
Abstract
MicroRNAs (miRNAs) are a class of genome-encoded small RNAs that post-transcriptionally regulate gene expression by repressing target transcripts containing partially or fully complementary binding sites.
Despite their relatively low number, miRNAs have been shown to directly regulate a large fraction of the transcriptome. In agreement with their pervasive role in the regulation of eukaryotic gene expression, miRNAs have been implicated in virtually all biological processes, including different pathologies.
The use of screening technologies to systematically analyze miRNA function in cell-based assays offers a unique opportunity to gain new insights into complex biological and disease-relevant processes. Given the low complexity of the miRNome and the similarities to small interfering RNA (siRNA) screening experimental approaches, phenotypic screening using genome-wide libraries of miRNA mimics or inhibitors is not, per se, technically challenging. The identification of miRNA targets and, more importantly, the characterization of their mechanisms of action through the identification of the key targets underlying observed phenotypes remain the major challenges of this approach.
This article provides an overview of cell-based screenings for miRNA function that were performed in different biological contexts. The advantages and limitations of computational and experimental approaches commonly used to identify miRNA targets are also discussed.
Introduction
MicroRNAs (miRNAs) are endogenous small noncoding RNAs, typically 20–22 nt in length, that post-transcriptionally control gene expression. 1 As part of a ribonucleoprotein complex, denominated miRNA-induced silencing complex (miRISC), which includes the essential proteins Argonaute (AGO) and TNRC6, miRNAs repress their target transcripts by inhibiting translation and promoting deadenylation, leading to degradation of target messenger RNAs (mRNAs).2,3
According to the latest release of miRBase (http://www.mirbase.org), the public repository of annotated miRNA sequences, a total of 28,645 miRNA hairpin precursors, corresponding to 35,828 mature miRNAs, have been identified in 223 species (miRBase release 21, June 2014); 2588 mature miRNAs are currently annotated in the human genome.
Despite their relatively small number, miRNAs are central players in the control of gene expression, estimated to regulate up to 60% of the human transcriptome. 4 Consistently, miRNAs have been shown to modulate virtually all biological and pathological processes analyzed, including development, 5 cellular differentiation, 6 proliferation, 7 apoptosis, 8 cancer, 9 and neurodegenerative 10 and cardiovascular 11 diseases.
From a functional genomics perspective, the often convergent action of miRNAs and the fact that the relatively low number of miRNAs modulate a large fraction of the transcriptome provide a unique opportunity to interrogate the genome through the development of phenotypic gain- or loss-of-function miRNA screenings, and thus gain mechanistic insights into complex biological and disease-relevant processes. As well, it provides the opportunity to identify genes involved in phenotypes that ensue from the simultaneous targeting of multiple genes; these are not accessible through classic genetic or RNA interference (RNAi) screening methods that rely on the phenotypic analysis of single mutations or knockdowns.
Modulation of intracellular miRNA levels can be achieved through transfection of synthetic miRNA hairpin precursors or duplex miRNA mimics (to increase miRNA levels), or of oligonucleotides, designated miRNA inhibitors that are designed to sequester mature miRNA sequences and thus decrease the availability of a particular miRNA. Different genome-wide libraries are available from various commercial sources. The design of miRNA mimics frequently involves chemical modification of the passenger strand to minimize their incorporation in the miRISC complex and thus better discriminate the phenotypic consequences of modulating each mature miRNA individually. Although the nature of these modifications is in most cases proprietary, this is an important point that should be considered. The use of miRNA hairpin precursors may be more representative of the physiological setting because these are processed by the cell machinery, but it renders analysis of the effect of each miRNA strand difficult to define because potentially both miRNAs strands can be active. MiRNA inhibitors are typically based on modified nucleotides, most notably locked-nucleic acids (LNAs), or, alternatively, they are based on proprietary designs that involve hairpin structures or other modifications for more efficient miRNA sequestration and increased stability. Alternatively, miRNA overexpression can be achieved from plasmid or viral vectors, which typically allow prolonged expression of the miRNAs; similarly to synthetic precursors, both miRNA strands can be active in this scenario. Although less common, miRNA sponges (i.e., decoy sequences expressed from plasmid or viral vectors containing multiple binding sites for a particular miRNA) can also be used to inhibit miRNA function. As a screening strategy, the use of plasmid- or viral-based libraries allows screening in a pooled format, circumventing the requirement of automated screening platforms required for arrayed screenings. The selection of screening reagents is often dictated by specificities inherent to the cell types under analysis and assay requirements.
The recent generation by the Kim laboratory 12 of a library of transcription activator-like effector nucleases (TALENs) to knockout 274 human miRNA loci constitutes a useful resource for future screening and validation studies.
Given the low complexity of the miRNome, and the similarities of reagents, experimental procedures, and methodologies with those applied for small interfering RNA (siRNA) screening, the development of cell-based phenotypic screenings for systematic analysis of miRNA function using genome-wide libraries of miRNA mimics or inhibitors is not, per se, technically challenging. However, in contrast to siRNA screenings in which the genes of interest are known, miRNA screenings must be followed by an extensive follow-up work aimed at the identification of the target genes of the selected miRNAs.
The process of miRNA target identification, in particular the identification of key miRNA targets responsible for specific phenotypes, is a complex process. The still-incomplete knowledge of the rules governing the binding between miRNAs and their target transcripts, the fact that each miRNA regulates a plethora of target mRNAs, the simultaneous targeting of individual transcripts by multiple miRNAs, and the redundancy of miRNA-mediated control of gene expression, particularly for miRNAs sharing common seed sequences, are important factors that contribute to this complexity.
