
Abstract
Fragment screening is becoming widely accepted as a technique to identify hit compounds for the development of novel lead compounds. In neighboring laboratories, we have recently, and independently, performed a fragment screening campaign on the HIV-1 integrase core domain (IN) using similar commercially purchased fragment libraries. The two campaigns used different screening methods for the preliminary identification of fragment hits; one used saturation transfer difference nuclear magnetic resonance spectroscopy (STD-NMR), and the other used surface plasmon resonance (SPR) spectroscopy. Both initial screens were followed by X-ray crystallography. Using the STD-NMR/X-ray approach, 15 IN/fragment complexes were identified, whereas the SPR/X-ray approach found 6 complexes. In this article, we compare the approaches that were taken by each group and the results obtained, and we look at what factors could potentially influence the final results. We find that despite using different approaches with little overlap of initial hits, both approaches identified binding sites on IN that provided a basis for fragment-based lead discovery and further lead development. Comparison of hits identified in the two studies highlights a key role for both the conditions under which fragment binding is measured and the criteria selected to classify hits.
Keywords
Introduction
Fragment-based lead discovery (FBLD), often also called fragment-based drug discovery (FBDD), is evolving as a robust method to identify hit molecules for drug development.1–3 An increasing number of compounds derived from fragment-based methods have progressed into clinical development targeting a variety of targets, including enzymes such as proteases, kinases, and polymerases (reviewed in Murray and Blundell 3 ).
FBLD methods use low molecular weight compounds (“fragments”) to probe pockets in the target protein. Fragments were initially described by a “rule of three”: having a molecular weight of less than three hundred Daltons with up to three rotatable bonds, no more than three hydrogen bond donors or acceptors, and a calculated partition coefficient (clogP) of three or less. 4 Modern fragment libraries now contain compounds much smaller than 300 Da to maximize the chemical diversity of a library. As fragments generally bind weakly to the target, sensitive biophysical techniques are required to identify a binding event, and multiple different biophysical assays are often used in conjunction to confirm binding.
The biophysical techniques that are used most commonly (either individually or in combination) to identify and confirm fragment hits are nuclear magnetic resonance (NMR), surface plasmon resonance (SPR), thermal shift assays, and X-ray crystallography. Each technique provides different information about compound binding and complements the other methods.
NMR is a powerful and sensitive method for hit detection, and a number of distinct experimental methods can be used for FBLD. For example, researchers at Abbott, who pioneered the FBLD approach with “SAR by NMR,” 5 used a number of different NMR techniques—initially reporting screening using 15 N-heteronuclear single-quantum coherence (HSQC) experiments, which required 15 N-labeled protein. In a subsequent study, they used a WaterLOGSY method 6 that did not require labeled protein and could be undertaken at lower protein concentrations. A strength of the 15 N-HSQC method is that it identifies whether binding has occurred and also the location of the fragment binding site in a single experiment. A disadvantage of this method is the need for significant amounts of 15 N-labeled protein. For saturation transfer difference nuclear magnetic resonance spectroscopy (STD-NMR), 7 the protein is magnetized via spin diffusion, and the magnetization is transferred selectively to protein-bound ligands and detected on the free ligand following dissociation. This approach offers similar advantages to the WaterLOGSY method, although STD-NMR and WaterLOGSY do not provide any direct information about the binding location, so secondary screening techniques are required.
SPR offers another sensitive means of identifying hits. Proteins are immobilized on a surface where interactions between the compound and protein are measured through changes in the reflection angle of polarized light. Although no structural information is obtained, a benefit of this technique is that the binding affinity of the compound and, in certain cases, kinetic data in the form of both on-rates and off-rates can be determined.
Although several companies, including ActiveSight, Plexxikon, Zenobia, and DeCODE, have used X-ray crystallography for both fragment screening and structural characterization, X-ray crystallography is most often used following the preliminary screen to obtain detailed information about protein-ligand interactions. In many cases, crystallography can effectively identify very weak protein-ligand interactions, but a range of factors may prevent the formation of protein/fragment complexes. First, the protein crystallization conditions may differ significantly from the biologically relevant environment (e.g., the crystals may only grow at a pH well removed from physiological pH). The most common FBLD approach, soaking, involves incubating preformed protein crystals in a liquor containing the fragment and assumes that the fragment will diffuse into the crystal and bind to the protein molecules therein in the same way that it would bind to protein molecules in solution. In this case, crystal contacts may prevent access to binding sites, or the crystal lattice may prevent conformational change in the protein necessary for binding. Co-crystallization, growing protein crystals in the presence of compound, can overcome these issues but is sometimes difficult and may require extensive crystallization screening for each compound. This may be due to conformational changes in the protein upon ligand binding, disrupting crystal contacts or changes in the system from addition of solvents such as DMSO that are commonly used to solubilize fragments.
Herein we report the independent efforts of our two groups to find novel inhibitors of the HIV-1 integrase core domain (IN) using a fragment-based screening approach.8–10 IN is an essential protein in the life cycle of HIV. 11 The IN protein occurs as a dimer in vitro (K dimer of 67.8 pM) and is believed to act as either a dimer or a tetramer (a dimer of dimers) in vivo. 12 The first drug targeting IN, raltegravir, was granted Food and Drug Administration (FDA) approval in late 2007 for patients who were failing other anti-HIV treatments and further approval in 2009 for previously untreated patients. Raltegravir contains a di-carbonyl moiety that coordinates to the divalent metal ion that is present at the active site of IN; its mode of binding was visualized by crystallography in the related Prototype Foamy Virus IN.13,14 Mutations in IN that cause resistance against raltegravir have already emerged. These mutations also result in cross-resistance to other IN inhibitors that are progressing through the clinic, which bind to the same site as raltegravir and act via a similar mechanism. 15 Consequently, drugs that can target other sites on IN and that do not have the same resistance profiles to currently available drugs are of interest and would complement the current IN inhibitors used in the multidrug treatment regimes. Several potential inhibitor binding sites have been reported for IN (reviewed in Al-Mawsawi and Neamati 16 ), including the active site, the LEDGF binding site,17,18 the fragment binding pocket (FBP), 9 the sucrose binding pocket, 10 and a binding site located adjacent to the active site flexible loop (residues 140–149).8,19
The two screening campaigns used closely related, 500-compound fragment libraries purchased from Maybridge (Cornwall, UK). In Approach A, STD-NMR was used as a primary screen. Compounds were initially screened in pools consisting of 10 fragments, and the hits were then retested as single compounds. Confirmed hits from the individual STD-NMR experiments were advanced into crystallization trials, and binding in solution was confirmed by recording HSQC spectra of 15 N-labeled IN in the absence and presence of the fragment. In Approach B, the fragment library was screened individually at a single concentration using SPR, and initial hits were confirmed by further dosage response studies also using SPR and subsequently put into crystallization trials. We compare and contrast the results of the two studies and look at the implications for other FBLD projects.
