
Abstract
The ubiquitous AAA+ ATPase p97 functions as a dynamic molecular machine driving several cellular processes. It is essential in regulating protein homeostasis, and it represents a potential drug target for cancer, particularly when there is a greater reliance on the endoplasmic reticulum–associated protein degradation pathway and ubiquitin–proteasome pathway to degrade an overabundance of secreted proteins. Here, we report a case study for using fragment-based ligand design approaches against this large and dynamic hexamer, which has multiple potential binding sites for small molecules. A screen of a fragment library was conducted by surface plasmon resonance (SPR) and followed up by nuclear magnetic resonance (NMR), two complementary biophysical techniques. Virtual screening was also carried out to examine possible binding sites for the experimental hits and evaluate the potential utility of fragment docking for this target. Out of this effort, 13 fragments were discovered that showed reversible binding with affinities between 140 µM and 1 mM, binding stoichiometries of 1:1 or 2:1, and good ligand efficiencies. Structural data for fragment–protein interactions were obtained with residue-specific [U-2H] 13CH3-methyl-labeling NMR strategies, and these data were compared to poses from docking. The combination of virtual screening, SPR, and NMR enabled us to find and validate a number of interesting fragment hits and allowed us to gain an understanding of the structural nature of fragment binding.
Introduction
p97 has an essential role in regulating protein homeostasis;1,2 however, because it acts on only a subset of protein degradation pathways, it is a potential drug target for new cancer therapies. 3 Functional p97 is hexameric, with each protomer consisting of three major domains that could be targeted to inhibit overall function. These domains include two highly conserved Walker A and B motif-containing ATPase domains (D1 and D2) and an N-terminal domain that interfaces with the D1 domain. The D2 domain exhibits strong ATPase activity in vitro4,5 and has been a primary target for high-throughput screening (HTS) for p97 inhibition. Both reversible (DBeQ, ML240, and ML241) and irreversible (3,4-methylenedioxy-β-nitrostyrene Syk inhibitor III and NMS-859) inhibitors have been identified as potent, adenosine triphosphate (ATP)-competitive inhibitors of p97 ATPase activity in vitro and inhibitors of p97-dependent degradation in cells.7–10 The activity of the D1 domain has been controversial, with recent work indicating that it is an active ATPase that can be inhibited differentially from D2. 5 The N-domain functions as a protein–protein interaction site for binding adaptor proteins that direct p97 to particular cellular pathways. 1 There are therefore multiple potential binding sites for small molecules.
The N, D1, and D2 domains are highly dynamic and intimately connected,11–14 making allosteric inhibition possible. The large size of the enzyme, combined with these intrinsic motions, adds additional challenges for drug discovery. One novel class of allosteric inhibitors (NMS-873) was recently reported to bind an interprotomer site proximal to the D1–D2 linker region.7,15 Binding of NMS-873 prevents necessary conformational changes within p97 by freezing the D2 domain in an adenosine diphosphate (ADP)-bound conformation. Identifying and targeting additional allosteric sites (e.g., the N–D1 interface, within the D1 domain, or in the D1 interprotomer region) might also prevent p97 activity by locking the enzyme in a particular conformation.
Here, we describe a fragment-based ligand discovery (FBLD) strategy to identify compounds that bind anywhere within the N and D1 ATPase domains, against which (to our knowledge) no inhibitors have yet been identified. Compared to HTS, FBLD uses smaller compounds (10–20 heavy atoms) that can more efficiently sample larger regions of chemical space. 16 Although, in general, fragments bind relatively weakly, they can achieve high ligand efficiency (defined as ΔG/heavy atom). In addition, fragments are more likely to explore different subsites within a given binding pocket, allowing for subsequent fragment linking and merging strategies to yield new chemotypes. 17 FBLD approaches rely on sensitive biophysical techniques, including ligand-detected nuclear magnetic resonance (NMR) experiments, such as saturation transfer difference (STD) 18 and 2D heteronuclear correlation–based chemical shift perturbation NMR methods that can provide specific structural data about ligand binding. 19 Surface plasmon resonance (SPR) is another sensitive, high-throughput technique for identifying and quantifying binding parameters for fragment hits. 20 Finally, X-ray crystallography is commonly used to describe in detail the fragment pose and binding pocket; however, obtaining a crystal of ligand-bound p97 is capricious, and co-structures have yet to be reported.
In this work, we report a case study for using fragment-based ligand design approaches against the large and dynamic ND1 hexamer ( Fig. 1 ). A screen of a fragment library was conducted by SPR and followed up by NMR, two complementary biophysical techniques. We also ran a prospective virtual screen and compared the docking scores of fragments for the different sites we probed, and we evaluated the scores of the hits identified by SPR and NMR targeting these sites. This screening effort yielded 13 validated fragments that bind to either one or two sites in ND1–p97. We then used competition assays and isotopic methyl-labeling NMR strategies to obtain structural data for three fragments that bind the nucleotide site. Together, our results suggest that ND1 has several potential binding sites. Targeting different sites could elucidate the allosteric mechanisms by which p97 transmits information about adaptor binding and ATP turnover, and could lead to the development of anticancer therapies. The ND1 ligands identified here provide starting points to advance toward lead compounds through chemistry based on either their crystal structures or, in their absence, the testing of analogs (“SAR by catalog”) by SPR and NMR.
