
Abstract
Kir7.1 is an inwardly rectifying potassium channel that has been implicated in controlling the resting membrane potential of the myometrium. Abnormal uterine activity in pregnancy plays an important role in postpartum hemorrhage, and novel therapies for this condition may lie in manipulation of membrane potential. This work presents an assay development and screening strategy for identifying novel inhibitors of Kir7.1. A cell-based automated patch-clamp electrophysiology assay was developed using the IonWorks Quattro (Molecular Devices, Sunnyvale, CA) system, and the iterative optimization is described. In total, 7087 compounds were tested, with a hit rate (>40% inhibition) of 3.09%. During screening, average Z′ values of 0.63 ± 0.09 were observed. After chemistry triage, lead compounds were resynthesized and activity confirmed by IC50 determinations. The most potent compound identified (MRT00200769) gave rise to an IC50 of 1.3 µM at Kir7.1. Compounds were assessed for selectivity using the inwardly rectifying potassium channel Kir1.1 (ROMK) and hERG (human Ether-à-go-go Related Gene). Pharmacological characterization of known Kir7.1 inhibitors was also carried out and analogues of VU590 tested to assess selectivity at Kir7.1.
Introduction
Inwardly rectifying potassium (Kir) channels display the unusual property of conducting potassium ions more effectively at potentials negative to the reversal potential for potassium (Ek) than at potentials positive to Ek. The underlying basis of this inward rectification is believed to result from blockade of the channel pore by intracellular substances such as magnesium ions and polyamines. 1 Kir channels play an important role in establishing resting membrane potential (RMP) and, under physiological conditions, contribute a persistent outward K+ conductance, which in some cases is under the control of ligands such as adenosine triphosphate (ATP). This permits Kir channels to play a key role in the regulation of the stimulus and duration of action potentials in electrically excitable cells and the establishing of electrochemical gradients in epithelia.
There are seven subfamilies of Kir channels (Kir1–7), which can be classified into four groups based on function. Kir7.1, classified as a K+ transport channel, is encoded by the KCNJ13 gene. Mutations in the KCNJ13 gene have been linked to snowflake vitreoretinal degeneration. 2 Kir7.1 shares only ∼38% homology with its closest paralogue, Kir4.2, and is expressed on the membrane of various epithelial cells. 1 The Kir7.1 channel displays several uncommon properties compared with other inwardly rectifying potassium channels. These include low sensitivity to barium and cesium, low single-channel conductance, no internal block by magnesium ions, and a relative insensitivity to tetraethylammonium (TEA), 4 aminopyridine (4-AP), and external potassium concentration.3,4
Postpartum hemorrhage (PPH) is defined as the loss of 500 mL or more of blood from the genital tract within 24 hours of the birth of the baby. 5 It is estimated to affect 10% of births and is a major risk of fatality in the developing world. The most common cause of PPH is uterine atony—a loss of tone in the uterine musculature. During normal delivery, the uterus contracts vigorously after delivery of the baby and placenta. This final tonic contraction is critical to stem bleeding from the placental bed. The current treatment for PPH is the prophylactic use of oxytocics. 5 However, based on mortality and hysterectomy rates, these fail to affect acute PPH outcomes compared with placebo. Moreover, native signaling pathways can become desensitized if oxytocics are used for labor induction.
Uterine contractility is profoundly influenced by the voltage-dependent control of calcium entry. Kir7.1 expression in the myometrium has been shown to alter during gestation in humans and mice, 6 and these changes in expression correlate positively with changes in the observed resting membrane potential of the myometrium. Under normal circumstances, the myometrium becomes depolarized to near threshold near birth, 7 increasing the probability of action potential firing and initiating a subsequent contraction. Knockdown of Kir7.1, using lentiviral-delivered microRNA (miRNA), significantly increased contractile force and duration, whereas overexpression led to decreased contractility, an effect mediated by a direct effect on membrane potential. 6 In addition, administration of the known Kir7.1 inhibitor VU590 8 rapidly depolarized resting membrane potential and led to an increase in contractility. 6
It was the aim of this work to identify inhibitors of Kir7.1 that may be of therapeutic potential in the treatment of postpartum hemorrhage. By targeting an alternative mechanism to that which current therapies focus on, cellular machinery that may have become exhausted or desensitized is bypassed. A novel cell-based IonWorks (Molecular Devices, Sunnyvale, CA) automated electrophysiology assay was developed and a low molecular weight compound library screened for Kir7.1 inhibitors. The IonWorks system was chosen as it offered a number of advantages 9 compared with fluorescence-based techniques, such as thallium flux or membrane potential dyes. Primarily, IonWorks, like standard electrophysiology techniques, allows the direct measurement of electrical currents while controlling the membrane potential of the cell, facilitating kinetic resolution of ion movement through channels. This, along with the ability to finely control leak subtraction, allows increased sensitivity of measurements, a major advantage for low-conductance channels such as Kir7.1. Preliminary assessments of potency and selectivity were used to triage and optimize compound profiles in addition to limited medicinal chemistry. Exemplar compounds were profiled for effects on mouse uterine contractility 6 to further validate a role for Kir7.1 regulation of resting membrane potential and postpartum hemorrhage.
Materials and Methods
Cell Culture
Chinese hamster ovary (CHO) cells were stably transfected, using Lipofectamine 2000 (Life Technologies, Carlsbad, CA), with vector pcDNA5/FRT (Life Technologies) containing human Kir7.1 (NM_002242.4). Cells were maintained in Ham’s F12 Glutamax (Life Technologies), supplemented with 10% fetal bovine serum (FBS; Life Technologies) and 200 µg/mL hygromycin (Life Technologies). Clones were selected functionally using the IonWorks automated patch-clamp electrophysiology assay described below. Cells were typically maintained at 37 °C with 5% CO2 and were not permitted to exceed 85% confluence. Prior to assaying, cells were moved to a 30 °C, 5% CO2 incubator (6–18 h after dissociation using 0.25% trypsin-EDTA solution; Life Technologies) and held for 90 to 120 h at this lower temperature prior to use.