Materials and Methods
The methods for Approaches A and B have been previously described by Wielens et al. 9 and Rhodes et al., 8 respectively, but will be summarized here briefly for comparison.
Mutagenesis
Four HIV-1 IN50–212 constructs were used for the X-ray crystallography experiments. The well-described solubilizing mutations C56S, W131D, F139D, and F185H were introduced into a truncated HIV-1 IN50–212 (NL-43 strain) sequence (INCORE4H) in a pET23b vector using oligonucleotide mutagenesis. In addition to the four solubilizing mutations, the second IN50–212 construct (INCORE4H123) contained changes at residue positions 123–127 from STTVK to GATVR. This second construct was more consistent with that used by Chen et al. 20 (PDBID 1EXQ) and crystallized in a different space group to INCORE4H. The third construct, INCORE3H, contained the mutations C56S, F139D, and F185H only in the IN50–212 (NL43-strain) construct. A fourth construct (F185H), denoted INCOREF185H, was also prepared as described by Maignan et al. 21
IN expression and purification
For Approach A NMR experiments, INCORE4H123 was subcloned into a plasmid vector encoding an N-terminal His6-tag. All mutations, tags, and cleavage sites were confirmed by DNA sequencing. His6-INCORE4H or His6-INCORE4H123 was purified using a HisTrap column (GE Healthcare, Piscataway, NJ). The His6-tag was removed with thrombin and purified further over a HisTrap column. Fractions containing IN were pooled and dialyzed overnight against 25 mM HEPES (pH 8.0), 500 mM NaCl, 5 mM imidazole, 10% (v/v) glycerol, and 1 mM dithiothreitol (DTT) before being concentrated to 7 to 10 mg mL–1. INCORE4H123 was stored in 25 mM Tris-HCl (pH 8.5), 50 mM NaCl, 3 mM CHAPS, and 5 mM DTT at 4 14;°C at a protein concentration <2 mg/mL, under which conditions it was stable for several months.
For Approach B, both His6-INCORE4H and His6-INCORE3H were purified on a HisTrap column. The protein was subsequently buffer exchanged using a desalting column (PD-10) into 40 mM Tris (pH 8.0), 250 mM NaCl, 30 mM MgCl2, and 5 mM DTT and concentrated to 5 mg/mL using an Amicon spin concentrator (Millipore, Billerica, MA). The protein was used immediately after concentration as it was observed to precipitate upon storage at 4 14;°C, despite being soluble at room temperature.
Fragment libraries
Fragment libraries for Approaches A and B were both purchased from Maybridge, albeit at slightly different times (late 2006 for A vs. early 2007 for B). For Approach A, stocks of each fragment were made by diluting the individual fragments (25 mg) in 2H6-DMSO (200 µL) to give a concentration of approximately 660 mM, based on the average molecular weight of compounds in the library (189 Da). For Approach B, stocks of each fragment were made by diluting the individual fragments (30 mg) into 500 µL of neat DMSO to give concentrations of approximately 200 to 600 mM and were then further diluted with DMSO (v/v) for both the SPR and crystallography experiments.
NMR Spectroscopy
Reference spectra
1 H NMR spectra for individual fragments in the absence of protein (1 mM in 50 mM phosphate buffer [pH 7.0], 100% 2H2O) were collected at 10 14;°C on a Bruker-Biospin Avance 800-MHz spectrometer fitted with a cryo-probe and sample changer (Bruker-Biospin, Billerica, MA).
Saturation transfer difference (STD) NMR
Fragments were tested for binding to IN as mixtures of 10 compounds per sample. 7 Briefly, a fragment cocktail (10 µL in 2H6-DMSO) was diluted with 540 µL INCORE4H123 (100 µM) in 25 mM Tris-HCl (pH 8.5), 50 mM NaCl, 3 mM CHAPS, and 5 mM DTT, to which was added 60 µL 2H2O, giving a final concentration of each fragment of ~1 mM. NMR data were collected at 800 MHz on a Bruker Avance spectrometer (Bruker-Biospin) at 10 14;°C. Saturation of the protein resonances was achieved by a 5-s train of Gaussian pulses centered at −1 ppm. For the reference spectra, a similar saturation pulse was applied 20 14;000 Hz off-resonance. A 20-ms spin-lock period was employed before acquisition to allow the residual protein signal to decay. Results were analyzed using TOPSPIN (Bruker BioSpin) by comparison of the STD spectra with 1D spectra of the individual compounds. Fragments that gave a positive STD signal in the cocktail were retested in the STD-NMR assay as individual compounds.
15 N-HSQC spectra
Where individual compounds gave a positive STD result, binding was confirmed by recording 15 N-HSQC experiments on uniformly 15 N-labeled INCORE4H123 (0.15 mM) in the presence of single fragments (1 mM). Compounds were regarded as positive hits if chemical shift perturbations were observed in the HSQC spectrum upon addition of the fragment. Spectra were recorded at 600 and 800 MHz on a Varian Inova 600 or Bruker Avance 800 (Bruker-Biospin), respectively, both of which were equipped with cryogenically cooled probes.
Surface Plasmon Resonance
Immobilization of proteins
His6-INCORE3H and His6-INCORE4H proteins were immobilized onto a CM5 sensor chip using standard amine-coupling chemistry at 25 14;°C. HBS-P+ (10 mM HEPES, 150 mM NaCl, 0.05% [v/v] Tween-20, pH 7.4) was used as the running buffer. The carboxymethyl dextran surface was activated with a 7-min injection of a 1:1 ratio of 400 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC)/100 mM N-hydroxysuccinimide (NHS). His6-INCORE3H and His6-INCORE4H were diluted in 10 mM sodium acetate (pH 5.0) to 20 µg/mL and coupled in separate flow cells with a 7-min injection. Remaining activated groups were blocked with a 7-min injection of 1M ethanolamine (pH 8.5). Plasmodium falciparum AMA1 recombinant protein, used as an unrelated reference protein, was immobilized on the same chip using a similar method as for the IN proteins except that AMA1 protein was diluted to 50 µg/mL in 10 mM sodium acetate (pH 4.5). Protein immobilization levels typically achieved were as follows: His6-INCORE3H ~10 14;200 RU, His6-INCORE4H = 11 14;500 RU, and AMA1 (62.5 kDa) = 9 700 RU.