Notwithstanding, a detailed understanding of the phenotypes elicited by miRNAs requires, ultimately, the extensive and detailed characterization of their targeted transcripts. In this article, we provide an overview of miRNA functional screening studies performed to date, and discuss the progress and challenges associated with target identification strategies.
High-Throughput Screenings for miRNA Function
A number of functional cell-based screenings aimed at elucidating the role of the miRNome in different biological processes and disease-relevant scenarios have been performed to date. These include the study of cell proliferation and cancer, infection by different pathogens, signaling, stem cell maintenance and differentiation, and cardiovascular biology ( Table 1 ; cf. Ref. 13 ).
High-Throughput Screenings for miRNA Function.
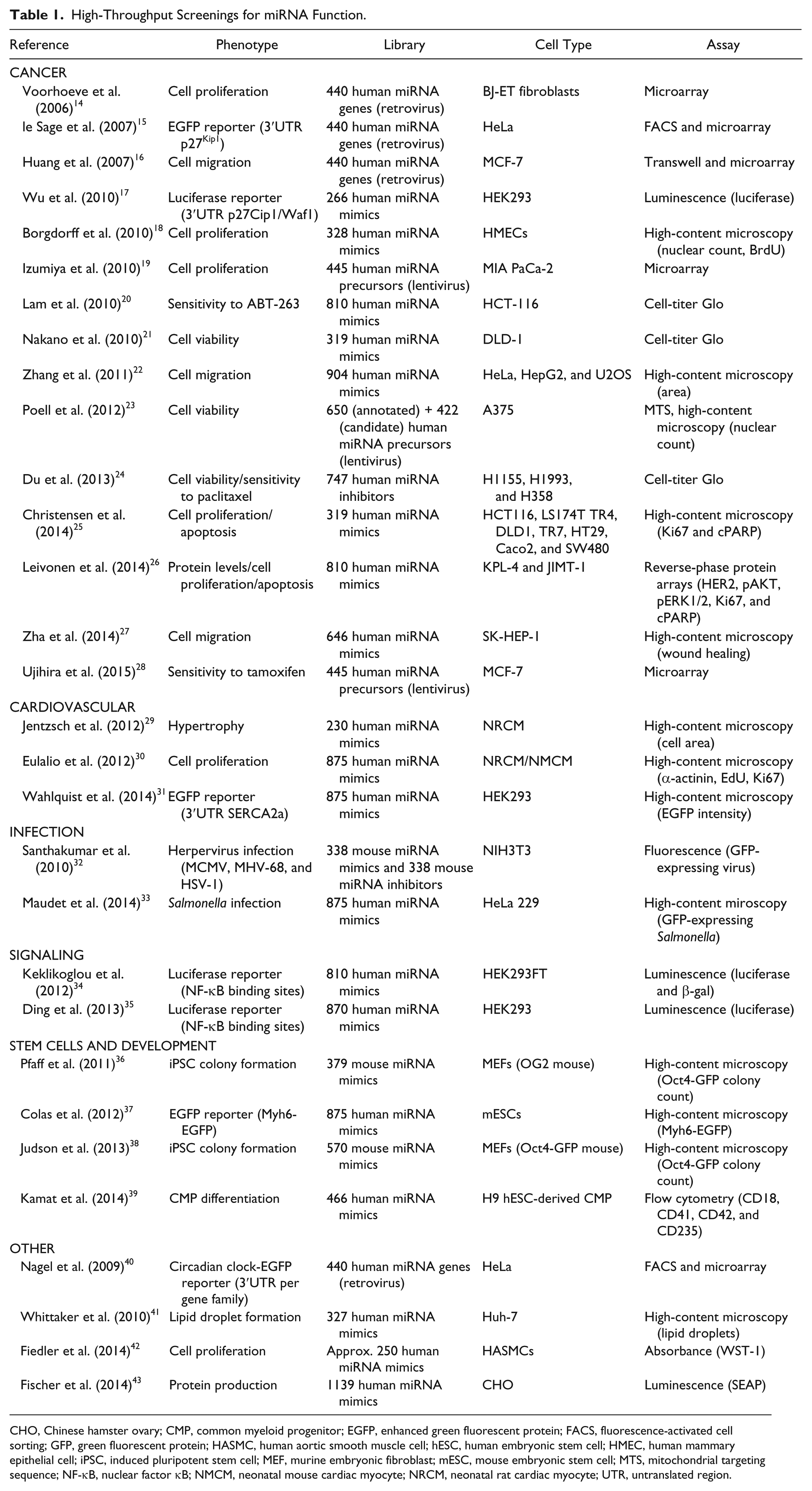
CHO, Chinese hamster ovary; CMP, common myeloid progenitor; EGFP, enhanced green fluorescent protein; FACS, fluorescence-activated cell sorting; GFP, green fluorescent protein; HASMC, human aortic smooth muscle cell; hESC, human embryonic stem cell; HMEC, human mammary epithelial cell; iPSC, induced pluripotent stem cell; MEF, murine embryonic fibroblast; mESC, mouse embryonic stem cell; MTS, mitochondrial targeting sequence; NF-κB, nuclear factor κB; NMCM, neonatal mouse cardiac myocyte; NRCM, neonatal rat cardiac myocyte; UTR, untranslated region.
