleave the 5′ nucleotide flap, which allows for final sealing of the DNA gap by DNA ligases. 13
Downregulation of these vital endonucleases, APE1 and FEN1, has been shown to result in stalled BER and sensitization of cells to base-damaging alkylating agents.14–16
Nucleotide Excision Repair Endonucleases
In contrast to BER, nucleotide excision repair (NER) involves removal of damage to bases through endonuclease excision of short strands of oligonucleotides ( Fig. 2 ). While BER recognizes damage from nonbulky adducts caused by alkylation, deamination, and oxidation, NER identifies specific bulky adducts. The most common bulky adducts are cyclobutane pyrimidine (CPDs) and photoproducts (6-4PPs), caused by UV light. There are two subpathways of NER, global genome (GG) and transcription-coupled (TC). GG-NER addresses lesions across the whole genome, whereas TC-NER specifically targets damage to transcribed DNA.17,18 The heterodimer endonuclease complex “excision repair cross complementing group 1 xeroderma pigmentosum complementation group F” (ERCC1-XPF) and the endonuclease “xeroderma pigmentosum complementation group G” (XPG) are essential for endonuclease activity during NER and are involved in both GG-NER and also TC-NER. 19 Following recognition of DNA damage, such as the presence of CPDs, by XP group A (XPA) and XP group C (XPC) and unwinding of DNA by helicases, such as XP group B (XPB) and XP group D (XPD), the endonuclease ERCC1-XPF complex is recruited.
ERCC1 protein forms a heterodimer with XPF, and it is in fact only XPF that performs nuclease activity. Specific knockdown of ERCC1 has been shown to result in loss of XPF expression and nuclease activity.20–22 ERCC1 is essential for correct positioning and binding to DNA and in recruitment of other NER proteins. The nuclease activity of XPF proceeds to cut in the 5′ direction upstream of the damaged strand, resulting in a DNA flap containing the damage. A single strand of undamaged DNA is left behind, which is used as a template to quickly synthesize a complementary strand by DNA polymerase. 23 DNA polymerase synthesis machinery partially fills in the gap prior to cleavage at the 3′ end, performed by the endonuclease known as XPG, after which completion of the repair process occurs. In this way, NER is tightly regulated and an empty gap in DNA during repair is avoided ( Fig. 2 ).24,25
The heterodimer ERCC1-XPF also plays a role in interstrand crosslink (ISC) repair and double-strand break (DSB) repair caused by exogenous sources such as radiation and chemotherapy, notably platinum-based therapy.26–28 ERCC1-XPF acts independently of NER when involved with ISC repair. ERCC1-XPF–deficient cells have been shown to be hypersensitive to cross-linking agents. Following the formation of an ISC in DNA, replication is stalled and the replication fork collapses, resulting in a DSB. The DSB creates a substrate for XPF to cleave and remove the interstrand cross link (ICL) from one of the strands. How the remaining adduct of an unhooked ICL is removed remains unclear.