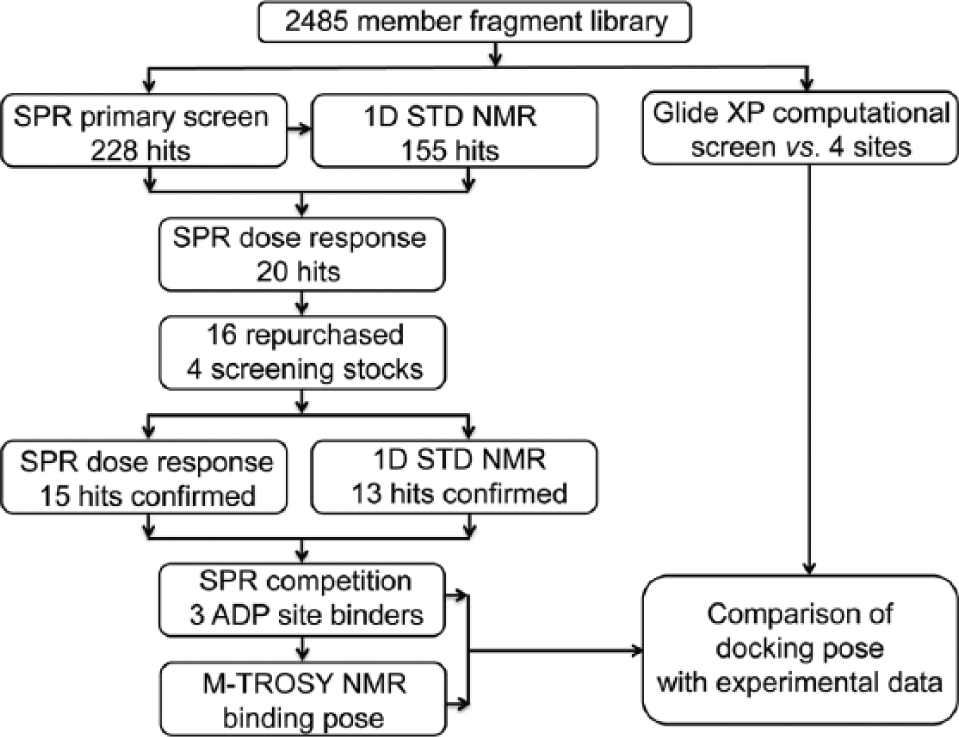
Roadmap of the fragment-based drug discovery approach applied to the p97 ND1 domains.
Materials and Methods
Fragment Libraries
The UCSF Small Molecule Discovery Center (SMDC) fragment library includes 2485 compounds (Life Chemicals, Munich, Germany; Maybridge, Loughborough, UK; and Asinex, Winston-Salem, NC) with median values for molecular weight of 207 Daltons, heavy atoms n = 15, and a partition coefficient (ALogP) of 1.5. On average, the number of rotatable bonds is two, and the number of hydrogen bond donors and acceptors averages one and two, respectively. All fragments were included in the virtual screen.
Sixteen of the 20 confirmed SPR hits were available for repurchase. The remaining four compounds (11, 14, 15, and 19) were reconstituted as dry powder from the NMR screening stocks. To confirm compound identities and estimate purity, the molecular weights of the 20 compounds were determined by liquid chromatography–mass spectrometry (a Waters 2790 separation module and a Waters micromass ZQ single quadrupole mass spectrometer equipped with a Waters 2996 photodiode array detector, a Waters 2424 ELS detector, and an XBridge C18 column). All 20 compounds exhibited the expected molecular weight; estimated purity is listed in
Virtual Screening
Three-dimensional coordinates for each fragment were generated by the LigPrep module (Schrödinger Release 2013-1: LigPrep, version 2.6; Schrödinger, New York, NY) in the Schrödinger Maestro software suite (Schrödinger Release 2013-1: Maestro, version 9.4; Schrödinger) using the OPLS 2005 force field. 21 Ionization states were generated at pH 7.0 ± 0.5 pH units, and tautomers were generated using Epik. Specified chiralities were retained, and one low-energy ring conformation was allowed. At most, four tautomers per ligand were generated. A total of 3115 structures was generated.
The coordinate file for p97 ND1 domains (PDB: 1E32) was prepared in the Maestro Protein Preparation “Wizard” to remove waters, assign bond orders, and add hydrogens. Orientations of side chain amide and hydroxyl groups in the protein, and the protonation state and tautomers of histidine, were also optimized. A receptor grid box of 30 × 30 × 30 Å with an inner box of 10 × 10 × 10 Å was generated for each of four binding sites as follows: the nucleotide-binding site was centered on ADP; a triangle site centered on the centroids of residues Asn199, His384, and Met158; the arginine fissure site centered on coordinates (48.52, 22.54, 21.78); and the N-domain site centered on (17.39, −8.28, 15.37).
Fragment docking was completed using the Glide eXtra Precision (XP) protocol (Suite 2012: Glide, version 5.8; Schrödinger) 22 with flexible ligand sampling, nitrogen inversions, ring conformation sampling, penalizing nonplanar amide torsions, Epik state penalties, and no enhancement of planarity for conjugated π groups. Van der Waals radii of ligand atoms were scaled to 0.8 (default), and a partial charge cutoff of 0.15 was used. Postdocking minimization was performed using 10 poses per ligand, with a convergence threshold of 0.5 kcal/mol. All other settings were set to defaults. Strain correction terms were not applied. One pose per input ligand conformation was exported. Structure visualization was carried out with PyMol (version 1.5.0.4; Schrödinger).
Molecular Cloning, Protein Overexpression, and Purification
Human p97 constructs, including ND1 (residues 1–458) and N-terminal AviTagged ND1, were cloned as described. 6 A C-terminal AviTagged construct of the N-domain (residues 1–213) was also cloned into the modified N-terminal 6xHisTag pET15b vector. 6 All constructs were verified by Sanger sequencing.
To obtain site-specific biotinylated proteins for SPR, AviTag–p97 constructs were expressed and purified using a combination of affinity and size exclusion chromatography, as previously described. 6
For methyl-TROSY (transverse relaxation optimized spectroscopy) HMQC (heteronuclear multiple quantum coherence) NMR experiments, [U-2H; Alaβ-13CH3; Ileδ1-13CH3; Leuδ-13CH3, Valγ-13CH3]-labeled p97 ND1 (13CH3-ILVMA p97 ND1) was expressed in Rosetta 2(DE3) cells (EMD Millipore, Billerica, MA). An initial culture grown in Luria broth (LB) was used to inoculate M9 media (100% H2O) and grown at 37 °C to an OD600 of 0.5. Cells were pelleted by centrifugation, resuspended in 100% D2O M9 media, and cultured at 37 °C until the OD600 reached 0.5. At this point, isotopically 13C-labeled Ile, Leu, Val, Ala, and Met precursors (50 mg 2-ketobutryic-4-13C,3,3-d2 acid; 100 mg 2-keto-3-methyl-13C-butyric-4-13C,3-d acid; 250 mg L-methionine-(methyl-13C); and 100 mg L-alanine-3-13C,2-d) were added. After 30 min, expression was induced with 1 mM IPTG followed by overnight culture at 20 °C. 13CH3-ILVMA p97 ND1 was purified as described for unlabeled AviTagged ND1, with the exception that phosphate buffered saline (PBS), 0.5 mM Tris (2-carboxyethyl) phosphine (TCEP) was used during the size exclusion chromatography step.