Automated Patch Clamp
Automated patch-clamp electrophysiology experiments were carried out using the IonWorks system as described. 9 Using a population patch-clamp (PPC) mode, 10 an ensemble average of the current from up to 64 cells per well was recorded. The external recording solution comprised 4 mM NaCl, 50 mM RbCl, 90 mM KCl, 10 mM HEPES, 1 mM MgCl2, and 1.8 mM CaCl2 at pH 7.3. The internal solution comprised 90 mM K-gluconate, 40 mM KCl, 10 mM NaCl, 3.2 mM EGTA, 5 mM HEPES, and 3.2 mM MgCl2 at pH 7.3. All buffer constituents were obtained from Sigma-Aldrich (St. Louis, MO). Electrical access was achieved using amphotericin (200 µg/mL) in the internal solution to obtain the perforated patch-clamp configuration. A brief holding potential period of 1 s at 0 mV was followed by a leak subtraction step to +10 mV, and then the voltage protocol was employed where cells were initially held for 1 s at −130 mV before a ramp to +60 mV was applied over a 1-s period. This was then followed by a 200-ms step from −30 to 0 mV. An identical voltage protocol was applied before (predrug) and 5 to 7 min after (postdrug) application of test compounds.
Screening and Data Analysis
Test compounds were prepared from 100-mM (100% DMSO) stocks and diluted in external buffer (33.3 µM final assay concentration, 0.33% DMSO). High and low controls were defined with Ba2+ (3.3 mM final assay concentration) and buffer alone (0.33% DMSO), respectively. During primary screening, VU590 (100 µM final assay concentration) was included on all screening plates. Compound solubility was also visually assessed.
Using the IonWorks protocol, mean current amplitudes of the step period at each end of the voltage ramp were routinely measured (–130 to +60 mV). To avoid contamination by offset or leak subtraction errors, the Kir7.1 activity was determined by subtracting the current at 0 mV from that at −130mV, thus determining a rectification value. Analysis of the data was performed using IonWorks software v.2.0.4.4, Microsoft Excel Fit 5 (v5.0; Microsoft, Redmond, CA), and GraphPad Prism (v5; GraphPad Software, La Jolla, CA). The effects of compounds were quantified by first dividing the Kir7.1 current amplitude in the presence of compound by the amplitude of the precompound current, multiplied by 100. This % inhibition value was then normalized to the effect of the time/vehicle (low) control change by dividing it by the average values for that test plate (see equation below). Eleven-point concentration-response curves were generated for compounds with a maximal concentration of 33 µM. IC50 values were determined using a four-parameter logistic fit (GraphPad Prism) from the individual values compiled across three recordings performed on a single day.
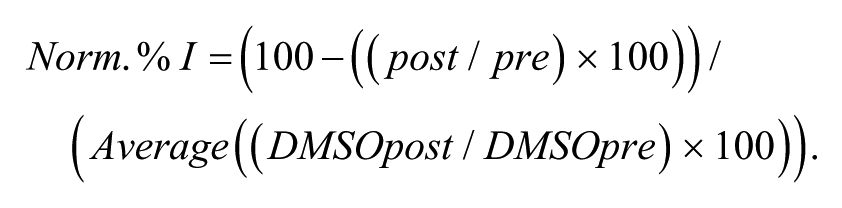
Selectivity Assays
Kir1.1 (ROMK) activity was measured using the FluxOR thallium assay (Life Technologies). CHO cells were transduced using Kir1.1 baculovirus (Life Technologies) at 10% v/v, overnight at 37 °C with 5% CO2. Cells were plated at 10k cells per well and incubated at 30 °C with 5% CO2 for 72 h. FluxOR assays were carried out as per the manufacturer’s guidelines. Plates were measured using the Flexstation II (Molecular Devices); activity was defined as fluorescence (normalized to prethallium addition baseline) at 180 s. hERG (human Ether-à-go-go-Related Gene) activity was measured using the Predictor hERG Fluorescence Polarization Assay (Life Technologies) as per the manufacturer’s guidelines.
Whole-Cell Patch-Clamp Electrophysiology—Voltage Clamp
A drop of CHO-Kir7.1 cell suspension was placed in a glass-bottomed Petri dish and mounted on the stage of an inverted microscope (IX51; Olympus, Tokyo, Japan). After settling (approximately 10 min), cells were perfused with bath solution at a rate of 1 to 2 mL/min−1 at 37 °C. Patch pipettes were fabricated (Model P-87; Sutter Instruments, Novato, CA) from 1.5-mm glass capillaries and had a resistance of 2.0 to 4.0 MΩ when filled with pipette solution (containing 140 mM KCl, 1.1 mM EGTA, 0.06 mM CaCl2, 10 mM HEPES, and 2 mM MgCl2, adjusted to pH 7.2 at 25 °C with NaOH). Liquid junction potential was zeroed prior to seal formation. Transmembrane currents were recorded with an amplifier (Axopatch 700b; Axon Instruments, Molecular Devices, Sunnyvale, CA) using the perforated patch configuration of the whole-cell patch-clamp technique. 11 The antibiotic amphotericin B (720 µg/mL) was used to perforate the cell membrane. Series resistance was compensated after membrane perforation. Currents were elicited by stepping to a range of potentials between −150 and +80 mV from a holding potential of −60 mV. Voltage protocols were delivered via a Digidata 1440a computer interface using pCLAMP 9.0 software (Molecular Devices).