Endonucleases in Repair of DSBs
There are two main modes of DSB repair in mammalian cells: homologous recombination (HR) and nonhomologous end joining (NHEJ). HR requires a homologous sequence of DNA at the break region to act as a template to ensure error-free repair ( Fig. 3 ),29–31 whereas NHEJ does not use a template but directly processes end terminals of DNA and initiates ligation. Therefore, NHEJ results in restoration of damaged DNA but may not preserve original sequence fidelity. In this way, NHEJ is a far more error-prone pathway compared with HR.32,33
The DNA repair protein meiotic recombination 11 (MRE11) is essential in HR in mammalian cells and is thought to also play a minor role in NHEJ, although its specific role remains unclear.34–37 MRE11 has numerous mechanisms of action in HR, including exonuclease activity, endonuclease activity, and recruitment of other complexes, including ataxia-telangiectasia mutated (ATM). MRE11 has two DNA binding domains, one for double-stranded DNA and one for single-stranded DNA. In HR, the MRE11-RAD50-NBS1 (MRN) complex has distinct endonuclease activity and preferentially cleaves the 3′ single-strand DNA overhang generated by resection of double-stranded DNA to allow DNA synthesis to progress ( Fig. 3 ). Furthermore, it has recently been shown that inhibition of the endonuclease activity of MRN blocks HR and induces cells to resort to the more error-prone NHEJ pathway for DSB repair. 38
Additional endonucleases recently described in DNA repair include FANCD2-associated nuclease 1 (FAN1) and SLX1-SLX4 endonucleases. FAN1 is involved in the processing of DNA ICLs. 39 SLX-SLX4 endonuclease has a role in Holliday junction resolution during homologous recombination. 40
DNA Repair Endonucleases as Prognostic and Predictive Biomarkers in Cancer
It is becoming clear that expression of DNA repair enzymes in tumors is a valuable prognostic and predictive tool. Loss of DNA repair activity in previously healthy cells often results in genomic instability and subsequently carcinogenesis. Therefore, downregulation of DNA repair genes is often observed in numerous cancer types. However, to complicate matters further, upregulation of DNA repair activity is also a common feature associated with development of resistance to DNA-damaging therapy, such as radiotherapy and chemotherapy. Furthermore, in recent years, much focus has been put on germline mutations and single-nucleotide polymorphisms (SNPs) in DNA repair genes, which result in a predisposition toward genomic instability and the development of cancer.41–46
APE1
Recently, several studies in human tumors have demonstrated a promising prognostic role for APE1 expression in cancer development.47,48 For example in several cancer types, including, head and neck cancer, ovarian cancer, osteosarcoma, lung cancer, and melanoma, low APE1 expression has been shown to be a marker of better prognosis and positive response to both chemotherapy and radiotherapy.49–53 In a screen of 103 patients with non–small cell lung cancer, more than 70% of patients displayed high APE1 expression. APE1 was expressed in both the nucleus and also cytoplasm in the majority of tumor samples in contrast to normal healthy lung tissue, where APE1 staining was mostly seen only in the nucleus. Patients with high APE1 expression had a significantly shorter disease-free survival time and overall survival time. Seventy-three patients in the study received cisplatin treatment, and of those patients who developed resistance, more than 80% expressed high levels of APE1. Preclinical studies in the lung cancer cell line A549 demonstrated that inhibition of APE1 using small interfering RNA (siRNA) technology sensitized cells to cisplatin treatment.
In contrast, in some cancers, low APE1 expression was actually seen to have a poorer prognosis. In adult patients with glioma, for example, low nuclear expression of APE1 was associated with decreased overall survival in patients who received the DNA alkylating agent therapy tomozolomide but not in patients who had not received therapy. Similarly, in patients who had received radiotherapy, low APE1 expression was significantly associated with decreased overall survival but was not significant in predicting survival in patients who had not received radiotherapy. 54 Furthermore, the significance of APE1 expression in breast cancer appears to be complex. Initial studies suggest that high APE1 expression indicates a poor prognosis in breast cancer,55,56 whereas a more recent study conducted in a large cohort of breast cancers showed that low APE1 expression in estrogen receptor (ER)–positive tumors correlated with an aggressive phenotype and poor response to hormone therapy. 57 The clinicopathological significance of APE1 in patients appears to very much depend on tumor type and therefore must be approached with caution.