Surface Plasmon Resonance
SPR biosensor experiments were performed using a Biacore 4000 instrument (GE Healthcare, Little Chalfont, UK). CM5 sensor chips were prepared essentially as described, 6 with the exception that proteins were immobilized to high levels (5000–14,000 resonance units) in buffer [10 mM HEPES (pH 7.5), 150 mM NaCl, 0.005% Tween 20] by injecting 100–400 µg/mL protein for 1–10 min at 25 °C.
For the primary screen, fragment stocks (5 mM in DMSO, in 384-well plates) were diluted 20-fold with buffer [25 mM Tris (pH 7.5), 150 mM NaCl, 20 mM MgCl2, 1 mM DTT, and 0.005% Tween 20] resulting in a final concentration of 250 µM in 5% (v/v) DMSO buffer. Compounds were screened at 20 °C using a flow rate of 30 µL/min, and association and dissociation times of 60 s. ADP (20 µM) and buffer were injected every 10 cycles as positive and negative controls, respectively. A solvent correction curve was completed at the beginning and end of the run to account for buffer and DMSO mismatches. Data were double-referenced, scaled, and normalized as described. 20 Hits were defined as having a response unit (RU) greater than 2.5 standard deviations over the baseline, and desirable sensorgrams characterized as a square-shaped trace with fast on- and off-rates.
Fragment hits were confirmed in dose–response using a concentration series ranging from 19.5 µM to 1.25 mM and 2-fold dilutions. Binding of ADP (0.027 – 20 µM; 3-fold dilutions) and/or adenosine monophosphate (AMP; 12.5–400 µM; 2-fold dilutions) was measured at the beginning and end of each run to assess the quality of the protein surface. Raw sensorgrams were reduced, solvent-corrected, and double-referenced using the Biacore 4000 software, and fragments were classified as either well-behaved compounds or promiscuous binders. 23 The repurchased fragments and four compounds from the screening stocks were reassayed in duplicate (12.5–800 µM; 2-fold dilutions) for binding to the N and ND1 p97 constructs to confirm binding specificity and to obtain binding affinities. Sensorgrams were imported into Scrubber2 (Biologic Software, Campbell, Australia) for fitting. The stoichiometry of ADP binding to ND1 monomer was assumed to be 1:1; the experimental ratio (typically, 0.8:1) was used to define the fraction of active ND1 protein. This active fraction was then used in stoichiometry calculations. Responses were normalized for molecular weight (MW) using Eq. 1, and were fit to a 1:1 or 2:1 binding model (ND1), or a 1:1 binding model using a maximum response equivalent to the theoretical maximum response (TRmax; Eq. 2) for the N-domain.
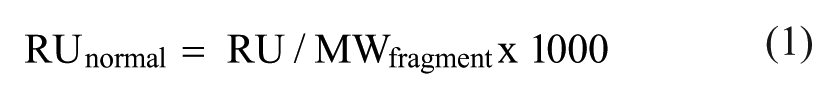

To determine if any of the repurchased fragments bound in the nucleotide-binding site, the Biacore 4000 was used to simultaneously measure the binding of the repurchased fragments (250 µM) to ND1 in the standard SPR screening buffer and in an SPR screening buffer containing 100 µM ADP. AMP (250 µM) was run as a positive control. Fragments that bound competitively in the nucleotide-binding site were expected to show a reduction in binding in the ADP buffer.
NMR Spectroscopy
STD NMR spectroscopy
All 1D STD NMR spectra 18 were acquired on a Bruker AVANCE DRX500 MHz spectrometer at 296.8 K (referenced to 4% CH3OH–CH3OD using a coefficient of 1.0183) with a 5 mm Bruker QCI Cryoprobe ( 1 H, 13C, 15N, and 31P) with actively shielded Z-gradients and a BACS-60 automated sample changer (Bruker Biospin, Billerica, MA). Samples (500 µL) containing 500 µM fragment (or less if the compound had limited solubility) and 50 µM unlabeled ND1 protein were prepared in Norell XR-55 glass NMR tubes (Norell, Marion, NC) in 100% D2O PBS (pH 7.5), 1 mM NaN3, and 1 mM DTT, 0.5% d6-DMSO (Norell, Marion, NC). 10 µM disuccinimidyl suberate (DSS) was added as an internal 1H chemical shift reference, and compound reference spectra were acquired prior to addition of protein. The “stddiffgp19.3” pulse program from the Bruker pulseprogram library (TopSpin 1.3 pl10) was used for data acquisition with an excitation-sculpting double gradient echo-based 3-9-19 composite pulse WATERGATE-5 element for water suppression. IconNMR and a BACS-60 sample changer were used to automate data collection, and Topspin 3.1 (Bruker Biospin) was used for data analysis. Further details of experimental parameters can be found in the Supplementary Material.
STD NMR ADP competition experiments
The NMR STD experiment (described above) was used to measure the binding of ligand (500 µM) to ND1 (50 µM) in the presence and absence of ADP (250 µM) in the standard NMR buffer (see above). AMP was used as a positive control.
Methyl-TROSY HMQC NMR spectroscopy
NMR experiments were performed on an 800 MHz Bruker AVANCE-I spectrometer with a TXI Cryoprobe equipped with an actively shielded Z-gradient at 298.2 K. Samples were buffer-exchanged into 100% D2O PBS buffer by repeated ultrafiltration, concentrated to 200 µM (in 200 µL volume), and placed in a Shigemi advanced NMR microtube (part no. BMS-005TB; Shigemi, Allison Park, PA). Two-dimensional [13C, 1H]-HMQC methyl correlation experiments 24 on 13CH3-ILVMA ND1 were acquired with 86* and 768* complex points in the 13C and 1H dimensions, respectively, with maximum acquisition times of 20 (t1) and 53 ms (t1), respectively. Presaturation was introduced into the experiment for water suppression. A relaxation delay of 1.5 s was used, along with 128 scans, giving rise to an acquisition time of 10 h. All 2D HMQC-TROSY spectra were processed with NMRpipe, converted to Azara format (pipe2azara), and imported into CCPN Analysis2 for data analysis. A 1% (v/v) d6-DMSO control (no fragment) did not recapitulate the observed shifts or broadening patterns seen for nucleotide or fragment binding (data not shown).