Fragment screening
The 10-mM fragment stocks were diluted a further 20-fold in 1.05 × HBSICP-P+ (50 mM HEPES, 150 mM NaCl, 0.05% Tween-20, pH 7.4) screening buffer to obtain 500-µM fragment concentrations in 1 × HBSICP-P+ buffer containing 5% (v/v) DMSO. Screening experiments were run at 20 14;°C with 1 × HBSICP-P+ running buffer supplemented with 5% (v/v) DMSO. Compounds were screened using a 96-well format with an association and dissociation time of 60 s each. To assess the stability of the protein surface and to allow for accurate ranking of selected fragments, the best hit selected from the first 96-well screen was used as a positive control for the subsequent 96-well plates. Experiments were normalized to a Rmax of 100 RU using a normalization formula of (MWcontrol/MWsample) · (100/Rmax, control). 22 Hits that generated an SPR signal for IN that was two times larger than the response on the reference (AMA1) protein were considered as selective binders of IN. Hits were further ranked based on their normalized response against IN. A concentration series in 2-fold dilutions (31.25–500 µM) in 1 × HBSICP-P+ buffer/5% DMSO under the same conditions as described above was used to confirm hits.
Data processing
Raw sensorgram data were reduced, solvent corrected, and double referenced using the Scrubber 2 software package (BioLogic Software, Campbell, Australia). Where appropriate, the binding affinity of the compounds was fit to a 1:1 steady-state affinity model.
Cross-validation of SPR hits
A subset of the fragments that were identified as hits in the initial SPR screen was subsequently reevaluated by both STD-NMR and SPR. For the NMR experiments, STD spectra were recorded using a slightly modified version of the protocol described above. Samples for STD contained single fragments (300 µM) and INCORE4H (5 µM) in HEPES buffer (50 mM) containing NaCl (150 mM), Tween-20 (0.05% v/v), and 1% DMSO at two pH values of 7.4 and 8.5. SPR data were also acquired using the same buffer conditions, with INCORE3H using the protocol described above. The fragments were initially tested at a single concentration (100 µM), and where a positive response was obtained, data were acquired for a concentration series in 2-fold dilutions (6.25–200 µM) containing 2% DMSO.
X-ray Crystallography
Crystallization
For Approach A, a 2-µL drop of 8 mg mL−1 INCORE4H protein solution containing 25 mM HEPES (pH 6.5), 500 mM NaCl, and 5 mM DTT was mixed with an equal volume of reservoir solution that contained 1.4 to 1.8 M (NH4)2SO4, 100 mM sodium citrate (pH 5.6), and 5 mM CdCl2. Crystallization was performed by the hanging drop vapor diffusion method at 22 14;°C. Crystals of INCORE4H123 were generated in a similar way, except the preferred reservoir solution was 1.8 M (NH4)2SO4, 150 mM sodium citrate (pH 4.6), and 5 mM CdCl2. Crystals of INCORE4H123 and INCORE4H grew up to 300 microns in the longest dimension within a few days and crystallized in the P32 and P212121 space groups, respectively. Crystals of INCOREF185H were grown as described by Maignan et al. 21
For Approach B, purified His6-INCORE3H protein was concentrated to about 5.5 mg/mL in 40 mM Tris (pH 8.0), 250 mM NaCl, 30 mM MgCl2, and 5 mM DTT. All crystallization was performed at the CSIRO Collaborative Crystallisation Centre (Parkville, Victoria, Australia) at 20 14;°C. Drops were set up in SD-2 (IDEX Corp., Lake Forest, IL) sitting drop plates using a Phoenix robot (Art Robbins Industries, Sunnyvale, CA) with 50 µL of crystallant in the reservoir and droplets consisting of 200 to 300 nL of the reservoir and equal volumes of the protein sample. Final crystallization conditions were as follows: 100 mM sodium acetate at pH 5.0 to pH 5.5 and 1.2 to 1.5 M (NH4)2SO4. The bipyramidal crystals appeared and grew to a final size of 50 to 200 microns in the longest dimension within a few days in space group P31. The crystals were cryo-protected with a cryo-solution consisting of 100 mM sodium acetate (pH 5.0), 1.5 M (NH4)2SO4, and 25% ethylene glycol.
Soaking experiments
For Approach A, stock solutions of fragments were made by dissolving each fragment in ethanol to 20 mM. Crystals of INCORE4H or INCORE4H123 were transferred to a drop containing 4 µL reservoir solution and 1 µL compound stock solution and incubated for 2 h to overnight. Prior to cryo-cooling, crystals were transferred to mother liquor containing reservoir solution, 4 mM fragment solution, and 25% (w/v) sucrose or 20% (v/v) glycerol. The X-ray diffraction data were collected on either the GMCA-CAT 23-IDB or 23-IDD beamlines at the Advanced Photon Source (Argonne, IL) or the MX beamlines at the Australian Synchrotron (Clayton, Victoria, Australia).
For Approach B, fragments in neat DMSO were added to the cryo-solution so that the DMSO-fragment solution was at 5% (a 1/20 dilution), and then 1.2 µL of this was added to drops containing crystals. Twenty-four to 48 h later, the crystals were taken to the MX1 beamline at the Australian Synchrotron for data collection. MicroLoops (MiTeGen, Ithaca, NY) were used to gently remove the crystal from the drop, and the crystals were cryo-cooled in the cold nitrogen stream of the beamline.