FEN1
Preclinical studies in vivo have shown that FEN1 deficiency results in altered DNA repair, a higher rate of mutations, and a greater predisposition to cancer development. Mice harboring a mutation to the nuclease active site of FEN1 developed significantly higher numbers of tumors, including lymphoma, lung, kidney, liver, and stomach, due to increased genetic instability.58–62 However, in human studies, FEN1 has frequently been shown to be overexpressed in a number of tumors, including breast, ovarian, colorectal, stomach, lung, and kidney cancers, and is associated with resistance to therapy due to increased DNA repair capacity.50,57,63,64 It has recently been shown in a large cohort of patients with breast cancer that high FEN1 expression correlated to an aggressive phenotype and decreased overall survival. 63 FEN1 protein staining was observed in both the nucleus alone, cytoplasm alone, and both nucleus/cytoplasm. Unlike observations in APE1, localization of FEN1 did not have a significant effect on prognosis, although staining observed in both compartments had the worst prognosis. In the same study in a cohort of 156 patients with ovarian cancer, high FEN1 expression was associated with higher grade tumors and a poor overall survival probability. Again, the poorest outcome was seen in tumors expressing FEN1 in both the nuclear and cytoplasmic compartment. Taken together, this study demonstrates that FEN1 expression is a promising biomarker for both breast and ovarian cancers and may prove a novel target for therapeutic inhibitors.
ERCC1-XPF and XPG
The role of ERCC1-XPF and XPG in ISC and DSB repair in response to ISC formation following platinum-based chemotherapy has been shown to be significant, rendering these NER proteins promising predictive markers of platinum-based treatment in cancer. 65 Preclinical studies in several cancer cell lines, including ovarian, cervical, and lung, support the hypothesis that overexpression of ERCC1 correlates with resistance to platinum-based therapies.66–69 In a large-scale study into lung cancer treatment, it was shown that patients with ERCC1-negative tumors benefited significantly more from cisplatin treatment, whereas patients with ERCC1-positive tumors did not respond to treatment and had a poorer prognosis. 70 Furthermore, an investigation in colorectal cancer demonstrated that patients with low expression of ERCC1 had a significantly greater median survival in comparison to those expressing high levels of ERCC1, although there was no correlation with response to 5-fluoracil/oxaliplatin treatment. 71 However, in gastric cancer, ERCC1 has been shown to be an indicator of response to this type of therapy. Patients with low expression of ERCC1 were more likely to respond to 5-fluoruracil/oxaliplatin treatment compared with patients with high ERCC1 expression and hence had a longer overall survival. 72 An in vitro study using the osteosarcoma cell line U2OS demonstrated that knockdown of XPF and XPG expression resulted in sensitization of cells to both cisplatin and oxaliplatin treatment. 73 In a study of 90 ovarian cancer patient samples, XPG overexpression has been shown to be associated with poor overall survival compared with patients exhibiting low XPG expression. 74 In addition, in gastric cancer, expression of XPG is associated with progression from benign atrophic and superficial gastritis to gastric cancer. 75
There are a number of preclinical studies demonstrating that high expression of ERCC1-XPF is associated with platinum resistance in several cancer types, but in patients receiving non-platinum-based ERCC1-XPF and XPG, overexpression is in fact an indicator of improved overall survival and response to treatment. 76 The drug trabectedin is approved in Europe and the United States for treatment of soft tissue sarcoma (STS) and also ovarian cancer. Patients with STS expressing high levels of ERCC1-XPF and XPG have been shown to exhibit increased overall survival and a greater response to trabectedin treatment compared with patients with low expression. 76 In addition, cell lines derived from XP patients that are deficient in NER proteins, including XPF and XPG, have been shown to be resistant to trabectedin. 77 The observations that high expression of NER proteins predicts a greater response to trabectedin treatment are the opposite to observations in cisplatin, whereby overexpression of NER activity was associated with resistance to platinum-based DNA-damaging agents. 78 The exact mechanism of action of trabectedin in the context of NER activity remains unknown, but it has been suggested that trabectedin-induced adducts that are not effectively repaired trap NER proteins, resulting in the formation of cytotoxic complexes, and therefore NER-deficient cells are protected from the effects of trabectedin. 79
In light of the preclinical studies performed in ERCC1-XPF expression, there are several clinical trials currently under way investigating whether ERCC1-XPF expression status can be used to determine treatment decision. A phase II trial in advanced/metastatic esophageal and gastric cancer is currently investigating whether high ERCC1 expression is able to predict response to platinum-based DNA-damaging agents and determine which patients will benefit from receiving dual therapy. 80 A phase II trial in pancreatic cancer is also investigating ERCC1 expression as a marker for platinum-based chemotherapy treatment response following tumor resection. 81
In addition to its promising use as a prognostic and predictive marker in cancer, the data acquired from preclinical and clinical studies of ERCC1-XPF expression in cancer clearly support the notion that it may be a promising anticancer drug target.