The involvement of miRNAs in human disease, cancer in particular, was first inferred from studies on chronic lymphocytic leukemia (CLL) and the observation that the genomic region encoding the mir-15a/miR-16-1 cluster is frequently deleted in CLL patients.44,45 Not surprisingly, cancer constitutes a major focus of screening campaigns, including those based on the modulation of miRNA levels. The first study to address the role of miRNAs in the pathogenesis of cancer through an unbiased functional screening was performed by Voorhoeve and collaborators. 14 In this pioneering work, the authors constructed a retroviral vector library expressing the genomic regions corresponding to 400+ human miRNAs and screened for miRNAs able to overcome p53-dependent growth arrest in response to oncogenic RASV12. Using a pooled screening approach, the authors established that miR-372 and miR-373 can act as oncogenes, contributing to cellular transformation in the presence of wild-type p53. The Agami laboratory has subsequently used this retroviral-based miRNA expression library in other studies, which led to the identification of miRNAs involved, for example, in the regulation of the p27Kip1 tumor suppressor, 15 as well as in tumor invasion and metastasis. 16 More recent works explored the possible involvement of miRNAs in a wide range of malignancies by analyzing the effect of miRNAs on cancer cell migration,22,27 viability and proliferation, and/or apoptosis of pancreatic, 19 colorectal,21,25 melanoma, 23 and breast 26 cancer cell lines, as well as the susceptibility of cancer cells to different pharmacological agents.20,24,28 These screenings use different experimental strategies, ranging from synthetic arrayed libraries of miRNA mimics, precursors, or inhibitors to pooled lentiviral-based miRNA libraries.
The application of phenotypic high-throughput screenings to dissect the molecular events underlying the interaction of host cells with viral and bacterial pathogens has been largely dominated by siRNA screenings (e.g., HIV-1,46,47 influenza virus,48,49 West Nile virus, 50 Listeria monocytogenes, 51 Mycobacterium tuberculosis, 52 and Salmonella Typhimurium 53 ). Currently, there are only a few studies addressing the crucial regulatory roles of miRNAs in these processes. Through the combined screening of mouse miRNA mimic and inhibitor libraries, Santhakumar and coworkers 32 identified miRNAs with broad antiviral activity against virus representatives of all three herpesvirus families, specifically herpes simplex virus 1, murine cytomegalovirus, and murine gamma herpesvirus 68. Although it is well established that bacterial pathogens cause extensive changes in the miRNome of host cells,54–56 until recently the role of miRNAs during infection by bacterial pathogens had not been addressed on a systematic, genome-wide scale. To tackle this issue, we performed a phenotypic high-content screening using a library of miRNA mimics to identify novel modulators of infection by S. Typhimurium, 33 an extensively studied Gram-negative pathogen and a main causative agent of foodborne illnesses. In this study, we described the miR-15 family as important restriction factors for Salmonella infection. We also showed that miR-15 family members are downregulated by Salmonella to ensure efficient infection of host target cells.
Using a luciferase reporter assay in HEK293 cells, Keklikoglou and collaborators 34 performed an unbiased miRNA screening for novel regulators of nuclear factor (NF)-κB pathway activity. This screening identified the miR-520/373 family as a strong inhibitor of NF-κB signaling through direct regulation of RELA. Using a similar strategy, Ding and coworkers 35 also identified miRNAs controlling the NF-κB pathway. Although the authors of this study focused on the characterization of miR-15 family members, members of the miR-520/373 also showed a significant effect.
Modulation of stem cell maintenance and differentiation is a tightly regulated process orchestrated by a multitude of signals and molecular players, and therefore it is not surprising that miRNAs also play an important role therein. 57 Most likely, due to the technical challenges associated with the manipulation of these cells in conditions amenable to high-throughput studies, the number of functional screenings addressing the role of miRNAs in the context of stem cell biology is limited. To evaluate the contribution of miRNAs to the differentiation of embryonic stem cells (ESCs) into multiple hematopoietic lineages, Kamat and collaborators 39 performed a flow cytometry–based miRNA screening using a library of 466 human miRNA precursors that was electroporated in human ESC (hESC)-derived common myeloid progenitors (CMPs). This analysis identified, among other miRNAs, miR-105 as a potent enhancer of megakaryocyte production. To screen for miRNAs involved in the control of endoderm to mesoderm fate, Colas and collaborators 37 used a differentiating transgenic mESC line expressing enhanced green fluorescent protein (EGFP) under the control of the Myh6 cardiac-specific promoter. Using this approach, the authors uncovered a fundamental role of the let-7 and miR-18 families in the process of germ layer specification. Pfaff and collaborators 36 addressed the potential of a library of 379 murine miRNAs to improve the early phases of generation of induced pluripotent stem cells (iPSCs) from mouse embryonic fibroblasts (MEFs), by cooperating with the human transcription factors Oct4, Klf4, and Sox2 expressed ectopically from a polycistronic lentiviral construct. Among other miRNAs identified in this study, the miRNA family comprising miR-130, miR-301, and miR-721 was shown to significantly increase iPSC formation through downregulation of the transcription factor Meox2. A very similar screening strategy was applied by Judson and collaborators 38 using a library of 570 mouse miRNA mimics.
Functional miRNA screenings have also been conducted in the context of cardiovascular research, focusing on different aspects of cardiomyocyte physiology. To identify miRNAs regulating cardiomyocyte hypertrophy, Jentzsch and collaborators 29 performed a high-content screening in neonatal rat cardiomyocytes using 230 conserved miRNA precursors, revealing a number of miRNAs not described previously as anti- or pro-hypertrophic. We have applied a functional high-content screening approach to investigate the potential role of miRNAs in the control of proliferation of rat and mouse cardiomyocytes, using EdU incorporation and Ki67 positivity as phenotypic readouts. 30 From the 875 tested miRNAs, we identified several miRNAs able to dramatically increase the proliferative capacity of neonatal and adult cardiomyocytes in vitro, and promote cardiac regeneration after myocardial infarction in mice. To understand whether miRNAs could control heart contractility, Wahlquist and collaborators 31 performed a high-throughput functional screening to identify miRNAs that target the sarcoplasmic reticulum calcium ATPase SERCA2a, and therefore are potentially responsible for its downregulation and impaired calcium uptake during heart failure. Using a reporter construct composed of the 3′UTR (untranslated region) of SERCA2a fused with EGFP, the authors determined that several miRNAs reduced EGFP expression, and they characterized miR-25 as a strong regulator of SERCA2a expression. Of note, administration of an antagomiR specific to miR-25 improved cardiac function and survival of a mouse model of established heart failure.