Data collection
All diffraction data were collected using the BLU-ICE interface 23 and indexed with Mosflm 24 or XDS 25 and scaled with XDS or Scala. 26 Structures were solved by molecular replacement with AMoRE 27 or Phaser 28 as previously described.8–10 In Approach A, compounds were built in MarvinSketch (ChemAxon, Cambridge, MA) and parameterized using Monomer Library Sketcher in CCP4. 26 In Approach B, 3D models of the fragments were generated and placed in density using AFITT (OpenEye Scientific Software, Sante Fe, NM). Refinement was completed using Refmac5 29 and Phenix.refine, 28 from the CCP4 26 and PHENIX 28 suites, respectively. Manual model building was performed using Coot. 30 The quality of the final models was evaluated with Molprobity. 31 Final refinement statistics and Protein Databank accession codes are shown in Table 1 .
Crystallography Statistics
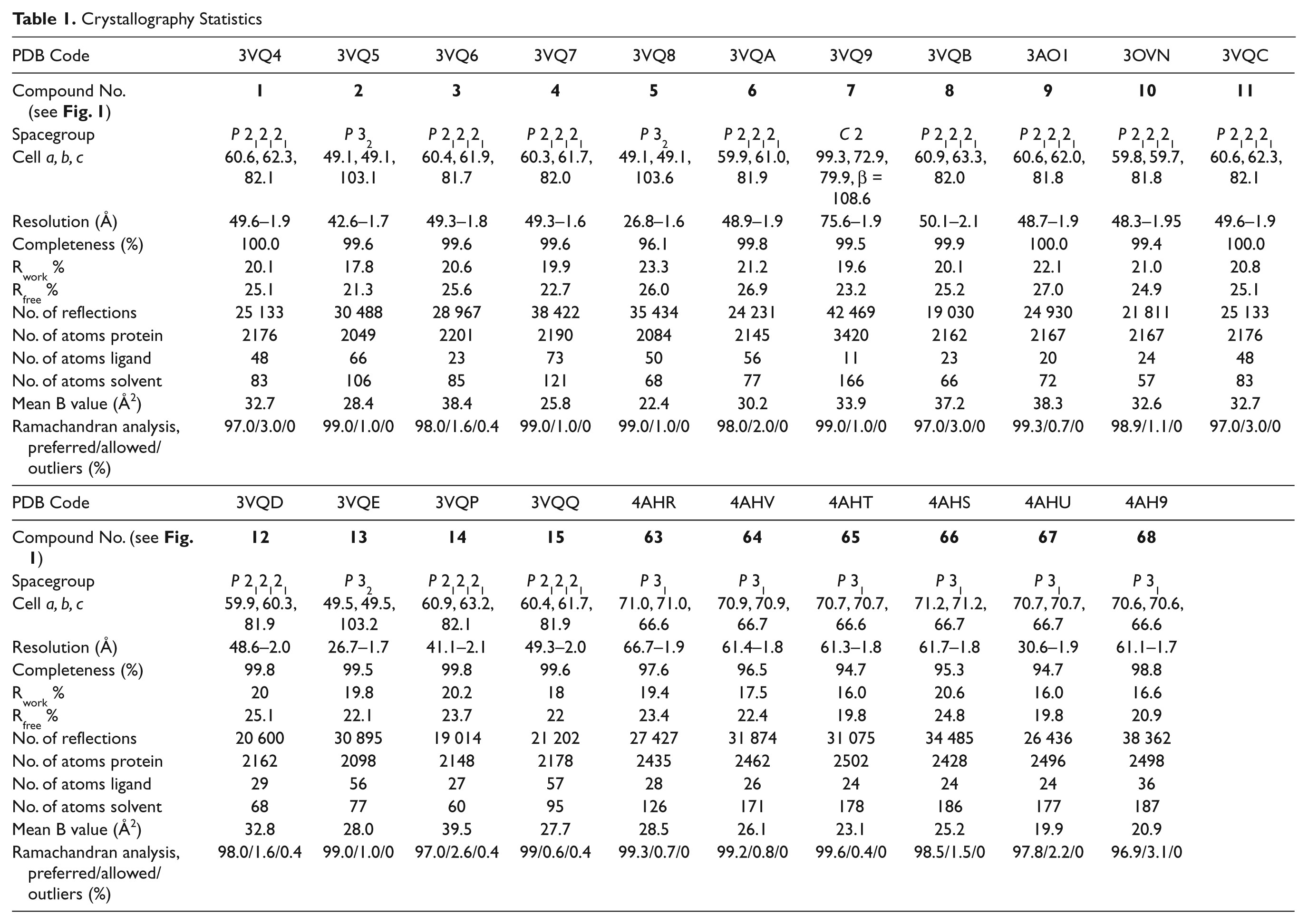
Figures were prepared with PyMOL (http://www.pymol.org). Chemoinformatics and analysis of chemical library properties were undertaken using Instant JChem (ChemAxon), CDD (Burlingame, CA), and the Benchware 3D Dataminer Suite (ACDLabs, Toronto, Ontario, Canada).
Results
Two independent groups embarked on an FBLD campaign targeting HIV-1 IN using different approaches. Approach A used NMR as the primary screening tool, whereas in Approach B, SPR was used for preliminary screening ( Table 2 ). In both cases, hits were then investigated by crystallography. Compounds from both approaches that resulted in an IN/fragment complex are shown in Figure 1 .
Identification of Fragment Hits during Screening

SPR, surface plasmon resonance; STD-NMR, used saturation transfer difference nuclear magnetic resonance spectroscopy; NA, not applicable.