MRN Complex
Loss of expression of the MRN complex is frequently observed in a number of cancer types, including colorectal, gastric, endometrial, breast, and bladder cancer.82–86 Impaired activity of MRN can occur due to loss of expression of any of the three proteins, MRE11-RAD50-NBS1, and therefore has a detrimental effect on MRN activity. Mutations in the coding region of MRE11 leading to loss of expression are regularly observed in colorectal cancer, whereas in breast cancer, loss of Nijmegen breakage syndrome 1 (NBS1) expression is more frequently observed. Furthermore, a change in the localization of NBS1 from the nucleus to the cytoplasm has been associated with a more aggressive phenotype in breast cancer. RAD50 has also been shown to be underexpressed in breast cancer, although not as frequently as NBS1 or MRE11. 87 In mice, overexpression of RAD50 has been proven to have an antitumorigenic effect. 88 Loss of the MRN complex in cancer also has protumor effects due to failure to recruit the DNA damage sensor ATM, which is essential for recruitment of numerous DNA repair proteins. 89
Germline Mutations and SNPs in DNA Repair Genes
Germline mutations are often found in the coding region of DNA repair genes. 41 In the context of DNA repair, probably the most well-characterized inherited germline mutations are those found in BRCA1/2 genes, resulting in a high predisposition toward the development of several cancer types.42–46 Others include germline mutations observed in the NER genes XPF and XPG. Individuals harboring these mutations have the rare syndrome XP.19,90,91 Patients have extreme photosensitivity and are at a 1000-fold increased risk of developing skin cancer.
In addition to germline mutations, a number of SNPs have been identified in human DNA repair genes.92–94 SNPs occur throughout the genome and at a much higher rate compared with mutations. The role of SNPs in DNA repair genes may serve as promising prognostic or predictive markers in a number of ways. For example, polymorphisms may result in suboptimal DNA repair and subsequently alter tumor biology, serving as a prognostic indicator. Alternatively, defective DNA repair due to SNPs may affect a tumor’s capacity to repair damage induced by therapeutic agents and therefore may predict a patient’s response to therapy. 3 Furthermore, the presence of certain SNPs has been reported as markers of a predisposition toward development of cancer. For example, in APE1, Ağaçhan and colleagues 95 reported that the amino acid substitution variant D148E resulted in a significant increase in lung cancer susceptibility. A frequent polymorphism observed in the XPF gene, rs1800067, which results in an arginine-to-glutamine transition at codon 415, is thought to reduce ERCC1-XPF activity by altering protein interactions and subsequently resulting in diminished NER and a higher risk of cancer. 96
DNA Repair Endonucleases as Targets for Cancer Therapy
In addition to surgical intervention, chemotherapy and radiotherapy remain the two chief treatment options available to patients with cancer. The efficacy of these agents is directly related to their capacity to induce DNA damage in cancer cells.97,98 Cells have a number of options available to them following DNA damage, including induction of apoptosis, cell cycle arrest, and DNA repair. Subsequently, cancer cells often overexpress DNA repair proteins, which promote cell survival and therefore negatively affect treatment efficacy and development of resistance. Significant progress in understanding how cells respond to DNA damage has been made in recent years, which in turn may have profound clinical benefits. Furthermore, a number of high-throughput, robust screening techniques have successfully identified potential novel inhibitors of DNA repair proteins to be taken forward.99,100 The studies discussed here thus far suggest that targeting DNA repair endonucleases in combination with DNA-damaging agents may be a promising anticancer strategy.