To identify miRNAs able to increase recombinant protein production in Chinese hamster ovary (CHO) cells, the prototype mammalian cell line for production of biopharmaceuticals, Fischer and collaborators 43 performed a miRNA screening using a library of 1139 human miRNA mimics. In this study, the miR-30 family was recognized as a potent enhancer of protein production in CHO cells. These miRNAs have a strong biotechnological value because they potentially can be exploited to optimize the performance of CHO cells in numerous applications.
Other examples include a miRNA screening for lipid droplet formation, 41 smooth muscle cell proliferation and vascularization, 42 and regulation of the circadian clock. 40
Although the majority of the screenings described in this section were designed as systematic genome-wide strategies, when considered collectively the heterogeneity of the number of miRNA probe sets representing the miRNome is evident. This reflects the continuous discovery of new miRNA sequences in recent years, driven mostly by developments in RNA-sequencing methodologies; a total of 2588 human miRNA mature sequences are currently annotated in miRBase (release 21), whereas only 988 were annotated in 2009 (release 13.0) and 470 in 2006 (release 9.0).
Computational Approaches for the Identification and Analysis of miRNA Targets
Understanding the role of miRNAs in a given biological process is dependent on the identification of its target mRNAs. An overview of the most commonly used computational approaches currently available to identify and characterize miRNA–mRNA interactions is provided here.
Tools for miRNA Target Prediction
During the past decade, a number of miRNA target prediction tools have been developed, based on algorithms grounded on the sequence complementarity between miRNAs and target transcripts and the location characteristics of miRNA binding sites (reviewed in Refs. 58,59). Table 2 provides an overview of the distinguishing features of the most frequently used miRNA target prediction tools, which collectively constitute a powerful approach for identifying putative miRNA-regulated genes.
Commonly Used Computational Tools and Databases of Predicted and/or Experimentally Validated miRNA Targets.
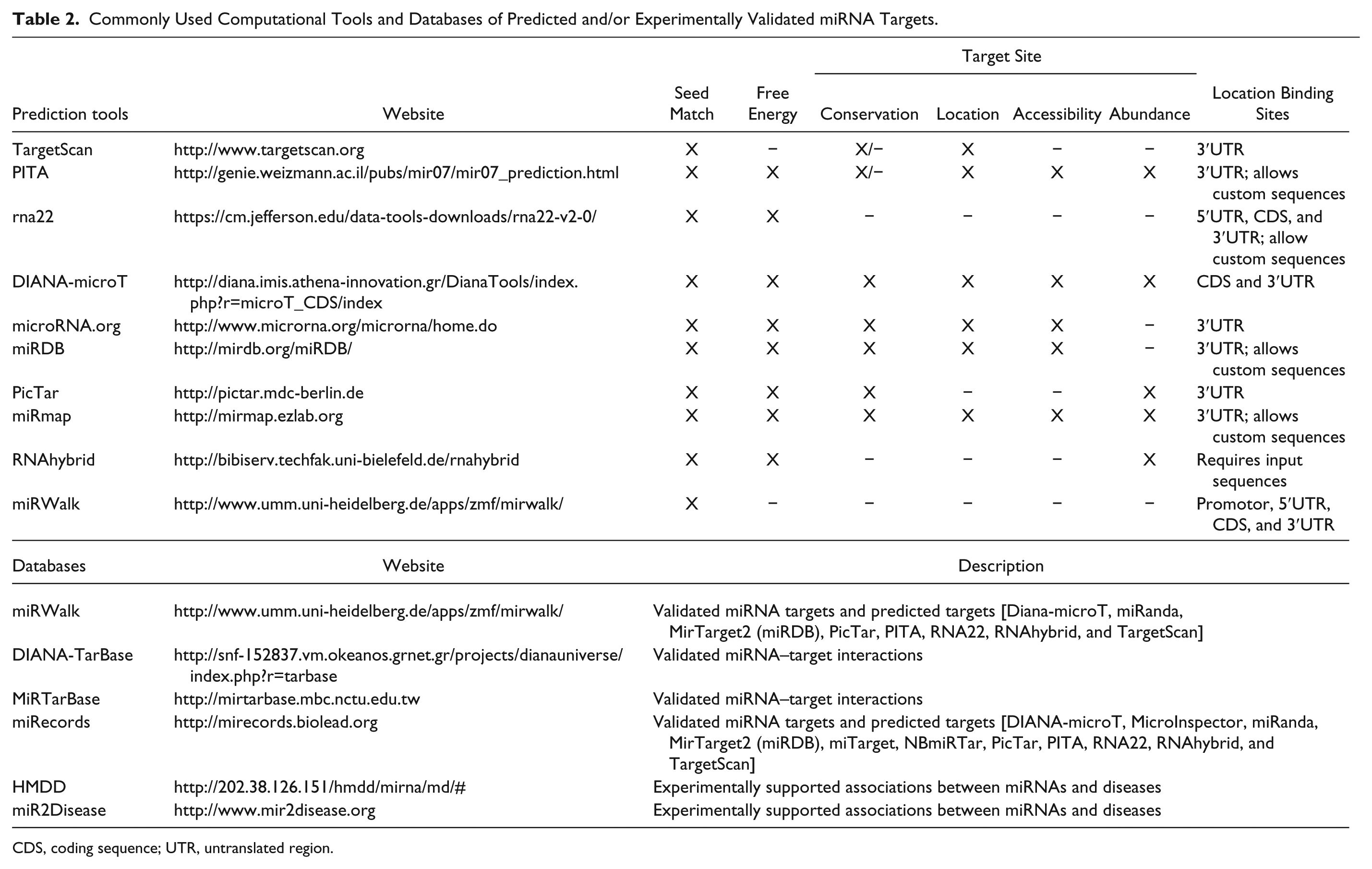
CDS, coding sequence; UTR, untranslated region.