Results
Generation of an Automated Patch-Clamp Assay for Kir7.1
Previous Kir7.1 screening strategies, using thallium flux as a measure of channel function, have used a mutation at the M125R residue of Kir7.1 to enable the generation of signals suitable for screening. 12 This methionine residue, located in the Kir7.1 pore region, gives rise to Kir7.1’s unique conductance properties. 4 The intention of this work was to use a more physiologically relevant sequence for screening (human wild-type Kir7.1) and to more directly measure channel conductance (compared with fluorescence-based techniques). Initially, the generated CHO Kir7.1 cells were also analyzed by standard whole-cell electrophysiology to ensure they exhibited the expected properties ( Fig. 1A ). Whole-cell recordings showed an inward current at negative voltages, characteristic of Kir7.1.
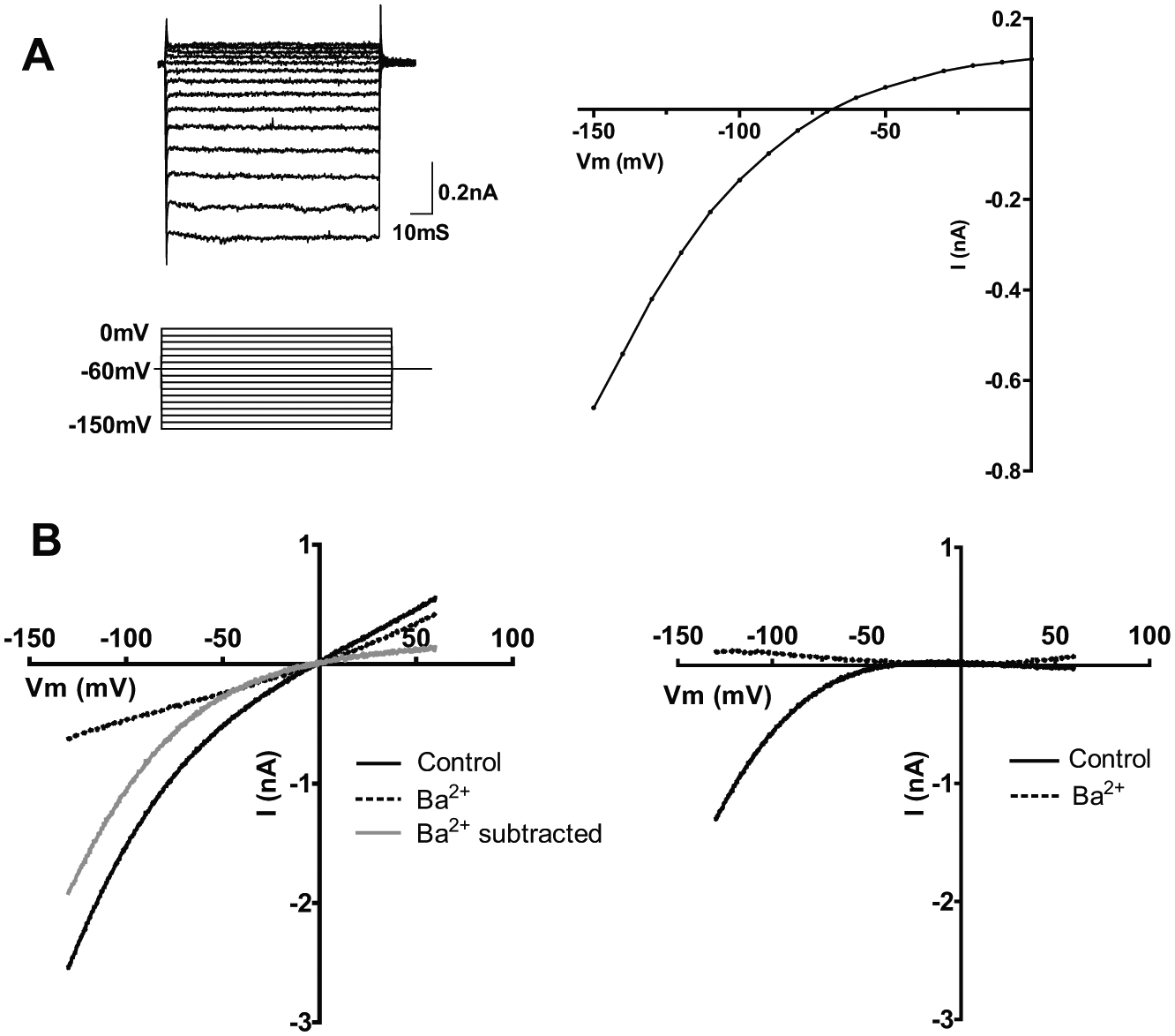
Electrophysiological characterization of a Chinese hamster ovary (CHO)–Kir7.1 cell line. (
IonWorks is an automated patch-clamp system that uses a planar, multiwell substrate (patch plate). Cells are positioned above a hole separating two fluid compartments in each well of the substrate. Voltage control and current recordings from the cell membrane are made subsequent to gaining access to the cell interior by applying a permeabilizing agent to the intracellular side. 9 Due to the relatively low seal resistances observed during automated patch-clamp recordings (megaohms compared with gigaohms), it is necessary to carry out “leak subtraction” to remove contaminating nonspecific signal. The strategy employed in this study was to take advantage of the inwardly rectifying nature of the Kir7.1 channel and introduce a leak subtraction voltage step from 0 to 10 mV, a potential range where little Kir7.1 current is observed using the described experimental conditions. Validation of this approach was achieved by comparing recordings with non-leak-subtracted data, where leak was determined by an additional application of the known Kir7.1 blocker, Ba2+ ( Fig. 1B ). The use of a positive voltage step to determine leak was an important factor when determining the parental cell line chosen. On the IonWorks platform, CHO cells exhibited the smallest outward current at these voltages, allowing a robust leak subtraction protocol. Importantly, wild-type CHO cells also showed no inward current, meaning there would be no interference on the recombinant Kir7.1 current being measured (data not shown).