Results
Virtual Screening
We began the exploration of p97 with a de novo search of the topology of the ND1 monomer by using the Sitemap
25
software to identify potential small molecule–binding sites (
Fig. 2A and 2B
; PDB: 1E32). This predicted five potential small molecule–binding sites (Sitemap scores, volumes, boundaries, and locations for these sites are listed in
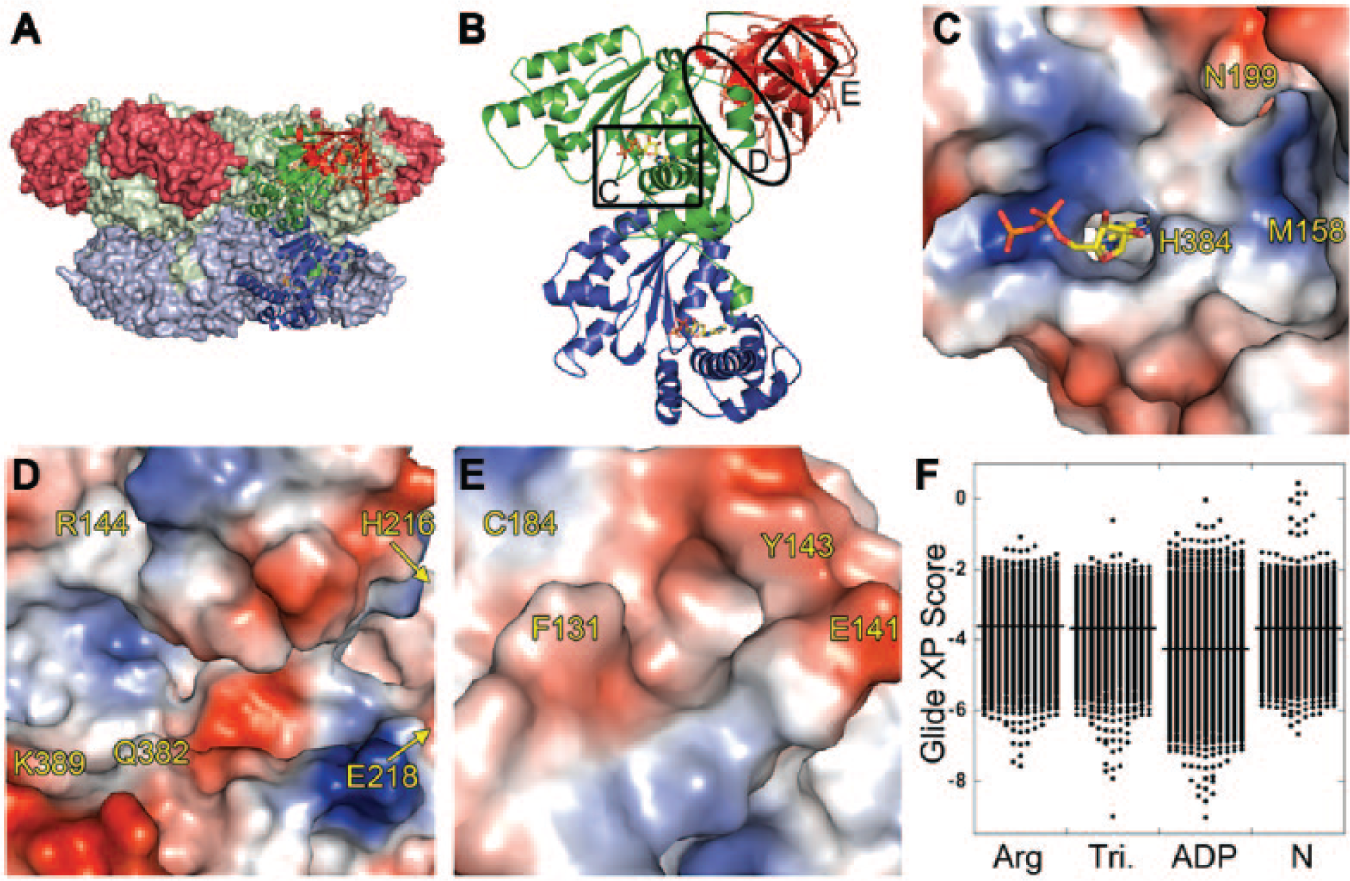
Locations and docking results for the four binding sites targeted in the ND1 domains of p97. (
Two more sites that were not identified by the computational search were also used for docking. A third site that might be targeted by fragments, located adjacent to the nucleotide-binding site, was identified by visual inspection ( Fig. 2C ). We named this site the triangle site because of its shape; it is bordered by three residues: Asn199, His384, and Met158. A cleft in the N-domain, also identified by visual inspection, was named the N-domain site ( Fig. 2E ) and selected as a fourth site to target in docking experiments.
The 2485-member SMDC fragment library (see Materials and Methods for a description) was docked using Glide XP against the four sites ( Fig. 2C–E ). Figure 2F shows the distribution of Glide XP docking scores by binding site. An ADP control docked to the nucleotide site with a Glide XP score of −10.3. The distribution of fragment scores at the nucleotide-binding site was skewed toward more favorable scores (better than −6) compared to the other three sites, with 211 fragments (6.7% of the library) having scores for the nucleotide-binding site in the −6 to −9 range. Only 31 (1%) and 51 (1.6%) fragments docked to the arginine fissure and triangle sites with scores better than −6, respectively. The N-domain site had the fewest docked poses with scores of −6 or better (10 fragments; 0.3%). We concluded based on these results that the nucleotide-binding site was the most likely, out of the sites we identified, to bind fragments. At this site, 4 of the top 10 ranked poses had a benzimidazole or indole moiety, consistent with the adenine-binding function of this site.