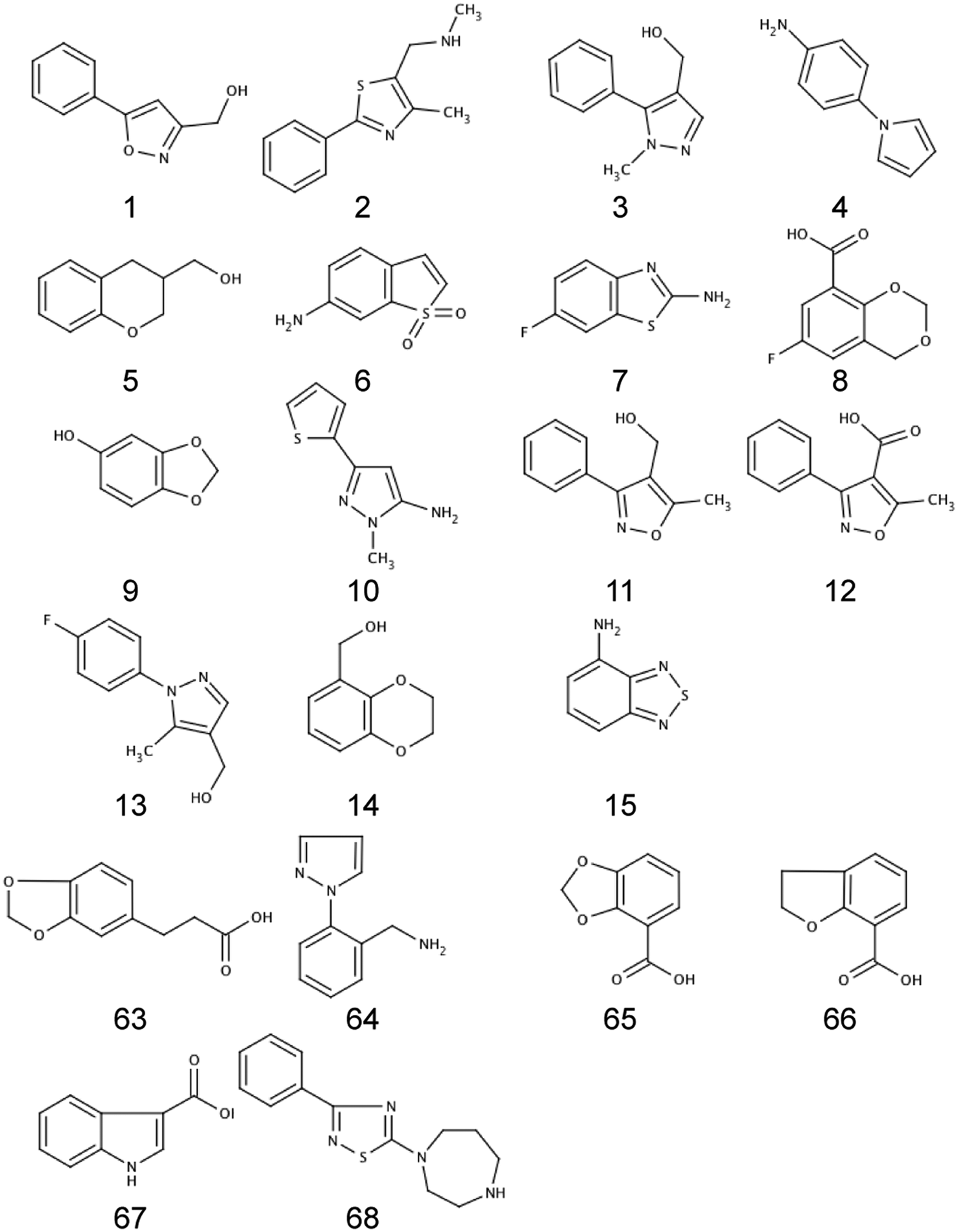
Compounds found to bind to the HIV-1 integrase core domain (IN) using a fragment-based lead discovery (FBLD) approach where a crystal structure of the fragment complex was obtained. Compounds
The Fragment Libraries
Two Rule-of-3 (Ro3) Diversity fragment libraries (Set A and Set B), each containing 500 compounds, were purchased from Maybridge about 6 months apart. The difference in purchase dates resulted in a content variation of approximately 10% between the two libraries (455 compounds in common between the two sets). Analysis of both fragment libraries showed that compounds had an average molecular weight of 189.0 Da and a calculated partition coefficient (cLogP) of 1.4. The average number of rotatable bonds was 1.8, the average number of hydrogen bond acceptors was 2.5 and 2.6, and the average number of hydrogen bond donors was 1.0 for libraries A and B, respectively. The libraries contained good diversity with 413 clusters identified (maximum cluster size of three compounds in four cases) using a Tanimoto cutoff of 85% similarity. Compounds typically comprised a 5,6 or 6,6 fused or linked ring system with one or two functional groups. Many are electron rich, containing two or more heteroatoms. Of those that contained a chiral center, it was assumed that both isomers were present.
Approach A
In Approach A, fragments were grouped into cocktails of 10 and screened against INCORE4H at a concentration of ~1 mM (based on an average molecular mass of 189 Da). Thirty-three cocktails contained positive responses, and from these, 84 compounds were identified in the preliminary STD-NMR screen. A second STD-NMR screen was performed where fragments that gave a positive STD in the mixtures were rescreened as singletons. This resulted in 51 compounds that gave a clear positive STD response and a further 11 fragments that gave a weak or very weak (borderline) response (