Kir7.1 is known to exhibit low conductance 3 ; hence, numerous strategies were employed to increase activity of the wild-type channel and generate a signal suitable for compound screening. Initial IonWorks experiments showed the CHO Kir7.1 cell line produced a current of approximately 0.1 nA. This was greatly enhanced by lowering the incubation temperature of the cells to 30 °C, and the mean current amplitude increased in a time-dependent manner, with cells incubated at 30 °C for 24 h showing a mean of 0.2 ± 0.04 nA and those incubated at 30 °C for 96 h showing a mean current of 0.7 ± 0.1 nA ( Fig. 2A ). Addition of sodium butyrate (1 mM) also caused an increase in the measured current, albeit to a lesser extent, while serum starvation had no effect. Potassium channels are known to exhibit increased conductance for rubidium relative to potassium, 13 and thus the addition of rubidium to the external buffer was hypothesized to further increase the measured current. A dose-dependent increase in current was observed by substituting K+ with Rb+ ( Fig. 2B ), with the addition of 70 mM Rb+ giving rise to an average current of 3.6 ± 0.3 nA, a fourfold increase compared with no Rb+-containing controls. Complete replacement of 140 mM K+ with 141 mM Rb+ in the external buffer caused a tenfold increase in signal. However, under these conditions, seal resistances were compromised, with successful recordings from only 6% of cells tested. Previous efforts to improve seal resistances, such as the addition of Cs-MeSO4, had proved ineffective (data not shown). An external solution containing 50 mM Rb+ and 90 mM K+ was used for screening and provided a greatly enhanced signal while maintaining seal resistance, translating to more successful recordings. This substitution of K+ to Rb+ did not significantly affect the apparent IC50 of the Kir7.1 inhibitor VU590 ( Fig. 2C ), and it is unlikely that ionic substitution with Rb+ would have any significant effect on osmolarity of the external solution.
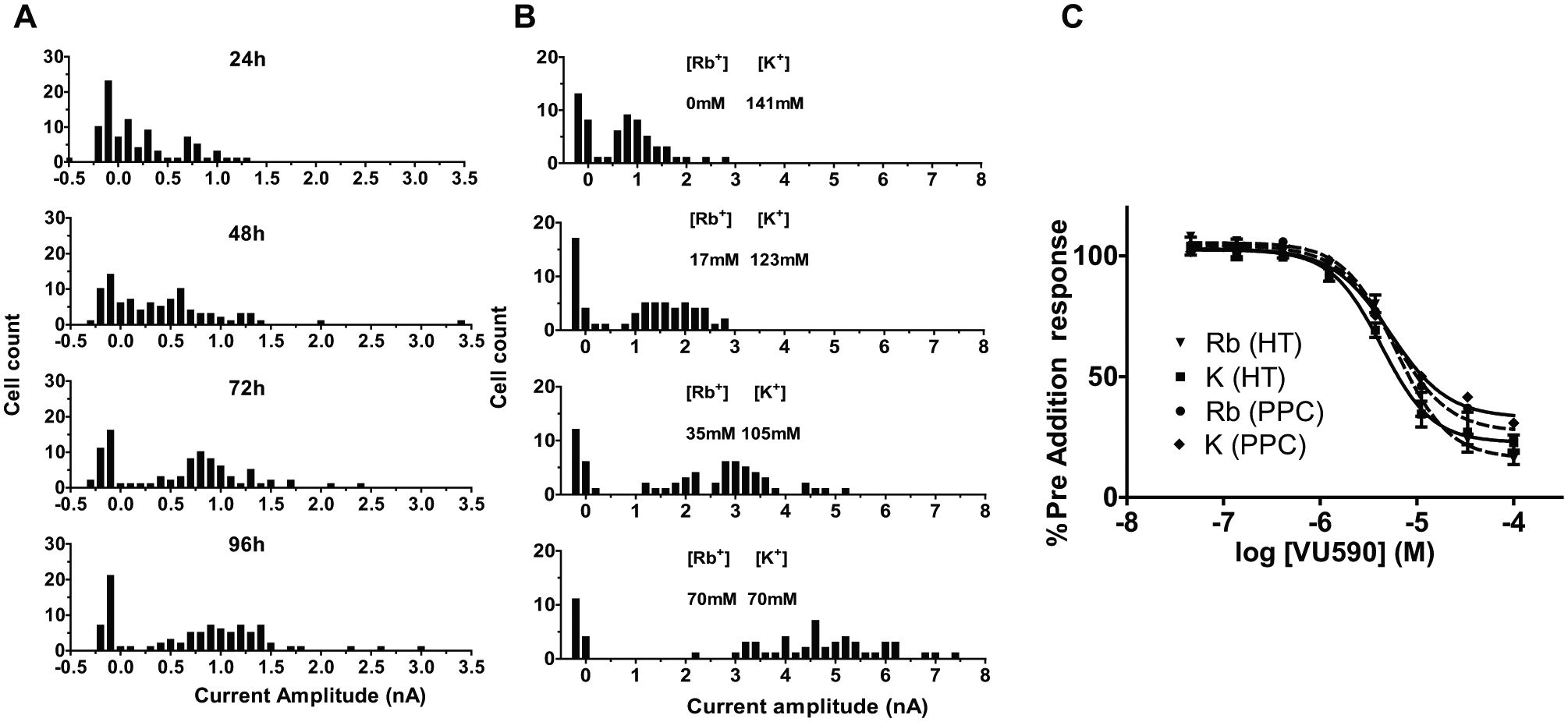
Optimization of Kir7.1 IonWorks assay. (
Despite successfully increasing the observed current in CHO Kir7.1 cells, populations appeared heterogeneous, with a subpopulation of cells showing a reduced current or no current. Initial optimization had used the IonWorks high-throughput (HT) format, which contains a single aperture per well and thus measures current from a single cell. In contrast, the PPC format contained 64 holes per well and thus averaged current from up to 64 cells. Preliminary experiments using the PPC format showed successful recordings (>0.3 nA current and successful seal formation, >20 MΩ) from 379 of 384 wells tested, with a Z′ of 0.72. In comparison, 227 of 384 successful recordings and a Z′ of 0.65 were observed using the HT format. Pharmacology of the HT and PPC systems was also compared using VU590, and comparable IC50 values were observed for the two systems ( Fig. 2C ). During optimization, mean currents were of a similar magnitude using the HT and PPC formats (~2 nA), and mean seal were resistances were higher (~100 MΩ) in the HT format. However, PPC mode was chosen due to the increased number of wells giving successful recordings. To assess its reproducibility and suitability for single-concentration screening, data from multiple runs were compared. Test plates were constructed containing DMSO controls, 3.3 mM Ba2+, and VU590 (2.5 µM, 5 µM, or 10 µM). Comparisons between experimental runs suggested excellent reproducibility (R2 = 0.95) and supported the choice of assay for high-throughput screening (HTS) ( Fig. 3A ).