SPR Primary Screen
The SMDC fragment library was screened by SPR for binding to the ND1 domains of p97. The screen was completed in two experimental runs in which N-terminally AviTagged ND1 was immobilized on a neutravin-coated CM5 sensor chip to ~14,000 and ~7000 RUs, respectively. Fragments were injected at a single concentration of 250 µM, with positive (ADP; 20 µM) and negative (buffer) controls included between each group of 10 fragments. The data were processed as described in the Materials and Methods section, and the results are shown as a scatterplot in Figure 3A . The average and standard deviation of the ADP positive controls and baseline were 98.9 ± 2.6 (n = 286) and 0.17 ± 8.2 (n = 2257), respectively, giving a Z′ value of 0.67 for the screen. Because ND1 was immobilized to a lower level in the second half of the screen, the binding response of the fragments was lower, resulting in more noise in the ADP positive control and baseline. Therefore, hits were defined as being greater than 2.5 σ above the mean assay response for each half of the screen, which was equal to a fractional occupancy of 16.7% and 23.4%, respectively ( Fig. 3A ). The sensorgram shapes for fragments with binding levels higher than this threshold were inspected individually (see Materials and Methods) to eliminate obvious promiscuous binders, resulting in 228 (9.2%) active fragments being selected for follow-up by both STD NMR and SPR dose–response analysis.
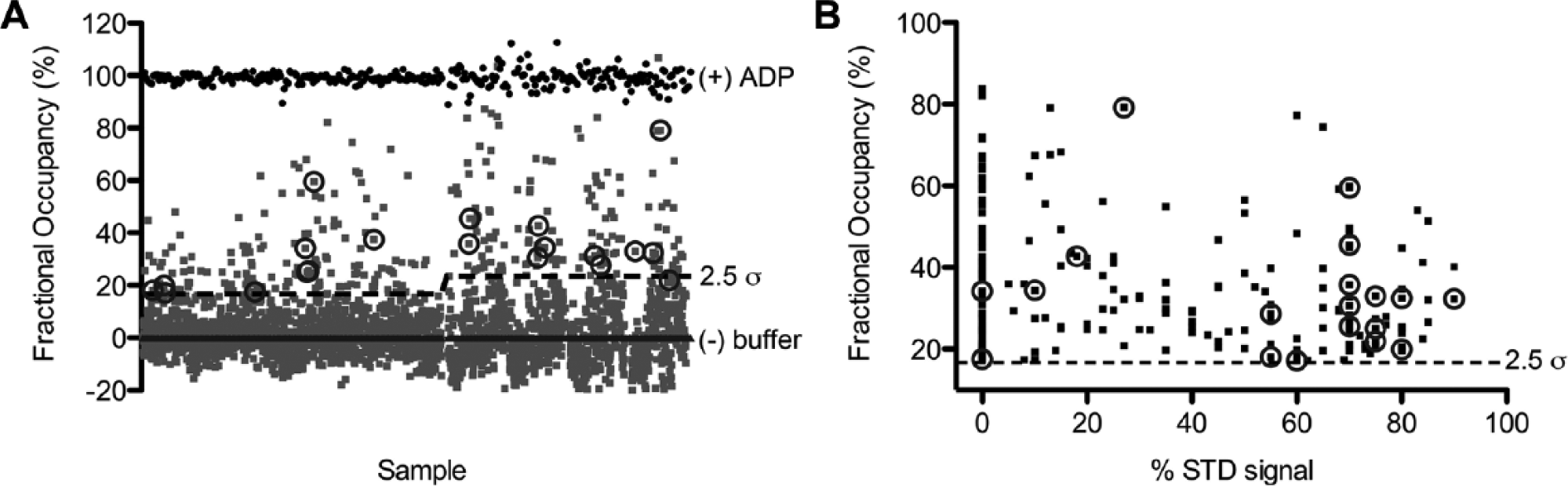
Surface plasmon resonance (SPR) and saturation transfer difference (STD) nuclear magnetic resonance (NMR) primary fragment screens against ND1. (
Confirmation of Active Fragments
STD NMR was used to follow up the 228 SPR-active fragments and identify possible false positives. STD NMR
18
experiments monitor ligand resonances and provide a direct measure of fragment binding to the ND1 domains (an example is shown in
SPR dose–response analysis
To identify false positives among the 228 SPR-active fragments, these fragments were tested in a dose-dependent manner using concentrations ranging from 19.5 µM to 1.25 mM in 2-fold dilution steps. In some cases, the fragments were visually insoluble in the assay buffer at the highest concentrations, and thus could not be investigated in the dose–response analysis. Twenty of the 228 active fragments (9%) bound with a stoichiometry <3 (stoichiometry is defined here as: ligands/protomer) and were defined as hits (circled in Fig. 3A ). The majority of SPR-active fragments that confirmed as “hits” had fractional occupancies in the range of 20–50% in the primary SPR screen (circled in Fig. 3A ). Three-quarters of the fragments (160; 70%) were categorized according to Giannetti 23 as promiscuous binders because they exhibited concentration-dependent aggregation, nonstoichiometric binding (stoichiometry between 3 and 5), or superstoichiometric binding (stoichiometry >5). Of the remaining fragments that did not reconfirm as hits, 30 showed weak or no binding, 13 were irreversible binders, and 5 bound nonspecifically to the reference surface. This step identified 20 compounds as ND1 hits to repurchase for confirmation.
Post hoc analysis of SPR and STD NMR
With both sets of biophysical data in hand for the 228 SPR-active fragments (SPR dose–response and STD NMR), a post hoc analysis of the data showed that SPR fractional occupancy did not correlate well with STD NMR values ( Fig. 3B ). In contrast, out of the 17 fragments that were sufficiently soluble for NMR investigation from the 20 compounds selected by SPR for repurchase, 14 showed a strong signal by STD NMR (>50%).