APE1 Inhibitors
Screening of small-molecule libraries has uncovered numerous potential inhibitors of DNA repair endonucleases, including APE1 inhibitors.53,100–103 Abbotts and colleagues 53 took four APE1 inhibitors previously identified and demonstrated that inhibition of APE1 activity in cutaneous melanoma cell lines resulted in accumulation of AP sites, increased DNA damage, and cell death. One of the APE1 inhibitors used in this study, CRT004876, has also been shown to potentiate the effects of ionizing radiation in lung cancer cell lines. 104 The known topoisomerase II inhibitors, lucanthone and hycanthone, have been shown to inhibit APE1 endonuclease activity by direct binding to APE1’s hydrophobic region without affecting redox activity. 105 In addition, treatment of radiotherapy-resistant glioblastoma cell lines with lucanthone has been shown to sensitize cells to radiotherapy. 106 Lucanthone has been shown to readily cross the blood-brain barrier in both the presence and absence of brain tumors in mice and rats. 107 In a small study of eight patients with brain metastases, lucanthone treatment in combination with radiotherapy resulted in a significant increase in tumor regression. 107 Lucanthone is now in phase II clinical trials for treatment of non–small cell lung cancer brain metastases in combination with radiotherapy. 108
The naturally occurring Bcl-2 homology 3 (BH3)–mimetic agent Gossypol has been shown to inhibit both the endonuclease activity and also the redox activity of APE1 through antagonizing Bcl-2, which directly interacts with APE1 through its BH domains. Therefore, overexpression of Bcl-2 results in downregulation of APE1 activity. 109 In combination with cisplatin, Gossypol has been shown to significantly decrease cell survival in vitro and tumor growth in vivo. 110 This study has led to a phase III clinical trial of Gossypol in patients with non–small cell lung carcinoma. 111 Patients whose tumors express high levels of APE1 are currently being treated with docetaxel and cisplatin in combination with Gossypol to determine whether blockade of APE1 expression in these patients sensitizes tumors to chemotherapy treatment. It is anticipated that in the coming years, APE1 inhibitors will be a viable treatment option for personalized therapy in patients exhibiting high levels of APE1.
FEN1 Inhibitors
Inhibitors of FEN1 are not as advanced in comparison to APE1 inhibitors, and there are currently no clinical trials registered for FEN1 inhibitors. However, several preclinical studies have identified potential small molecular inhibitors of FEN1. Chemiluminescent assays developed to determine FEN1 activity have uncovered a number of small molecules that are potent inhibitors of FEN1. 112 In this study, a previously reported inhibitor of FEN1, 3-hydroxy-5-methyl-1-phenylthieno [2, 3-d] pyrimidin-e-2, 4(1H, 3H) dione (PTPD), was confirmed to be a potent FEN1 inhibitor in the submicromolar range. The previously reported APE1 inhibitor, aurintricarboxylic acid (ATA), was also shown to be a potent inhibitor of FEN1 in the chemiluminescent assays.113,114 The specificity of these agents to FEN1 needs further investigation; however, it was shown that these compounds potentiate treatment with chemotherapies that induce DNA damage repaired by the BER pathway. They did not potentiate treatment with bleomycin, which does not activate the BER pathway, therefore suggesting their specificity to BER proteins, although further work is necessary.
A further four small molecules were identified as potent FEN1 inhibitors by Dorjsuren and colleagues 101 and are currently being screened in vitro. In 2013, AstraZeneca (London, UK) conducted a high-throughput screen of 850,000 compounds using a fluorescent DNA cleavage assay to determine FEN1 inhibition activity. 99 This screen resulted in the identification of 6261 compounds with FEN1 inhibition activity at an IC50 of less than 100 µM. 99
A study in colorectal cancer cell lines revealed that FEN1 inhibition by a number of small molecules had a synthetically lethal effect in cells deficient in the ubiquitin ligase CDC4. 115 Interestingly, in a recent study, curcumin, the active ingredient in turmeric, has been shown to restrict cell proliferation in breast cancer cell lines through inhibition of FEN1 activity. 116 It was shown that curcumin upregulates expression of the transcription factor NF-E2–related factor 2 (nrf2). Subsequently, nrf2 is translocated to the nucleus and binds directly to the FEN1 promoter, decreasing promoter activity.