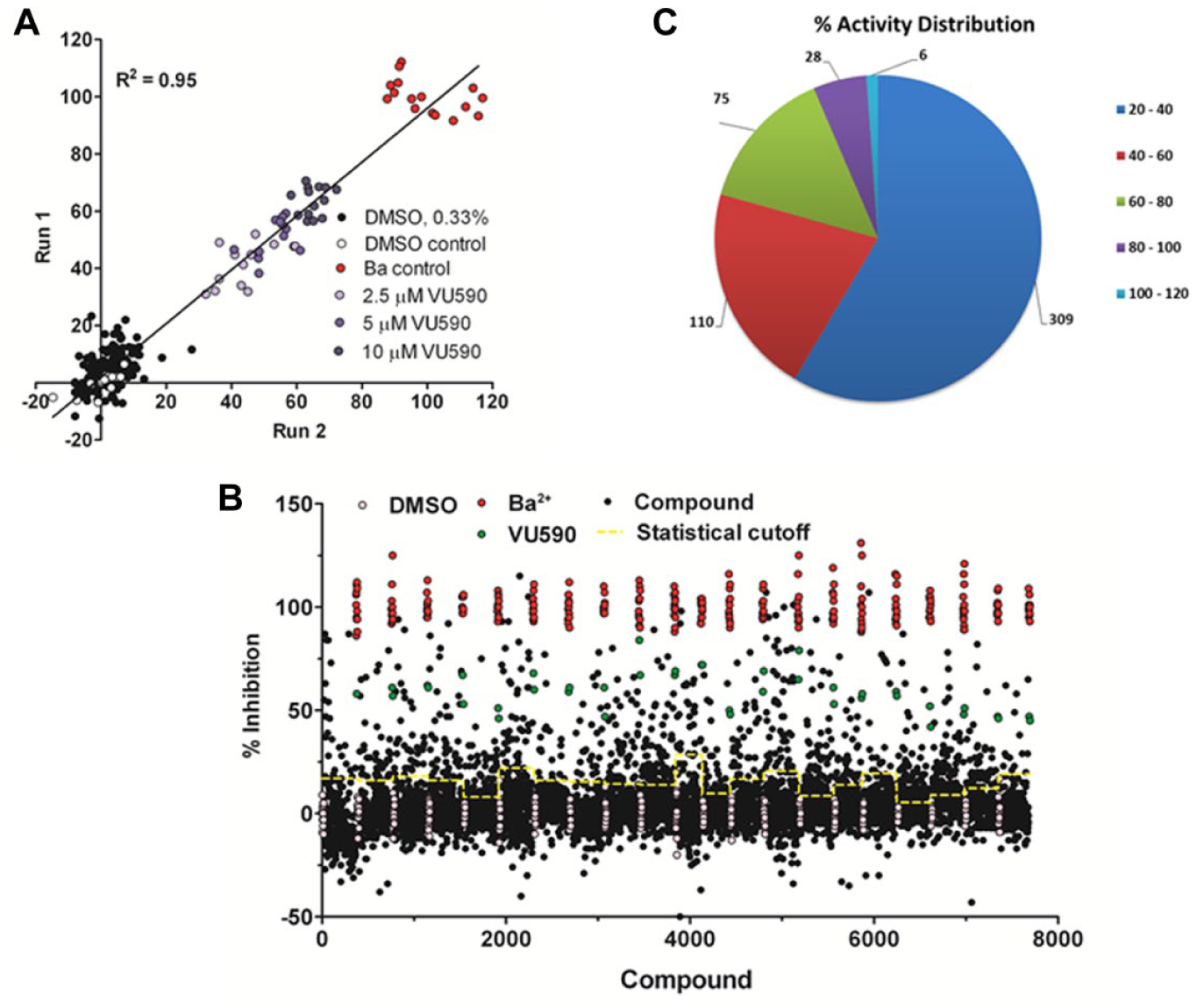
Kir7.1 population patch-clamp (PPC) IonWorks assay performance. (
Pharmacological Validation of Assay and Screening for Novel Inhibitors of Kir7.1
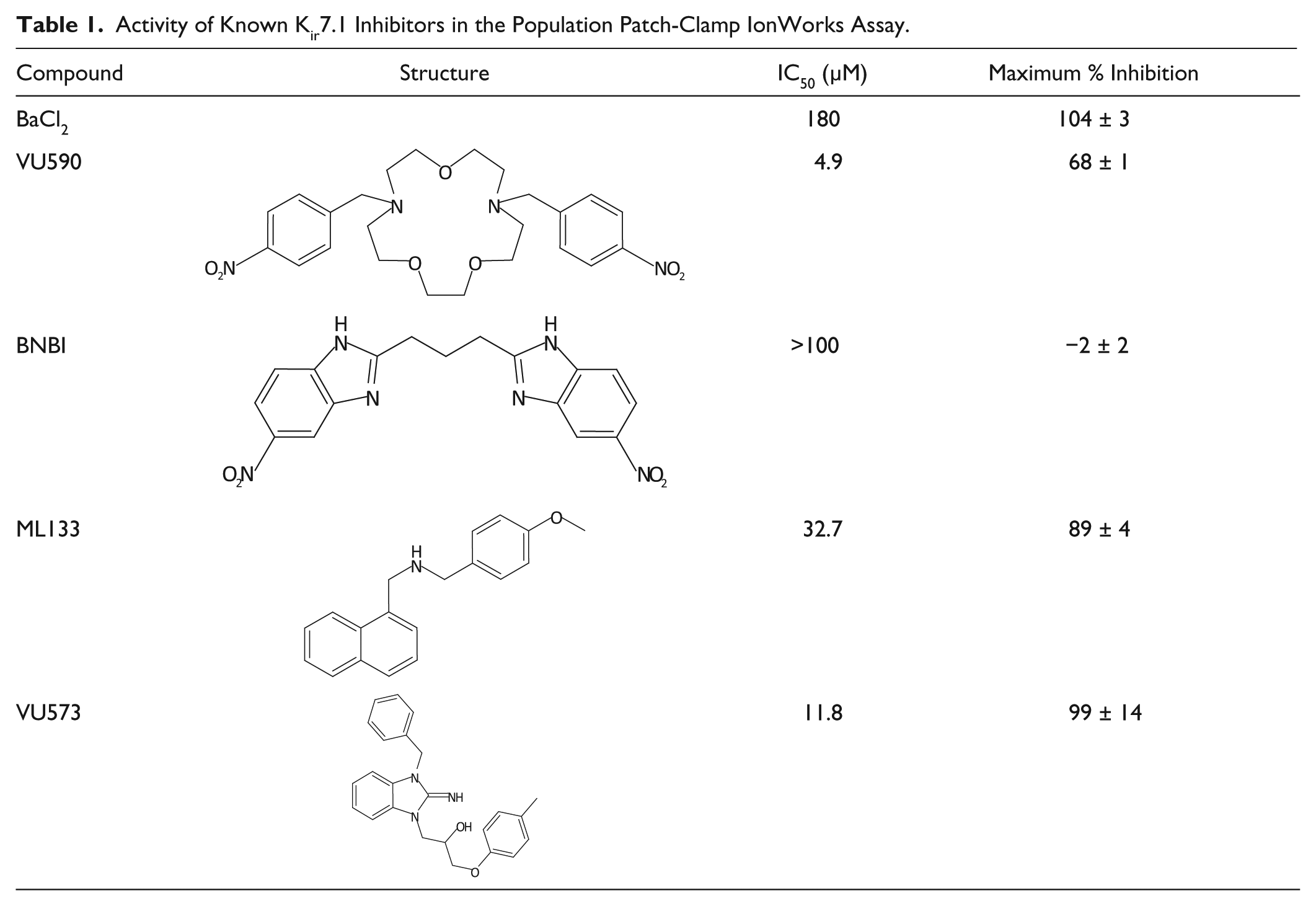
Two additional compounds were identified from the literature, VU573 12 and ML133, 14 which had been shown to have inhibitory activity at Kir7.1. These, along with VU590, were tested using the IonWorks assay ( Table 1 ). VU590 was the most potent inhibitor of Kir7.1, but complete inhibition was not observed. In agreement with previous data, another Kir1.1 inhibitor BNBI 15 showed no inhibition at Kir7.1. Eight analogues of VU590 (a potent sub-µM inhibitor of Kir1.1 8 ) were also tested and a range of IC50s at Kir7.1 observed ( Table 2 ), alongside differences in activity against Kir1.1 (ROMK). In addition, some of these analogues gave more complete inhibition of Kir7.1 (>90%) than that observed with VU590. Of particular note, MRTKIR-06 showed an IC50 of 4.2 µM at Kir7.1 but previously had been described as inactive (IC50 >100 µM) at Kir1.1. 8 These data, taken collectively, provided confidence that the assay could identify compounds with a range of potencies against Kir7.1 and that a divergent structure-activity relationship (SAR) versus other inwardly rectifying potassium channels was possible.
Activity of Known Kir7.1 Inhibitors in the Population Patch-Clamp IonWorks Assay.
Pharmacology of VU590 Analogues.