SPR Dose–Response Analysis and STD NMR of Repurchased Fragments
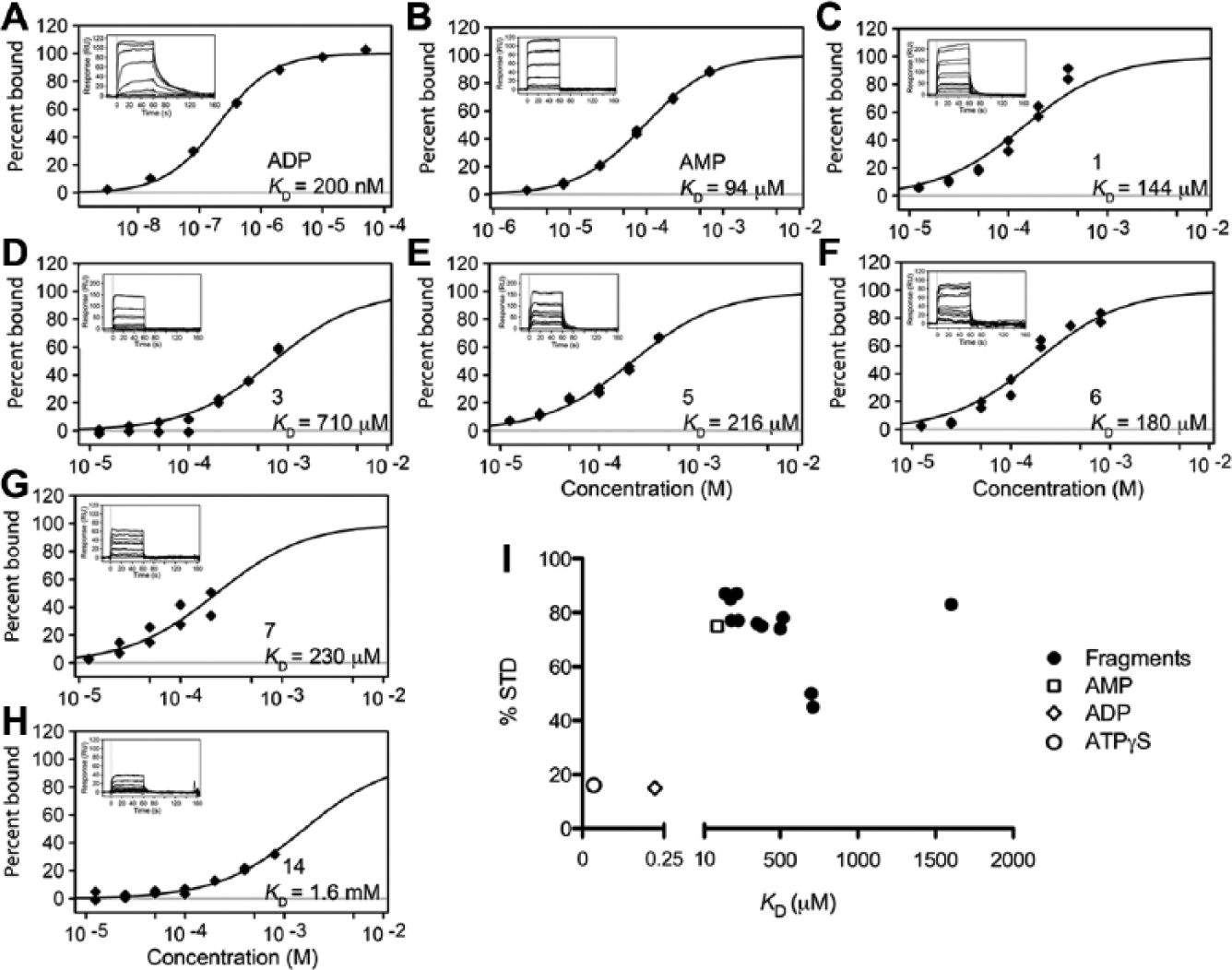
We repurchased and retested the 20 compounds identified by SPR as specific ND1 binders (16 repurchased and 4 from the NMR screening stocks). We also measured their binding to the N-domain alone. To monitor the ND1 protein’s binding capacity and stability, dose–response analyses of ADP and AMP controls were performed at the end of the SPR run. Both ADP and AMP controls bound with the expected affinities and kinetics (
Fig. 4A and 4B
). Three-quarters of the 20 retested fragments (15; 75%) reconfirmed as binding to the ND1 domains, with dissociation constants ranging from 150 to 1700 µM (
Table 1
). Examples of SPR dose–response data for fragments fitted either to a one-site model (
Fig. 4C–E
) or for a stoichiometry of N = 2 (
Fig. 4F–H
) are shown in
Figure 4
. Sensorgrams for the remaining fragments are shown in
Validation of fragment hits by surface plasmon resonance (SPR) and nuclear magnetic resonance (NMR). (
20 repurchased fragment hits; ND1 and N domain binding.

NB, no binding; ND, not determined owing to lack of solubility; SS, superstoichiometric binding (N > 3).
Log D octanol/water distribution coefficient calculated using Pipeline Pilot.
Classification of fragments as D1 nucleotide-site binding (Nuc), D1 binding (D1), or N-domain binding (N).
KD in micromolar. Reported errors are the standard error of the mean of the fit (n = 2).
Ligand efficiency, calculated as ΔG/(#heavy atoms) (kcal/mol*atom).
The largest observed percentage reduction in signal at the ligand resonances.
GLIDE XP score.
Although most fragments were found to bind to the ND1 construct with a 1:1 stoichiometry, we identified five fragments that bound to ND1 with a 2:1 stoichiometry (
Table 1
and
The 20 repurchased SPR hits were also retested for binding to ND1 by STD NMR (
Table 1
and
Figure 4I
shows the relationship between the affinity measured by SPR and the STD NMR value (at a ligand-to-protein ratio of 10:1). Except for the nucleotides (ADP and ATPγS), which have higher affinities and hence longer resonance times in the binding site, and one fragment with lower affinity (15; 1.7 mM), there is a rough trend correlating greater STD values with lower SPR dissociation constants (
Fig. 4I
). Although the STD NMR value does not predict the KD, strong STD NMR values were seen for fragments with affinities in the micromolar to millimolar range (other NMR techniques are more suited to investigate compounds with affinities in the sub-micromolar range).