In agreement with the notion that miRNA seed sequences (nt. 2-7/8 at 5′ end of mature miRNAs) are the major determinant of mRNA target recognition, 1 all prediction tools currently available are based on analysis of the complementarity between the miRNA seed region and putative miRNA targets. Distinguishing features include the weight attributed to different parameters, such as the evolutionary conservation of target sites, thermodynamic stability of miRNA–mRNA target heteroduplexes, and mRNA sequence characteristics outside the target site (by estimating the free energy cost to unfold the mRNA secondary structure that surrounds the target site). The use of evolutionary conservation of miRNA binding sites as a scoring criterion assumes that biologically relevant miRNA–target interactions are phylogenetically conserved, but this strategy should be considered with caution. Although this may prove useful for very well-conserved, fundamental biological processes, it precludes the identification of miRNA binding sites that are poorly conserved across species, and it cannot be applied to species-specific miRNAs. Of note, the RNA22 60 and mirWalk 61 algorithms disregard target conservation, whereas PITA 62 and TargetScan 4 include predictions with poor evolutionary conservation.
In an attempt to leverage the advantages and disadvantages associated with the different prediction algorithms, the miRWalk and miRecords databases are very useful resources. These databases compile the predictions obtained from widely used algorithms [miRWalk: Diana-microT,63,64 miRanda, 65 MirTarget2 (miRDB),66,67 PicTar, 68 PITA, 62 RNA22, 60 RNAhybrid, 69 and TargetScan;4,70–72 and miRecords: DIANA-microT, MicroInspector, 73 miRanda, MirTarget2 (miRDB), miTarget, 74 NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan], facilitating the comparison of the outcome of different tools for a particular miRNA of interest, as well as the selection of targets predicted by multiple algorithms.
Notwithstanding the relevance of the above-mentioned miRNA target prediction algorithms, there are important concerns to take into consideration regarding their application. First, these tools are generally not very stringent, generating predictions with high false-positive rates; and, potentially more important, even when taken together, prediction algorithms are characterized by a high degree of false-negative results. Second, most of the algorithms are designed to identify potential miRNA binding sites exclusively in the 3′UTR of target mRNAs. This constitutes a major limitation, considering the accumulating evidence demonstrating that a significant fraction of functional miRNA binding sites are located in the CDS and 5′UTR of mRNAs. Finally, because perfect seed base pairing is used as a defining feature by the majority of prediction tools, targets bearing noncanonical binding sites will be overlooked by these tools. In this context, it should be highlighted that the predictions generated by the RNA22 60 algorithm are not restricted to the 3′UTR and allow seed sequence mismatches. MiRWalk 61 and miRDB,66,67 although relying on stringent complementarity between the target and miRNA seed region, also allow the search for miRNA binding sites within 5′UTRs and the CDS, in addition to 3′UTR. The latest release of the Diana-microT algorithm also searches for miRNA binding sites within the CDS. 64
An additional point to consider when using some of these tools is the lack of regular updates, for example in implementing predictions for newly discovered miRNAs, as well as in reflecting changes to miRNA and/or mRNA sequences. This can be overcome partially by using the miRmap, 75 miRDB, RNA22, RNAhybrid, and PITA web interfaces that allow one to perform a target search using any user-provided miRNA sequence. Along the same line, the code used for TargetScan, Miranda, miRmap, and PITA predictions is available for download, giving users the possibility to run the algorithm locally for any novel miRNA of interest.
Databases of Validated miRNA–Target Interactions
Knowledge gathered during recent years has yielded a growing number of validated miRNA–target interactions. To render this information readily accessible to users, several databases have been released with the compilation of these data sets ( Table 2 ).
DIANA-TarBase was the first database released with the aim of cataloguing published, experimentally validated, miRNA–target interactions. 76 Currently, it contains more than half a million miRNA–target interactions, which have been manually curated from published data using 356 different cell types from 24 species. 77 In addition to the advantage of being a manually curated platform that is regularly updated, it provides detailed information on the experimental setup from which each target validation was derived, including experimental conditions such as cell and tissue type, binding site location, and primer sequences used for cloning experiments.
Other databases compiling validated interactions between miRNAs and target genes are MiRTarBase, 78 miRecords, 79 and miRWalk. 61 The database MiRTarbase provides the additional feature of showing expression profile information of both miRNAs and mRNAs in matched data sets obtained from public depositories. 78 Moreover, the correlation between miRNA and mRNA is statistically analyzed, allowing the user to assess the probability of significant anticorrelation, which may support a higher in vivo relevance of a particular interaction.
Two other databases, HMDD 80 and miR2Disease, 81 provide a collection of experimentally supported associations between miRNAs and different diseases. For example, each miR2Disease entry contains a description on the miRNA–disease relationship, as well as information on the miRNA expression patterns associated with different diseases and links to experimentally verified miRNA target genes. 81
Overall, these databases are useful resources, providing easy access to a wealth of information that can be used to streamline the validation and characterization of miRNA targets in different biological systems.