ERCC1-XPF Inhibitors
Although there is much focus currently on the prognostic and predictive value of ERCC1-XPF expression in cancer, there are very few identified inhibitors of ERCC1 or its co-protein, XPF. Disruption of the interaction between ERCC1 and XPF results in instability and reduced endonuclease activity. 117 A virtual screen of potential inhibitors of the ERCC1-XPF interaction using available crystal structures and subsequent in vitro studies in lung, breast, and colon cancer cell lines identified a novel inhibitor of the ERCC1-XPF interaction, NERI02. 118 The specificity of NERI02 remains unclear; however, it has been shown to act synergistically with both cisplatin and mitomycin C (MMC). This may be due to it binding directly with DNA and therefore potentiating platinum sensitivity. In addition, in lung and colon cancer, treatment with NERI02 sensitized cells to UV radiation. NERI02 has also been seen to act synergistically with the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in BRCA1 mutant breast cancer cells.
A further virtual screen and in vitro study investigating inhibitors of the interaction between ERCC1 and XPA has identified a novel compound that sensitizes colon cancer cells to ionizing radiation.119,120 A preclinical study has found that the kinase inhibitor sorafenib, which is currently used in clinic to treat advanced renal cell carcinoma, hepatocellular carcinoma, and thyroid cancer, indirectly downregulates ERCC1-XPF activity in head and neck cancer cell lines in a dose-dependent manner. 121 Combined treatment of cancer cell lines with sorafenib and radiotherapy resulted in significantly greater cell killing. In addition, sorafenib combined with radiotherapy treatment or cisplatin treatment in vivo significantly inhibited tumor growth.
MRN Blockade
Gene therapy studies in head and neck cancer cells in vitro and in vivo using the adenoviral mutant NBS1 protein have demonstrated that disruption of the MRN complex results in sensitization to radiotherapy and platinum-based chemotherapy.122,123 In addition to gene therapy–targeted disruption of the MRN complex, a forward chemical genetic screen of small molecules has also identified the MRN inhibitor “Mirin.” 124 Mirin was shown to disrupt the association of MRN with ATM and also the nuclease activity of MRN blocking efficient HR. However, Mirin is a relatively weak inhibitor, and efforts are under way to establish more potent inhibitors of the MRN complex. Since the MRN complex is vital in repair of radiation-induced DSBs, pharmacological targeting of MRN is a promising radiosensitizer.
Challenges to Development of Novel DNA Repair Inhibitors
Although APE1, FEN1, ERCC1-XPF, and MRN are promising targets for cancer therapy, the development of inhibitors for therapeutic application is still at an early stage. The “promise” of these agents to reach the clinic will be largely dictated by the physical and chemical properties of small molecules under development. Moreover, there are also concerns regarding the long-term toxicity of these agents either as monotherapy or in combination with cytotoxic agents such as chemotherapy and radiotherapy. In this context, selective targeting through synthetic lethality could be a promising approach for personalization of therapy.
Targeting Synthetic Lethal Relationships
Since the groundbreaking work investigating the synthetically lethal relationship between the BER polymerase PARP and BRCA1/2-deficient cells, efforts to bring an effective PARP inhibitor to the clinic have been overwhelming.125–127 BRCA1/2 mutant cells have defective DSB repair capacity and are therefore heavily reliant on the BER pathway to prevent the formation of DSBs. PARP is an essential component of the BER pathway in recognizing SSBs and recruiting repair proteins. Therefore, inhibition of PARP in HR-deficient cells is catastrophic. There are currently 111 registered clinical trials using a number of PARP inhibitors in solid tumors. The first-in-class PARP inhibitor, olaparib (Lynparza; AstraZeneca, London, UK), has now been approved for treatment of BRCA1/2 mutant platinum-sensitive ovarian cancer by both the US Food and Drug Administration and European Commission.