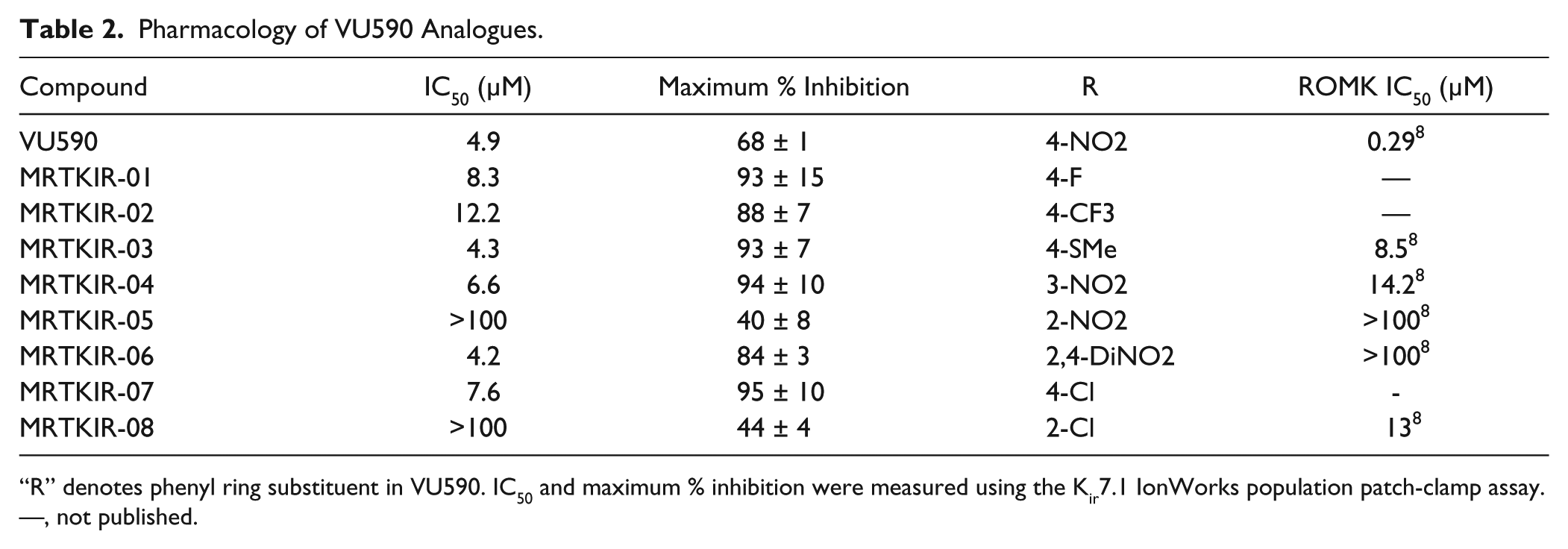
“R” denotes phenyl ring substituent in VU590. IC50 and maximum % inhibition were measured using the Kir7.1 IonWorks population patch-clamp assay. —, not published.
To identify novel inhibitors of Kir7.1, a focused set of 7087 compounds was screened. This comprised 3306 compounds identified as containing pharmacaphores linked to ion channel activity (MRC Technology ion channel set) and 3781 compounds of diverse structure (a subset of the MRCT diversity collection). Compounds were screened in singlicate at a final assay concentration of 33.3 µM (0.33% DMSO). HTS performance statistics were high ( Fig. 3B ), with an average Z′ of 0.63 ± 0.09, mean peak currents of 2.03 ± 0.23 nA, and mean seal resistances of 45 MΩ measured across all plates. Thirty-eight individual wells (0.5%) failed due to poor seal formation (<20 MΩ). As suggested by Z′ values, variance in the Ba2+- and DMSO-containing controls was low. The average standard deviation observed during screening was 6.67% ± 2.6% and 5.08% ± 1.8% for Ba2+ controls and DMSO controls, respectively. VU590 (100 µM) also showed consistent inhibition, with an average observed effect of 58.49% ± 9.6% across all plates tested.
A total of 519 compounds were initially identified as inhibiting Kir7.1 (reducing activity by >3 standard deviations, defined as 20%), and 219 compounds showed >40% inhibition of the Kir7.1-mediated current ( Fig. 3C ). After initial medicinal chemistry triage and confirmation of activity from resynthesized solid material, 36 of 41 compounds showed greater than 50% inhibition when rescreened at 33.3 µM. Concentration-response curve analyses identified a subset of compounds with IC50s less than 30 µM. Fourteen of these compounds were shown to have IC50s below 10 µM. The most potent was MRT00200769, which had an IC50 of 1.3 µM (95% confidence interval, 0.9–1.9 µM) and displayed a maximal inhibition of 85%. MRT00200769 was more potent and efficacious than VU590.
Hit Optimization, SAR, and Channel Selectivity
From screening, a number of promising compounds, including MRT00200769, were identified. A limited amount of iterative medicinal chemistry was subsequently carried out and IC50 values versus Kir7.1 generated ( Table 3 ). In the case of MRT00200769, limited structural modifications had a detrimental effect on the Kir7.1 activity. The conversion of the benzylic hydroxyl to a methoxy ether reduced the activity by approximately fourfold. Furthermore, the removal of the ortho methoxy group and the conversion of the central piperidine ring to a piperidone resulted in complete loss of activity.
MRT00200769 and Analogue Activity in Kir7.1 Population Patch-Clamp IonWorks Assay and Fluorescence Polarization–hERG Assay.