19
We would not have expected to observe even this loose correlation because many factors contribute to the measured STD value,
18
and this is probably due to the relaxation of the ligand protons being dominated by strong dipolar interactions with the protein protons in the bound state (due to the slow tumbling rate of the large ND1 324 kDa hexamer in solution; more details on physical factors contributing to these findings are given in the
ADP Competition Assays
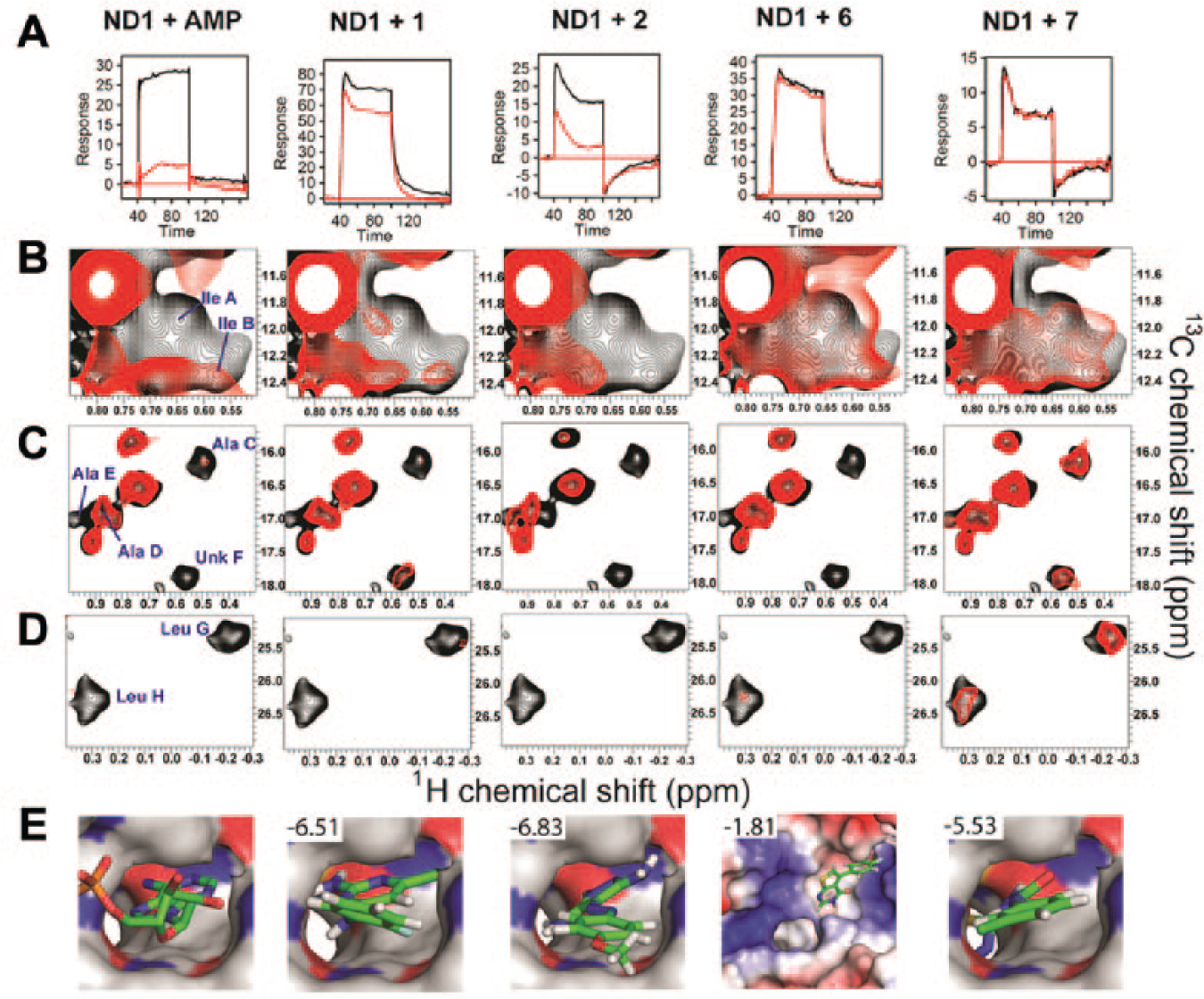
To investigate if the 13 validated fragments bound to the D1 nucleotide-binding site, we ran SPR competition assays in the absence and presence of a saturating amount of ADP (100 µM; KD = 200 nM). Fragments that bind in the nucleotide site should be displaced by the tighter binding ADP molecule, as is seen for the control AMP ( Fig. 5A ). By this measure, only three fragments 1, 2 ( Fig. 5A ), and 3 (data not shown) were found to bind the nucleotide-binding site. For the other 11 compounds (including 6 and 7), SPR traces were similar with and without ADP, indicating that they do not compete with nucleotide binding.
Mechanisms of fragment hit binding. (
Six of the 13 fragments were also tested using STD NMR with ADP competition (250 µM ADP). AMP was used as a positive control, and the STD NMR value decreased from 65% to 12% STD on ADP addition. Four compounds tested (4, 8, 9, and 13) showed little change in STD NMR values or the line widths of their NMR signals on ADP addition, indicating that they are not competitive with ADP. Fragments 1 and 2 showed a reduction in line broadening on addition of ADP, which would be consistent with moderate-affinity (~100 µM) binding in the nucleotide site. They did not, however, show significant changes in STD NMR values on ADP addition. These data are compatible with fragments 1 and 2 being displaced by ADP from a micromolar affinity nucleotide-binding site; however, an unchanged STD value may be due to binding to additional low-affinity (mM) sites, as indicated by the SPR stoichiometries (
Based on SPR binding, stoichiometry ratios, and ADP competition data obtained with the ND1 and N-domain proteins, we put forward a hypothesis classifying fragments as D1 nucleotide-site binders (Nuc), D1 binders (D1), or N-domain binders (N;
Table 1
and
Fragment Hit Binding Site Localization by Methyl-TROSY NMR
We used 2D [13C, 1H]-methyl-TROSY NMR methods (M-TROSY), which allow NMR studies of large protein complexes, to probe the sites of fragment binding. These experiments, in which precursors of Ile, Leu, Val, Met, and Ala amino acids with a terminal 13C-labeled protonated methyl group are incorporated into a highly deuterated background to produce [U-2H; Alaβ-13CH3; Ileδ1-13CH3; Leuδ-13CH3, Valγ-13CH3]-labeled p97 ND1 (13CH3-ILVMA ND1), can achieve excellent resolution and sensitivity for terminal side-chain methyl group signals, even in large protein complexes. 27 Previous work had shown well-resolved signals in M-TROSY spectra of the ND1 hexamer labeled with 13CH3-Ile and 13CH3-Ala, and identified chemical shift changes in alanine methyl groups in the N-domain of ND1 due to adaptor binding. 28 As the p97 ND1 domains form a symmetrical hexamer in solution, the nuclei in each ILVMA methyl group in individual protomers have identical chemical environments, giving rise to a single NMR signal for each residue in the protomer. This leads to a high-quality M-TROSY spectrum for the 13CH3-ILVMA-labeled p97 ND1 hexamer despite the large size of the complex (324 kDa). Although many of the NMR signals for 13CH3-ILVMA ND1 near the vertical centerline of the spectrum (~0.85 ppm in 1H) are overlapped, other signals are dispersed (owing to the proximity of methyl groups to aromatic residues), and these can be used as “reporter signals” to follow changes induced by ligand binding.