The discovery of the synthetically lethal relationship between BRCA1/2 and PARP has fueled investigations into whether targeting PARP in tumors with aberrations in other DNA repair genes could be an effective approach in BRCA1/2-proficient tumors. PARP inhibition using olaparib and veliparib has recently been shown to be effective as a single agent in ERCC1-low non–small cell lung cancer cells and also potentiate cisplatin treatment in these cells. ERCC1-low cells exhibited accumulation of DSBs, cell cycle arrest, and induction of apoptosis following PARP inhibitor monotherapy and in combination with cisplatin. In comparison, ERCC1-high cells did not exhibit the same cytotoxic effects following treatment. 128 A recent study has demonstrated that treatment of MRE11-deficient endometrial cancer cell lines with the PARP inhibitor BMN673 (BioMarin) results in significant cell death in vitro compared with MRE11-proficient cell lines. 84 In a screen of 430 endometrial tumors, MRE11 was underexpressed in 30% of cases, and therefore a synthetic lethal approach in these tumors in combination with PARP inhibition would be a feasible treatment option in nearly a third of all endometrial cancers. Subsequently, BMN673 is currently being trialed in a phase II clinical trial in advanced endometrial cancer with retrospective analysis of MRE11 expression in tumors. 129 It appears that PARP has a number of synthetic lethal gene relationships other than those previously reported with BRCA1/2. Therefore, the scope for using PARP inhibitors in tumors that underexpress DNA repair proteins such as ERCC1 and MRE11 is vast.
Although PARP inhibitors have proven to have remarkable promise in the field of personalized cancer therapy, there are some limitations to their use. The greatest issue is that there are 17 different variants of PARP known to have various functions in DNA repair pathways other than BER, in addition to roles in transcription, translation, and telomere maintenance. Therefore, specificity of inhibitors has proven to be highly problematic. Therefore, targeting other members of the BER pathway, such as APE1 and FEN1, in HR-deficient cells may prove to be just as effective and less problematic. Sultana et al. 130 recently provided evidence that targeting APE1 by small molecular inhibitors was synthetically lethal in BRCA-deficient cells. In a more recent study, inhibition of APE1 in cutaneous melanoma cell lines deficient in phosphatase and tensin homologue (PTEN) was significantly more sensitive to APE1 inhibition compared with PTEN-proficient cell lines. 53 PTEN is known to be involved in the antiapoptotic PI3K/Akt pathway and is also increasingly thought to be required for efficient HR. This study demonstrates the potential synthetic lethal applications of inhibition of an alternative BER protein, APE1, in tumors that are deficient in PTEN.
In conclusion, the mechanisms of DNA repair are essential in maintaining genomic stability in cells. A number of different pathways in place are capable of dealing with the different types of DNA damage. Defects in these pathways result in a greater risk of genomic instability and therefore a predisposition to cancer. Impaired DNA repair mechanisms in cancer cells provide an opportunity to exploit this weakness for therapeutic purposes due to their sensitivity to DNA-damaging agents. However, cancer cells often get around this vulnerability by using other repair pathways to deal with damage or by overexpressing DNA repair proteins. This allows cancer cells to tolerate DNA damage and subsequently puts them at risk of acquiring further genetic aberrations. Overexpression of DNA repair proteins is frequently associated with resistance to DNA-damaging therapeutics. Therefore, in the past decade, much emphasis has been on development of novel inhibitors of DNA repair proteins for monotherapy or in combination with DNA-damaging agents such as platinum therapy or radiotherapy. To this end, the first inhibitor of the BER protein PARP has been approved for use in cancers deficient in HR. PARP inhibitors are not alone, and numerous other DNA repair inhibitors currently in clinical trials will aid the transformation of current cancer therapy toward a more personalized approach. The role of DNA repair pathways and subsequent development of inhibitors of these pathways are hugely important in the field of cancer research and an important area of ongoing research.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.