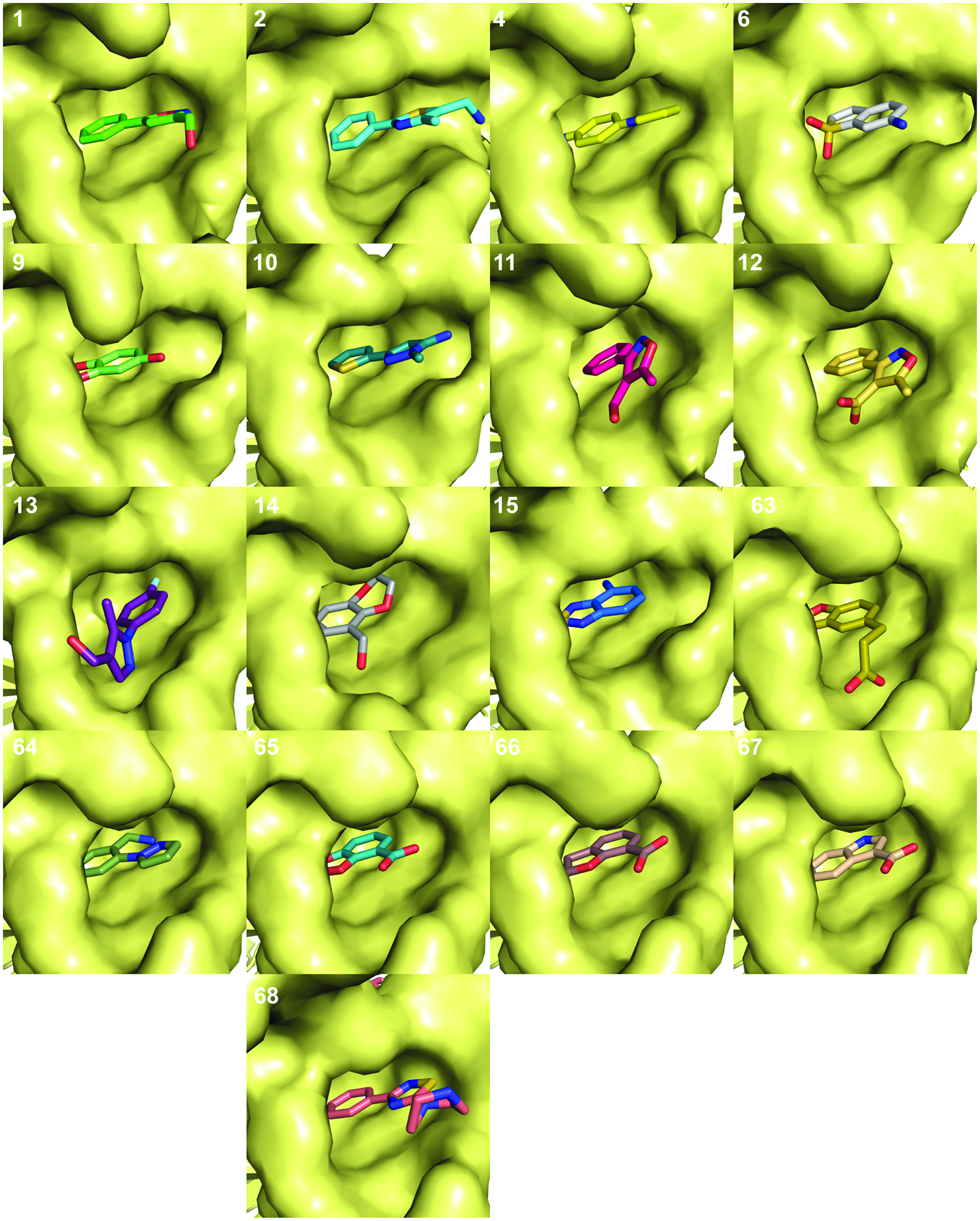
Crystal structures of fragments bound to the integrase core domain (IN) fragment binding pocket. The protein is represented as a surface with the ligands in stick form. The numbering refers to the compound numbers in Figure 1 . The coordinates for all IN/fragment complexes have been deposited in the PDB (see Table 1 for details). Structures of compounds bound to other sites on IN are not shown.
Approach B
In Approach B, compounds were individually screened by SPR. Compounds that gave a signal two times larger than the response to the AMA1 reference cell were considered hits. Hits were confirmed by SPR using a 2-fold dilution concentration series before entering crystallization trials (
Cross-Correlation of Results
The hits from the primary screen in each approach were compared, and the results of the comparison are summarized in
Figure 3
. No compounds were found to be confirmed hits in both screening methods. Three compounds (
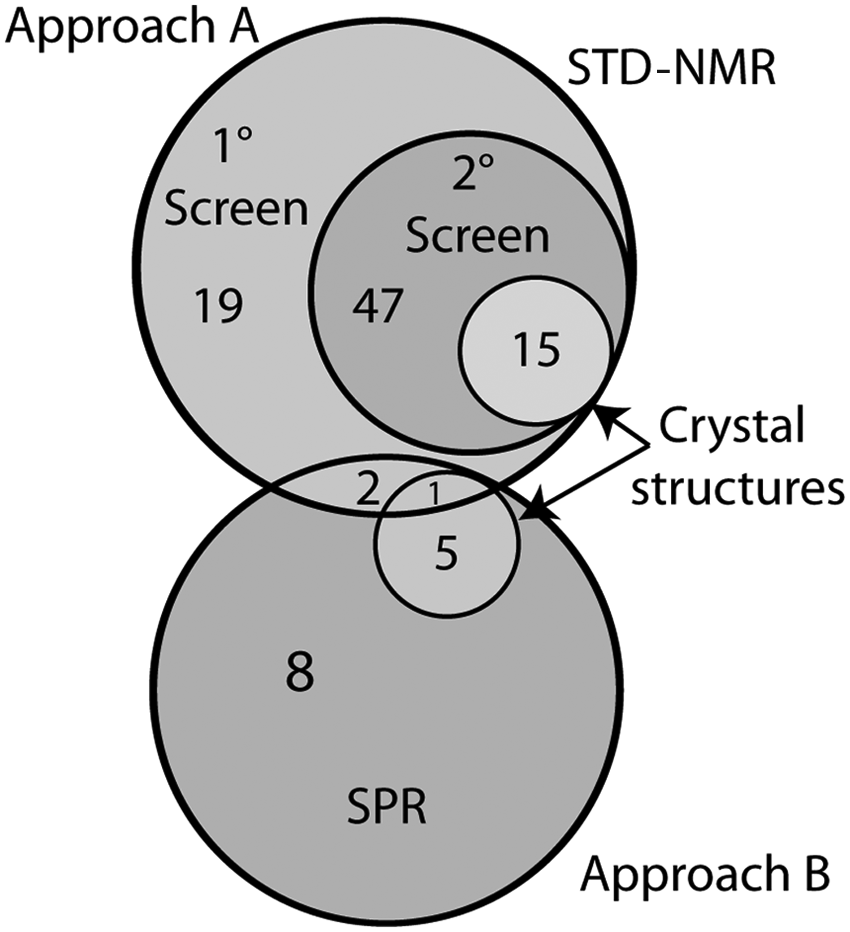
Venn diagram showing the numbers of compounds identified at each screening step and the overlap of hits found by the two approaches. STD-NMR, saturation transfer difference nuclear magnetic resonance spectroscopy.
Of the hits from Approach A that were confirmed by crystallography—eight (
Only one of the hits identified by SPR (
The same compounds were reevaluated in parallel using SPR. All of the fragments gave a positive response when tested at a single concentration, although one (
Of the six compounds from Approach B for which crystal structures were obtained, five were available for retesting. Of these, four (
The difference in pH had very little effect on the results of the STD-NMR assay—the same four compounds gave clearly positive responses at the two pH values.