Strategies for Experimental Identification of miRNA Targets
Despite the importance of the above-described computational tools to identify putative miRNA targets, these predictions suffer from a high degree of uncertainty, making experimental validation of these putative targets a crucial step. Of note, the identification of miRNA targets using the systematic experimental approaches described in this section has contributed decisively to the current knowledge of how miRNAs interact with their targets,82–85 for example the identification of miRNA binding sites that do not conform to the canonical seed complementarity, as well as the presence of binding sites outside the 3′UTR of target mRNAs.86–90
Analysis of Gene Expression following miRNA Modulation
Taking in consideration overwhelming evidence that miRNAs reduce the steady-state levels of the majority of the targeted transcripts,91–94 the identification of mRNAs that are downregulated upon ectopic miRNA expression, or upregulated upon miRNA inhibition, is a useful approach for the identification of putative miRNA-regulated transcripts. Global analysis of differential gene expression in the presence and absence of the miRNA of interest is typically performed by microarray or RNA sequencing. This approach is often combined with a bioinformatics analysis to determine the enrichment of particular miRNA seed sequence matches among the regulated transcripts.
This approach was first used to identify targets of miR-1 and miR-124, 95 which are miRNAs enriched in the muscle and brain, respectively. Remarkably, transfection of miR-1 and miR-124 into HeLa cells changed the gene expression profile toward that observed in muscle or brain, respectively. A significant enrichment of potential direct targets, bearing seed sequence matches, was found among the downregulated genes. Several other studies have successfully used this approach to identify miRNA target genes in various cellular contexts.91,93,94
A major limitation of this comprehensive, genome-wide approach is the inability to distinguish between direct and indirect miRNA targets. In fact, the comparison of the regulated transcripts with computationally predicted targets has, in certain cases, produced poor correlations.96,97 This can be explained by an enrichment for indirect targets, acting downstream of primary targets, or by the presence of direct targets with noncanonical miRNA binding sites. Nonetheless, it should also be considered that the supraphysiological increase of miRNA levels upon overexpression of a particular miRNA may lead to artifacts, such as an increase in the off-target effects and/or false positives. Although the disadvantages associated with miRNA overexpression can be circumvented by the converse use of miRNA inhibitors, this strategy also has inherent limitations: Namely, the inhibition of highly expressed endogenous miRNA as well as miRNAs belonging to the same family (which share a common seed sequence) can be technically challenging.
Strategies Based on miRISC Immunoprecipitation
Arguably, the most reliable strategy to identify miRNA targets is to isolate and identify miRNAs and miRNA targets associated with the miRISC, in native conditions. Indeed, the specificity of miRNA target identification can be significantly improved by the analysis of miRNA–target mRNA complexes isolated by immunoprecipitation of native miRISC 98 or epitope-tagged miRISC components,99–102 such as the AGO or TNRC6 proteins. This strategy often has been coupled with the modulation of expression of specific miRNAs to facilitate identification of their targets. Typically, the mRNAs captured in the precipitated fraction are identified using microarrays or RNA sequencing. For example, the Hannon laboratory used this approach to identify targets of miR-124a, using a myc-tagged AGO2. 102 Of note, among the 294 mRNAs specifically bound to AGO following miR-124a overexpression, the authors found a significant enrichment for miR-124a seed matches (67% contained a 6-mer seed sequence).
Despite the success of this approach, highlighted by the high degree of experimental validation of putative targets, there are shortcomings to consider. In addition to the discussed drawbacks associated with overexpression of miRNAs, overexpression of epitope-tagged AGOs has also been shown to introduce experimental artifacts, such as increased association with transfer RNAs 103 and global increase of endogenous miRNA production,104,105 with potential consequences to the profile of captured miRNA–target interactions. Moreover, it has been demonstrated that some Ago-immunoprecipitated mRNAs do not constitute true targets but are, rather, the result of artificial miRNA–mRNA associations occurring on lysis.102,106 An additional caveat is the fact that weak interactions between miRNAs and mRNA targets may be lost during the immunoprecipitation procedure.
These limitations were in part overcome by the introduction of procedural modifications that use ultraviolet (UV) radiation to crosslink RNA to RNA-binding proteins prior to immunoprecipitation with antibodies specific for miRISC components. This approach, referred to as HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation), was pioneered by the Darnell laboratory 107 and first applied to map AGO-bound miRNA–mRNA complexes in mouse brain, 86 and subsequently in C. elegans. 108 Briefly, the HITS-CLIP protocol starts with UV-crosslinking at 254 nm, followed by cell lysis and immunoprecipitation of miRISC with an AGO-specific antibody. Unbound RNA is subsequently digested by RNAse treatment, and the miRISC-protected RNA fragments are purified and ligated to 5′ and 3′ end linkers and processed for deep sequencing. Using this approach, genome-wide interaction maps for exogenously expressed miR-124 and for 20 other abundant miRNAs in the mouse brain have been successfully generated. 86 This approach has been applied with considerable success, revealing a previously unappreciated abundance of miRNA binding sites outside the 3′UTR of target mRNAs, as well as novel miRNA binding motifs.