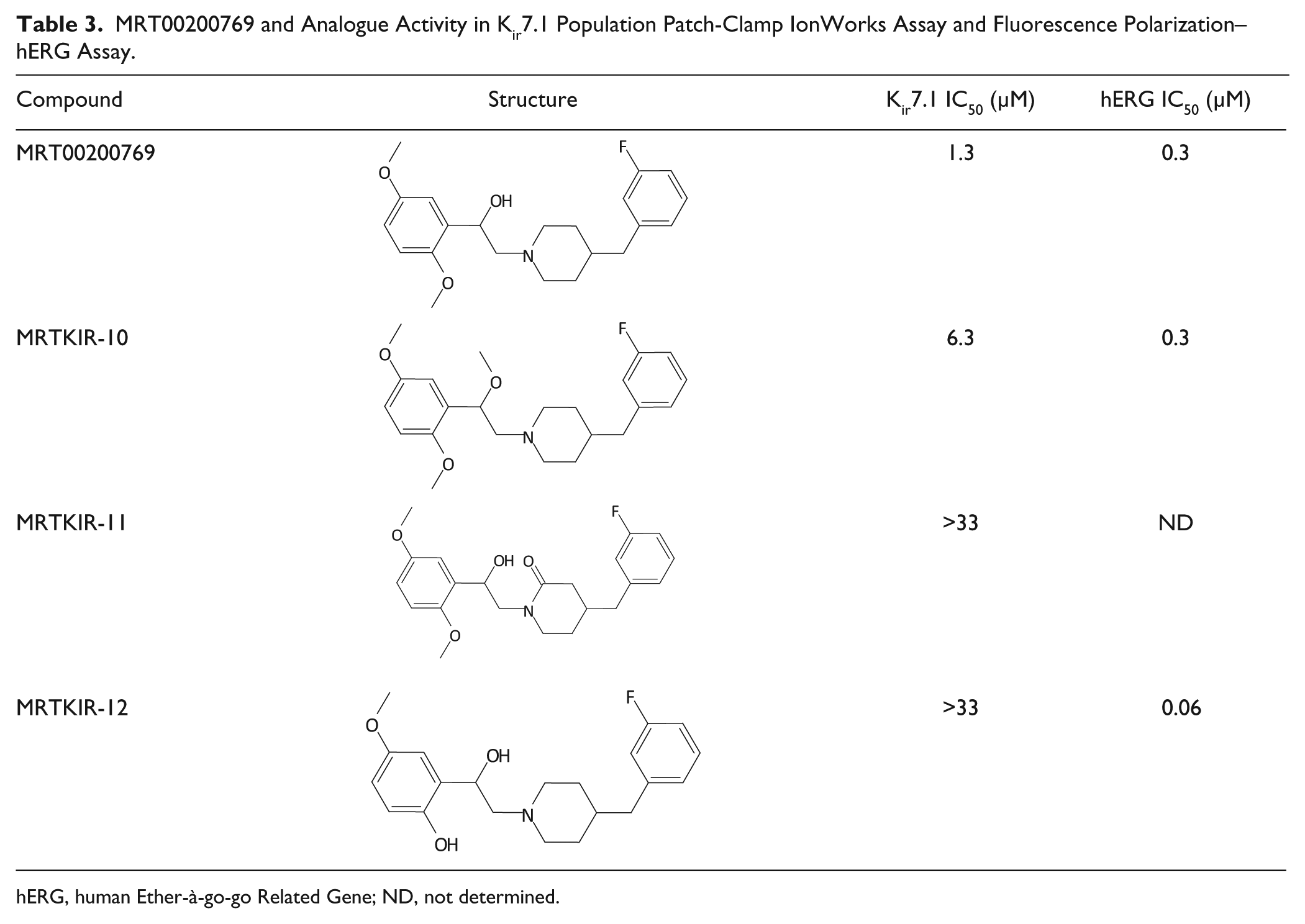
hERG, human Ether-à-go-go Related Gene; ND, not determined.
Preliminary cross-channel selectivity was also assessed for MRT00200769 versus Kir1.1 and hERG. Kir1.1, the best characterized of the Kir subfamily, is expressed in the renal tubule, where it critically regulates sodium and potassium balance. 16 Thallium flux assays have been described for Kir1.1, 8 facilitating an effective counterscreening assay. hERG is an important target for cardiac adverse effects, and its blockade is linked to acquired long QT syndrome and Torsades de Pointes. 17 Using a thallium flux assay, MRT00200769 was shown to have an IC50 of 38 µM at Kir1.1. MRT00200769 showed an IC50 of 1.3 µM at Kir7.1, and despite the different assay technology formats, this was highly suggestive of potential selectivity versus inwardly rectifying potassium channels. However, in a fluorescence polarization (FP)–hERG assay to check for potential block of the hERG channel, both MRT00200769 and the two analogues showed higher activity compared with that in the Kir7.1 assay ( Table 3 ).
Importantly, MRT00200769 was subsequently shown to stimulate profound, long-lasting contractions when administered to mouse myometrium tissue. 6 A similar effect was observed with VU590 but not BNBI, which does not inhibit Kir7.1. These data further validate the assay employed here for identifying clinically important molecules for PPH treatment.
Discussion
Kir7.1 represents a novel target in the treatment of postpartum hemorrhage and dysfunctional labor. 6 Current treatments, oxytocics, 5 fail to reduce acute outcomes and can be of limited use if pathways become desensitized due to medical intervention (e.g., labor induction). A further disadvantage of oxytocics is that they are thermolabile, restricting use in the developing world where PPH is a major risk of fatality. It is therefore hypothesized that a Kir7.1 inhibitor could be used alone and/or in conjunction with current treatments, conferring significant therapeutic advantages.
It has been demonstrated by lentiviral knockdown that inhibition of Kir7.1 greatly increased uterine contractile force and contraction duration. 6 The small-molecule Kir7.1 inhibitor VU590, initially identified in a Kir1.1 screening campaign, exhibited a similar phenotype in human myometrium and was significantly more active than tested oxytocics. However, VU590 also showed activity at Kir1.1, 8 and thus identification of novel small-molecule inhibitors of Kir7.1 proved attractive.
The assay described here is the first to use the native sequence of human Kir7.1 and the first Kir7.1 screen to use the automated patch-clamp platform. Automated patch-clamp assays have previously been described for Kir4.1, 18 Kir1.1, 15 and Kir2.1.14,19 These assays, which allow direct control of membrane potential, confer numerous advantages over fluorescence techniques, 9 potentially allowing the identification of novel chemotypes, with differing mechanisms of action, that may be missed using thallium flux or membrane dyes. This counterpoints the increased cost and time of automated patch-clamp systems, which likely result in smaller screening libraries.
In total, 7087 compounds were screened, and subsequent hit follow-up studies suggested divergent SAR with Kir1.1 and common pharmacopheric crossover with hERG. MRT00200769 was more potent than previously described Kir7.1 inhibitors and, unlike VU590, showed complete inhibition in the IonWorks assay. In addition, MRT00200769 and MRTKIR-06 showed greater selectivity for Kir7.1 compared with Kir1.1 than previously identified compounds. MRT00200769’s potential as a therapeutic is limited by its hERG activity, which we were unable to reduce in our initial medicinal chemistry. Of uppermost importance, however, was the observation that MRT00200769 induced long-lasting contractions in mice myometrium tissue. 6 These data, taken collectively, further validate a role for Kir7.1 in control of resting membrane potential and could provide novel therapeutic potential for PPH.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.