Discussion
Although ~90% of the fragments tested were identical for the fragment screening campaign from each group, we see that there is little overlap between the sets of hit fragments identified by the two approaches. Although some of the differences are easily understood, the lack of overlap between the two data sets is surprising, and it was initially thought that this may reflect in part the different conditions used in the two screening campaigns. The crystallization conditions were similar for the different proteins, all having precipitant solutions consisting of approximately 1.5M ammonium sulfate and a low pH acetate or citrate buffer (pH 4.6–5.6). In Approach A, CdCl2 was a component of the crystallization conditions, whereas in Approach B, MgCl2 was a component in the protein solution. Therefore, the initial selection of compounds from the screens done by NMR or SPR was probably the major contributor to the different results. A difference between the initial screens was the pH and buffer conditions under which the experiments were run; the NMR screen used Tris buffer at pH 8.5, and the SPR used HEPES buffer at pH 7.4. DMSO was present in both cases at about 1.5% for NMR and 5% for SPR, along with a small amount of detergent (CHAPS or Tween); NaCl was either 50 or 150 mM, and the fragment was present at either 1 or 0.5 mM (NMR and SPR, respectively). However, from the results of the rescreening hits by STD-NMR and SPR, we did not find any significant differences between the assays run at either pH (see
Selection Criteria
The criteria used in Approach A for progressing hit compounds into X-ray crystallography were less stringent than Approach B, as even very weak binders from the STD-NMR results were considered. This was to obtain a greater understanding of compound binding to IN and to maximize compound diversity for further fragment optimization at a later stage. No requirement for selective binding to IN was imposed, and this likely reflects the higher number of fragments that were progressed to crystallography from Approach A. In contrast, for progression into X-ray crystallography by Approach B, compounds had to show a preference to binding IN over the reference AMA1 protein as well as a clear dose-response curve from secondary SPR screening. The AMA1 protein was used in a reference cell as it had previously been shown to give reliable SPR data during fragment screening and it was unrelated to IN, so one could therefore eliminate promiscuous binders from the pool of binding fragments, leaving fewer compounds to screen via crystallography. Thus, some specificity was built in from the start of the project.
Although both approaches were successful in identifying fragments that bound and in generating structural data to support further development of the fragments, there was a surprising lack of overlap between the results of the two campaigns. In some respects, IN is an atypical target—in addition to the active site of the enzyme, there have been several other binding sites reported, as described above. In such a case, it might be anticipated that a higher than usual hit rate would be observed in the screen. This was the case in Approach A, where STD-NMR was used as the binding assay and a hit rate of ~17% was observed in mixtures of fragments, and ~13% of the fragments in the library gave a positive STD response when tested individually. In contrast, the primary screen from Approach B yielded a hit rate of only ~3%.
It is worth noting that in addition to the different biophysical techniques and solution conditions used to monitor binding, the criteria for defining a hit differed significantly in the two approaches. The addition of a selectivity filter in Approach B—such that fragments were only considered hits if the response against IN was twice as large as that observed for an off-target protein (AMA1)—removed half of the fragments for which crystal structures were subsequently obtained using the less stringent criteria of Approach A. Combined with the cases where DMSO mismatches were observed in the SPR assay, this provides an explanation for most of the fragment hits from Approach A not being classified as hits in Approach B. No such explanation was evident in the reverse comparison, where most of the compounds classified as hits in Approach B were present and identified as chemically correct in the NMR spectra of Approach A but did not give a positive STD result.
Rescreening a subset (12/15) of the Approach B hits in the NMR assay at a lower concentration identified four of these as hits. Eight of the 12 SPR hits were still classified as very weak/marginal or nonbinders by STD-NMR and would not have been followed up under the criteria of the initial screen. This may reflect a difference in the threshold for detecting binding under the conditions used for the different screening formats. In the case of the STD-NMR assay, it is apparent that screening of less complex mixtures at lower concentrations is likely to result in fewer false negatives that may arise due to insolubility or competition. Based on a hit rate of 17% observed in the NMR assay, most mixtures would contain two fragments that bound to IN, raising the possibility of competition, which may decrease the intensity of any observed STD.
Chemistry of Hits
Although the NMR and SPR screens failed to identify the same fragments as hits, some common chemical features are present in both sets of hits, for example, rings containing two oxygen atoms (
pH
It is worth noting that all the crystallographic studies were carried out at acidic pH (pH 4.6–5.6). However, during screening, the SPR was run at pH 7.4 and the STD-NMR at pH 8.5. For fragments
Binding Sites
Compounds were found to bind predominantly into the so-called fragment binding pocket (FBP), 9 although some of the fragments were also observed to bind at the LEDGF binding site (see below). Other binding locations were observed; however, these were typically isolated cases and involved contacts with neighboring proteins in the crystal lattice.
The IN dimer contains two equivalent FBPs and two equivalent LEDGF binding pockets, both located at the dimer interface of the INCORE domain. The FBP site is a small hydrophobic pocket adjacent to the LEDGF binding site. 9 There are two equivalent binding sites per dimer: FBP1 comprises residues A105, G106, R107, W108, P109, V110, K111, A133, G134, I204, T206, and I208 from Monomer A and Y83, W108, N184, F185H, K186, R187, S195, G197, E198, I200, and V201 from Monomer B. FBP2 has the equivalent residues donated by the opposite monomers. The FBP is conserved in other structures of related integrases from ASV, SIV, HIV-2, and BIV and has been suggested to be a suitable site for targeting by small-molecule inhibitors of IN.9,32 The LEDGF binding pocket was first visualized by Cherepanov et al. 33 The central residues in IN involved in interacting with the LEDGF IN binding domain are D167, Q168, A169, E170, H171, and T174 (monomer A) and T124, T125, and Q95 (monomer B) or similar residues on the opposite monomer for the second LEDGF binding site. LEDGF has an important role in the docking of the integration complex onto the host chromatin DNA. Disruption of this binding event significantly hampers or prevents integration, making it an excellent target site for allosteric drug development.
In addition to the complexes for which well-defined density was observed for the bound fragment, we obtained numerous examples of crystal structures where weak or partial density was observed in either the fragment or LEDGF binding sites. These results could be from multiple orientations of the fragment in the binding pocket or partial occupancy of the binding site. Although binding orientations could sometimes be inferred from better defined examples or closely related fragments, these fragments were not considered suitable structures for final models.
Availability of Binding Sites due to Crystal Packing
Slightly different protein constructs were used by the two groups during screening, and protein crystallization and soaking experiments were performed under different conditions. As a consequence, it is possible that crystal packing had an influence on the results obtained ( Fig. 4 ). The packing of the INCORE4H is such that one FBP and one LEDGF binding pocket are mostly obscured by a neighboring IN molecule in the crystal lattice. This resulted in the fragment being observed in only one of the two equivalent binding sites in the crystal structure in some instances. However, in the P32 (INCORE4H123) and P31 (His6-INCORE3H) space group structures, both fragment binding sites and LEDGF binding sites are unobstructed and available to bind fragments. In these cases, any bound fragment was found in both FBP sites and adopted the same binding mode. The three FBP sites were accessible for fragment binding in the C2 (INF185H) space group structure; however, all three LEDGF binding sites were blocked by crystal neighbors, so this construct was not considered further.