Major limitations associated with this method relate to the low efficiency of RNA–protein crosslinking by 254 nm UV light and the inability to accurately define specific target sites. 87 Moreover, a recent report based on detailed analysis of HITS-CLIP data has revealed that a significant portion of reverse transcription products terminate at the site of crosslinking and therefore are missing in HITS-CLIP data sets. 109
PAR-CLIP (photoactivatable ribonucleoside enhanced crosslinking and immunoprecipitation) is a modified method for isolation of protein-associated RNAs, based on a more efficient crosslinking method. 87 In this case, cells are cultured with photoreactive nucleosides, such as 4-thiouridine, that are incorporated during transcription (substituting uridine), followed by UV-crosslinking at 365 nm and immunoprecipitation. The higher efficiency of crosslinking achieved with this method largely improved RNA yields by 100- to 1000-fold compared to HITS-CLIP. Moreover, the reverse transcription of incorporated 4-thiouridine results in thymidine-to-cytidine transitions, predominantly in crosslinked sites. In subsequent bioinformatics analysis of the deep-sequencing data sets, this characteristic allows the accurate mapping of the interaction sites. PAR-CLIP has been performed with epitope-tagged AGO1-4 proteins in HEK293T cells, resulting in the identification of more than 17,000 enriched sequences. 87 Importantly, 9 out of 10 of the most enriched 7-mer motifs identified in the co-immunoprecipitated RNA matched the seed sequences of highly expressed miRNAs. It should be noted that issues such as potential toxicity derived from pre-incubation of cells or tissues with photoreactive ribonucleoside analogs, as well as different uptake efficiencies by different cell lines and tissues, have yet to be thoroughly addressed. 109
Another crosslinking immunoprecipitation method recently applied to the identification of miRNA targets is iCLIP (individual-nucleotide resolution CLIP).110–112 This method uses an alternative complementary DNA (cDNA) library preparation protocol designed to isolate all cDNA products, including the cDNAs that truncate at the crosslinking site, with a consequent increase of the RNA–protein binding sites and increased confidence in the identified target sites.
The recent development of the CLASH (crosslinking ligation and sequencing of hybrids) technique, 88 the first high-throughput method for the direct identification of RNA–RNA interactions, constituted a significant improvement in the portfolio of available methods for the identification of miRNA targets. This approach is similar to CLIP but includes a crucial additional ligation step, leading to the formation of miRNA–target mRNA chimeras. Therefore, this approach allows the identification with high specificity and confidence of miRNA binding sites in target mRNAs, without the need for computational predictions of putative binding sites. Using this approach, the Tollervey laboratory identified more than 18,000 miRNA–mRNA interactions in HEK293 cells. However, the efficiency of RNA–RNA ligation with the current protocol is low, with chimeric reads representing less than 2% of the overall sequencing reads, suggesting that only a fraction of miRNA–target interactions is being captured.
Another biochemical approach to enrich for miRNA targets is that based on the transfection of biotin-labeled miRNAs into cells and subsequent capture of miRNA–target mRNA complexes using streptavidin beads. 113 This method has been successfully applied in Drosophila 113 and mammalian cell lines,114–116 as well as in mouse embryonic stem cells. 114 For example, microarray analysis of the immunoprecipitation of biotinylated miR-10a from mouse E14 embryonic stem cells revealed that this miRNA targets the 5′UTR of ribosomal mRNAs, resulting in an increase of protein translation. 114 A similar approach has been applied for miRNA target discovery using digoxigenin-labeled precursor miRNAs. 117 An advantage of this approach over other techniques based on miRISC immunoprecipitation is the enrichment for targets of the biotin-modified transfected miRNA. However, as for other protocols described previously, caveats associated with miRNA overexpression should be taken in consideration, as well as concerns associated with the introduction of a tag in the miRNA, which may affect the profile and specificity of miRNA–mRNA binding.
Recently, a novel approach has been described that combines modified miRNA mimics, crosslinking, and a two-step purification of the protein–miRNA–target complexes to identify direct miRNA targets. This method, termed miR-CLIP (miRNA crosslinking and immunoprecipitation), is based on the use of miRNA mimics modified with photoreactive psoralen and biotin. 118 Following transfection of the modified miRNA mimic, cells are irradiated at 254 nm to crosslink RNAs to miRISC proteins, followed by irradiation at 365 nm to crosslink the psoralen-labeled miRNA to its targets. The crosslinked complexes are sequentially isolated by AGO2 immunoprecipitation followed by streptavidin-based purification. The captured targets are then identified by deep sequencing. This strategy has been applied successfully to the identification of miR-106a and let-7g targets. Advantages of this approach include the use of low concentration of the modified miRNA, potentially addressing the concerns associated with miRNA overexpression, and the two-step purification procedure that appears to provide greater enrichment of high-confidence targets when compared to the standard single-step protocol. Nonetheless, issues related to the modification of the miRNA mimics and the consequences for its incorporation into the miRISC complex still have to be thoroughly addressed.
Proteomic-Based Approaches
Taking into consideration that the ultimate result of miRNA action is a decrease of protein levels, proteomic analysis has also been used effectively to identify miRNA targets. Stable isotope labeling with amino acids in cell culture (SILAC) followed by mass spectrometry analysis has been used to assess the effect of specific miRNAs on the proteome.93,94,119 Briefly, cells overexpressing a miRNA and cells treated with a control miRNA are maintained in culture in two different media, one containing normal and the other heavy isotope-labeled amino acids. The difference in de novo protein synthesis is then quantified by mass spectrometry, comparing the peak intensities of heavy and light isotopes. The use of this approach has revealed that miRNAs can repress the expression of hundreds of proteins. Also, parallel measurements of gene expression correlated well with the protein levels measured using this technique.93,94 It should be noted that with the current technology, large-scale proteomic approaches are more limited in terms of depth of coverage than gene expression analysis.