A complete assignment of the ILVMA 13CH3 terminal groups in the ND1 hexamer is outside the scope of this work. However, we hypothesized that specific NMR signals were associated with terminal methyl groups of particular ILVMA residues near the ND1 nucleotide-binding site based on (
Using the set of reporter signals, we sought to locate the binding site(s) for several fragments with <250 µM affinity (1, 2, 6, and 7). Binding of these fragments to the ND1 domains was accompanied by changes in M-TROSY spectra (
Fig. 5
) that resembled AMP or ADP binding. For two fragments (1 and 2), the set of signals affected was very similar to those for the nucleotides. In particular, the signals for LeuG and LeuH, which in our hypothesis we associated to the CH3δ1,2 groups of Leu253, and both IleA and IleB (associated with the CH3δ1 groups of Ile380 and Ile383) showed significant broadening, consistent with them having a close proximity to the bound fragments. In contrast, the binding of two other fragments (6 and 7) affected only a subset of the reporter signals (
Fig. 5
), indicating that they bound differently to 1 and 2. A more detailed comparison of the spectra from nucleotides and fragments, and explanation of the M-TROSY NMR results, can be found in the
Comparison of the Virtual Screen and the Experimentally Validated Hits
Virtual screening performed prior to the biophysical experiments suggested that the nucleotide-binding site was the most likely, out of four potential sites that were examined, to bind fragments. However, we could only confirm, by ADP competition experiments and TROSY NMR, binding at this site for 3 of the 13 validated hit fragments. Two of these 3 fragments were among the top 100 docking hits (1: Glide XP score −6.5, rank 96; and 2: Glide XP score −6.8, rank 55), and the third received a reasonable score (3: Glide XP score −5.7, rank 326). The predicted docking poses of 1 and 2 are shown in Figure 5E . They show that the fragments attempt to satisfy polar interactions at the back of the pocket with the Gly207 backbone carbonyl O-atom. The chlorine atom from 1 partially fills the hydrophobic pocket created by Ile383. The remaining top docking hits for the nucleotide-binding site represent false positives; although some of these did show binding in the initial SPR screen, they were later eliminated due to nonstoichiometric binding.
The locations of the binding sites for the 10 other confirmed hit fragments remain unknown. The docking results at the three other sites that we considered generated very few fragment poses with good docking scores, including those for confirmed hits. Sitemap did not identify any other plausible binding sites in either the D1 or N domain. One possible explanation is that conformational changes, possibly associated with functional motions, could open other binding pockets that are not apparent in the available crystal structures.
Discussion
p97 is a large and dynamic protein machine. Data from electron microscopy (EM) 29 suggest large D1–D2 interdomain movements during the catalytic cycle; X-ray crystallography has also indicated N–D1 interdomain conformations. This dynamic behavior has significant consequences for developing approaches to inhibiting the enzyme. In addition to the conformational complexity, the large size of the protein and the rugged features of the surfaces of the three domains could combine to present a large number of potential sites that might bind small fragments and potentially be targeted.
A combination of SPR and NMR identified 13 confirmed ND1-binding fragments from a library of ~2500 compounds with affinities <1 mM (seven with affinities <250 µM), stoichiometric binding (N ≤2), NMR STD values >45%, and good ligand efficiencies (~0.3). Eleven fragments likely bind the D1 domain, and two the N-domain. Sitemap calculations and visual inspection were used to identify four possible binding sites for small molecules in the p97 protomer, but our studies were able to confirm binding at only two of these, where three fragments were found to bind in the nucleotide-binding site and two likely target the N-domain site. It is possible that additional sites in the ND1 domains remain undiscovered. Indeed, the identification of cryptic allosteric sites by computational methods is a challenging task and is the subject of active development. 30
As a screening and validation tool, STD NMR proved to be an effective confirmatory method, and, with a judicious choice of a cutoff level, it has the potential to exclude fragments that bind nonspecifically or with high stoichiometries. SPR hits with a strong STD value (>45% for this study) were much more likely to show stoichiometric binding in SPR dose–response studies (the assessment of “strong” STD value generally scales with protein size and shape). In this study, tighter-binding fragments generally gave larger STD values for most of the specific, reversible binders as long as the affinity was higher than the single-digit micromolar range. We would generally not expect the STD value to correlate with ligand affinity because many variables affect the value. STD NMR can also identify weaker binders, and these may not be identified as positives by our SPR assay.
Recent advances in NMR have enabled investigations of large protein complexes. 24 By applying high levels of deuteration together with 13CH3-ILVMA residue type-specific labeling and M-TROSY, we could probe fragment binding to the 320 kDa p97 ND1 hexamer. In general, the M-TROSY results correlated well with computational and ADP-competition experiments, allowing us to develop a hypothesis of how two fragments could bind in the D1 nucleotide site. In the absence of X-ray crystallography, high-field NMR can provide structural information describing weak binding fragments.
The combination of virtual screening, SPR, and STD NMR enabled us to discover and validate a number of interesting fragment hits. The docking, SPR, and M-TROSY NMR allowed us to gain an understanding of the nature of fragment binding. The structural information, for the fragments that we hypothesize bind in the nucleotide site, will help guide fragment hit-to-lead development and directions for chemical synthesis to improve potency.
Footnotes
Acknowledgements
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government. We thank John Irwin for help identifying potential docking sites to target.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MPJ is a consultant to Schrödinger LLC.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Chemical Biology Consortium Contract No. HHSN261200800001E.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.