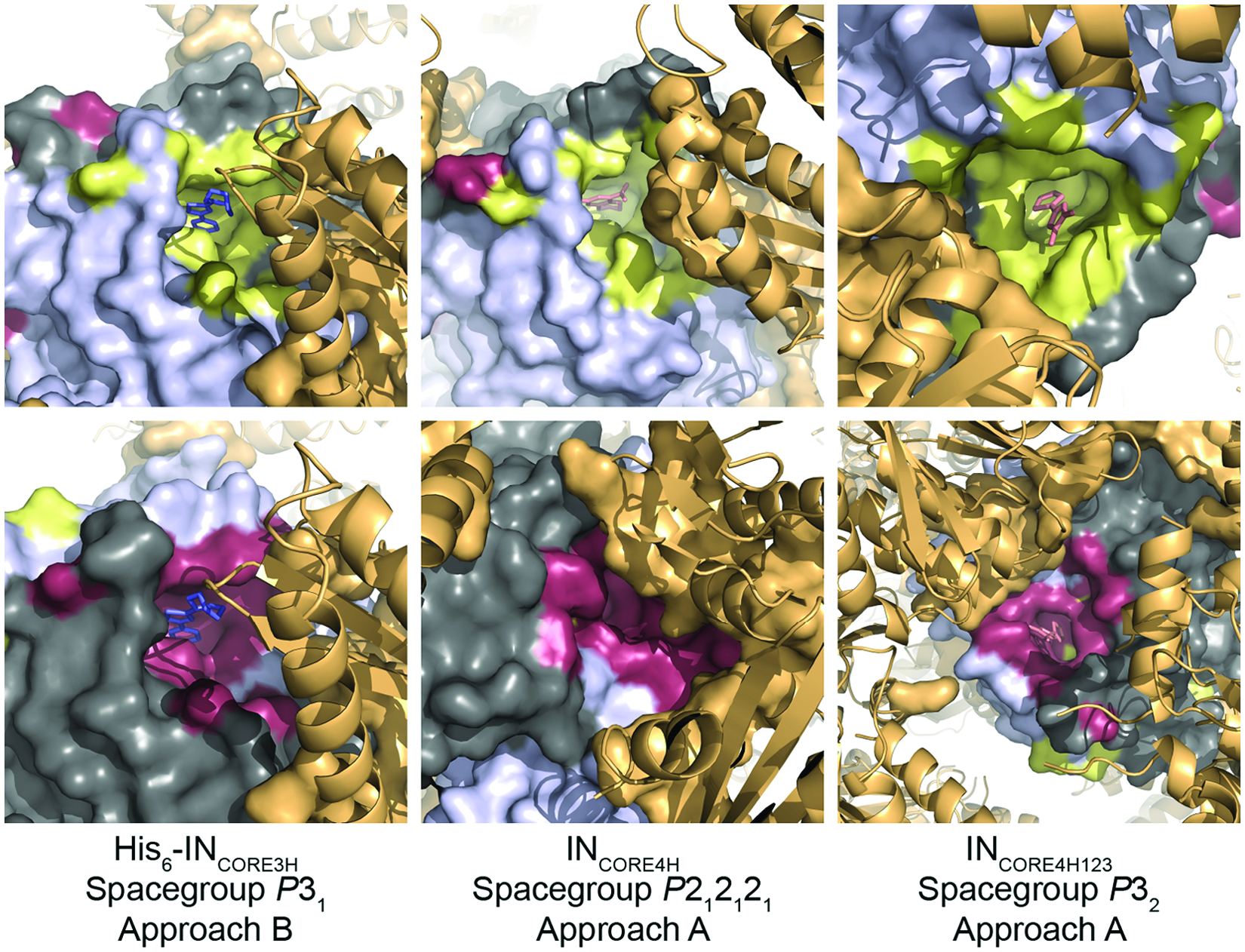
The effect of crystal packing on the fragment binding pocket. Several protein constructs were used in these studies. Each construct crystallized in a different space group, leading to variable accessibility of the fragment binding pocket and active site for soaking experiments. Integrase core domain (IN) dimers are shown with a Connolly surface in gray/dark gray, and fragment binding pockets are yellow or raspberry, respectively. Crystallographic neighbors are orange. Residues of the crystallographic neighbors within 5 Å of the fragment binding pocket are shown as an orange surface. In the case of INCORE4H, one fragment binding pocket (top, middle) is mostly blocked by crystal contacts.
This study highlights the sensitivity of different assay formats to the experimental conditions and assay design. It also reveals some of the strengths of different assay formats—for example, screening by STD-NMR allows the chemical authenticity of fragments to be established at the time of screening, whereas SPR allows relatively straightforward measurement of binding affinity. This reinforces the complementarity and value of using different biophysical assays for characterizing binding in the early stages of fragment screening campaigns. However, it also highlights the need to adopt a coordinated approach to analysis of the screening data—in the current example, the different selection criteria used to classify hits in the two approaches led to mutually exclusive sets of fragments. In our original screening, we saw no overlap of hits between the two approaches, and in such situations, one might elect to take both sets of hits forward. However, we showed on rescreening that there was significant overlap of hits between the two approaches. In this case, if one were to combine NMR and SPR data in the early stages of screening, it would allow the presence of a binding event to be detected, the chemical authenticity of the hit to be established, and an estimate of the binding affinity and therefore ligand efficiency of the fragment to be obtained.
Footnotes
Acknowledgements
This research was partly undertaken on the MX beamlines at the Australian Synchrotron and the GMCA/CAT beamlines at the Advanced Photon Source.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by an Australian Research Council Linkage Project grant (LP0775192) and a Commercial Ready grant (COM04229) from the Commonwealth of Australia, Department of Innovation, Industry, Science and Research. Infrastructure support from the NHMRC Independent Research Institutes Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Support Program is gratefully acknowledged. M. W. P. is an NHMRC Research Fellow.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.