A second proteomic approach applied to the identification of miRNA targets is two-dimensional fluorescence difference gel electrophoresis (2D-DIGE). In this technique, proteomic samples of cells in two different conditions (e.g., control and miRNA mimic/inhibitor transfected cells) are labeled with different fluorescent dyes and separated by isoelectric focusing and molecular-size gel electrophoresis. Proteins differentially expressed, recognized based on the fluorescent dyes ratio, are subsequently identified by mass spectrometry. This strategy was firstly applied to the identification of miR-21 targets in MCF7 cells. 120
As with the analysis of gene expression, artifacts associated with miRNA overexpression and the identification of both direct and indirect targets are inherent to these proteomic-based approaches.
Notwithstanding the relevance of the different methodologies described for the identification of miRNA targets, some of these approaches are technically challenging and require specific expertise and state-of-the-art instrumentation to be successfully applied. For example, the crosslinking-followed-by-immunoprecipitation approaches involve multistep protocols that are complex and time-consuming, whereas the SILAC experiments require access to specific equipment that is not available to many laboratories. Moreover, considerable bioinformatic expertise is required to analyze the large data sets produced by these genome-wide approaches. The challenges versus advantages of applying the different methodologies should thus be carefully considered. Altogether, the unbiased identification of miRNA targets, followed by confirmation of their biological relevance, is crucial for understanding miRNA function in various biological settings.
Validation of Putative miRNA Targets
Following the identification of putative miRNA targets through computational and/or experimental approaches, as described in this article, the first validation step typically involves analysis of the effect of modulation of miRNA expression (usually by overexpression or knockdown) on protein (by Western blot) and/or mRNA levels [by quantitative reverse-transcriptase PCR (qRT-PCR) or Northern blot] of the candidate targets. An inverse relationship between the protein/mRNA and the miRNA levels is expected for a true target.
In addition, to establish whether the identified miRNA targets are direct or indirect, direct binding of the miRNA to candidate mRNAs must be confirmed. The standard method for this type of validation study involves the use of reporter assays based on expression plasmids incorporating the 3′UTR or CDS of the candidate target gene downstream of a reporter gene sequence (e.g., luciferase or a fluorescent protein). This plasmid is then used in gain- or loss-of-function experiments by co-transfecting it with a miRNA mimic or inhibitor, respectively. Direct binding of the miRNA to the cloned sequence leads to downregulation of reporter gene expression, which constitutes proof of the existence of functional miRNA binding(s) site(s). Importantly, further confirmation must be provided by the identification of the miRNA binding site. Mutation or deletion of the candidate binding sequences, identified by inspection of the 3′UTR or CDS of the candidate target gene using different algorithms as described above, should lead to restored activity of the reporter. Although reporter assays remain the gold standard for validation of miRNA binding to target sequences, these assays are time-consuming, requiring the cloning of each of the transcripts of interest. In addition, they are dependent on various additional factors, including the cell type chosen, transfection protocol, and promoter strength.
Assessing the Biological Relevance of miRNA Targets
Despite the availability of many methods for miRNA target identification, pinpointing the targets that are relevant for a particular phenotype caused by a specific miRNA is still a difficult task. This is particularly important, because one miRNA can have hundreds of putative targets, but only a fraction of these might be relevant for the phenotype under study.
In this context, knockdown or genetic ablation of the candidate target gene should recapitulate the phenotype observed with the miRNA under study. Importantly, this can be performed at a global scale, by combining systematic approaches to identify miRNAs, such as gene expression analysis upon miRNA modulation, followed by RNAi screening to discover genes whose knockdown phenocopies the modulation of the selected miRNA. For example, Mavrakis and coworkers applied this approach to identify, among the putative miR-19 targets, the ones responsible for its oncogenicity phenotype. 121 We have also used this method to discover relevant targets of miR-199a-3p and miR-590-3p for their role in increasing cardiomyocyte proliferation, through screening of a custom siRNA library against 601 genes downregulated by these miRNAs. 30
Concluding Remarks
Although conceptually simple, functional genomic approaches based on the modulation of miRNA function constitute a powerful approach to unravel the role of miRNAs in complex biological and disease-relevant contexts. To fully explore their potential, screening efforts must be accompanied by an in-depth understanding of the intricate miRNA–mRNA regulatory networks, which are accessible only through a comprehensive identification and characterization of miRNA targets.
Although this remains a major challenge, the portfolio of existing tools for target prediction, databases of validated miRNA–mRNA interactions, and the continuously developed experimental strategies for improved miRNA target identification constitute a solid basis from which a wealth of relevant biological information can be derived.
The application of genome-editing technologies based on TALEN and CRISPR/Cas9 for screening and/or validation studies will likely contribute to further addressing important questions in miRNA biology.
Due to their intrinsic multitarget mechanism of action, the concept of exploiting miRNA modulation as a therapeutic strategy is also extremely attractive. Exciting results from phase 2 clinical trials using Miravirsen (Santaris Pharma, Copenhagen, Denmark), a miR-122 inhibitor developed for the treatment of hepatitis C virus (HCV) infection and the first miRNA-based product to advance to clinical trials, demonstrated that modulation of miRNA activity is feasible and can provide a therapeutic benefit. 122 MiRNA-based therapeutics are being actively developed for a number of other clinically relevant scenarios, including cardiovascular disorders and cancer.123,124
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MM is supported by the FIRB RBAP11Z4Z9 project from the Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Italy, and the FCT Investigator Programme IF/00694/2013 from the Portuguese Foundation for Science and Technology (FCT), Portugal. AE is supported by the Bavarian Ministry of Sciences, Research and the Arts in the framework of the Bavarian Molecular Biosystems Research Network (BioSysNet) and DFG project BR 4837/1-1, Germany.