
Abstract
The bacterial translational apparatus is an ideal target for the search of new antibiotics. In fact, it performs an essential process carried out by a large number of potential subtargets for antibiotic action. Moreover, it is sufficiently different in several molecular details from the apparatus of Eukarya and Archaea to generally ensure specificity for the bacterial domain. This applies in particular to translation initiation, which is the most different step in the process. In bacteria, the 30S ribosomal subunit directly binds to the translation initiation region, a site within the messenger RNA (mRNA) 5′-untranslated region (5′-UTR). 30S binding is mediated by the interaction of both the 16S ribosomal RNA and the ribosomal protein S1 with specific regions of the mRNA 5′-UTR. An alternative, S1-independent pathway is enjoyed by leaderless mRNAs (i.e., transcripts devoid of a 5′-UTR). We have developed a simple fluorescence-based whole-cell assay in Escherichia coli to find inhibitors of the canonical S1-dependent translation initiation pathway. The assay has been set up both in a common E. coli laboratory strain and in a strain with an outer membrane permeability defect. Compared with other whole-cell assays for antibacterials, the major advantages of the screen described here are high sensitivity and specificity.
Keywords
Introduction
The spread of multidrug-resistant bacterial strains has made the development of new antibacterial molecules urgently needed. A strategy that can be applied to the research of new antibiotics is to look for molecules that inhibit essential cellular processes. Translation has proven to be an excellent target in this respect; indeed, it is one of the few bacterial processes that are targeted by currently in use antibiotics. However, few structures/steps of the translation pathway are inhibited by extant antibiotics, and many potential drug targets have not yet been exploited. 1
Initiation is the rate-limiting step in translation. It is also the least evolutionary conserved step in this otherwise very conserved process. In particular, the initial interaction between the messenger RNA (mRNA) and the ribosome occurs through very different pathways in bacteria versus eukarya.2,3 In bacteria, the 30S small ribosomal subunit directly binds to the translation initiation region (TIR), an internal site within the mRNA 5′-untranslated region (5′-UTR) that encompasses the start codon and the upstream Shine-Dalgarno sequence. The ribosomal protein S1 has been specifically implicated in promoting the initial interaction between the mRNA and the 30S subunit.4,5 S1 is encoded by essential genes in distantly related bacteria such as Escherichia coli and Mycobacterium tuberculosis6,7 (TubercuList database), and it is widely conserved among bacteria. 8 Conversely, in mammals, the 40S ribosomal subunit lacks an S1 orthologue; the protein is absent also from mammalian mitochondria.
S1 has been shown to be involved in translation in Gram-negative and high GC-content Gram-positive bacteria.8–10 In E. coli, S1 is strictly required for translation of most mRNAs, 11 with the notable exception of leaderless mRNAs (i.e., starting with the AUG start codon), which enjoy a peculiar pathway of translation initiation.12–14 Evidence from different groups has shown that S1 is dispensable both in vivo and in vitro for initiation complex formation on leaderless transcripts.12,15,16 Leaderless mRNAs are very rare in E. coli. One of the few examples is represented by bacteriophage λ cI mRNA, which is naturally leaderless when transcribed from the PRM promoter.
Interestingly, the aminoglycoside antibiotic kasugamycin specifically inhibits the canonical pathway of bacterial translation initiation, whereas it does not interfere with leaderless mRNA expression. This antibiotic acts by destabilizing fMet-tRNAfMet binding to the 30S-mRNA preinitiation complex. 17 Moreover, kasugamycin induces the formation of 61S ribosomal particles in E. coli cells. The 61S ribosome lacks several 30S proteins, among which is S1, and is unable to translate leadered transcripts, but it is still proficient in leaderless mRNA translation. 18
We have designed a simple whole-cell fluorescent screen to identify specific inhibitors of the S1-dependent pathway of bacterial translation initiation. As this process is both essential and specific for bacteria, such inhibitors would constitute promising hits in the research of new antibacterial drugs.
Materials and Methods
Bacterial Strains and Plasmids
All the experiments were performed with the AS19 19 or DH10B 20 E. coli strains carrying either plasmid pGM991 or pGM999. To construct these plasmids, the eGFP ORF was amplified from pZR80-2 21 with oligonucleotides 2803 and 2804 (CTCGGTACCAGTAAAGGAGAAGAACTTTTCA C and CTCCTGCAGCTATTTGTATAGTTCATCCATGC), digested with Pst I–Kpn I, and cloned in pGM931, 22 obtaining pGM963. A DNA fragment encompassing the recA 5′-UTR and first nine codons was synthesized by PCR with oligonucleotides 2928 and 2929 (TCTGGTACCCAAC AGAACATATTGACTATCC and CACGGTACCTTTCTG TTTGTTTTCGTCGATAG) on MG1655 genomic DNA. The PCR fragment was digested with Kpn I and cloned in the Kpn I site of pGM963, obtaining pGM991. pGM999 was obtained by cloning in pGM963 a DNA fragment carrying the araBp promoter and the first 189 bp of phage λ cI open reading frame (ORF). The fragment was synthesized by three-step PCR as follows. Oligonucleotides 1349 and 2964 (CAAAGCGGGACCAAAGCC and ATGGAGAAA CAGTAGAGAG) on pBAD24-Δ1 23 were used for araBp amplification, as well as 2962 and 2963 (CTCTCTACTG TTTCTCCATATGAGCACAAAAAAGAAACC and CCC GGTACCGGCGTTATAAGCA TTTAATG) on pGM397 16 for the cI gene. The two partially overlapping PCR fragments were used as templates in a PCR reaction with oligonucleotides 1349 and 2963, obtaining the complete fragment, which was digested with Mlu I–Kpn I and cloned in pGM963 digested with the same enzymes. Transcription from araBp of pGM999 gives a leaderless cI-GFP mRNA.
Assay Conditions and Prestwick Chemical Library Screening
Overnight cultures of E. coli strains carrying either pGM991 or pGM999 grown in LD (10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl) supplemented with 100 µg/mL ampicillin were diluted to OD600 = 0.1 in M9 (0.1% NH4Cl, 1.6% Na2HPO4·12 H2O, 0.3% KH2PO4, 0.5% NaCl, 0.013% MgSO4, 0.001% CaCl2, and trace elements) supplemented with 100 µg/mL ampicillin and 0.4% glucose and incubated with aeration at 37 °C. Irrespective of the plasmid, AS19 strains exhibited a long lag phase, and the OD600 did not significantly increase in the first 3 h upon dilution, whereas the DH10B derivatives made one to two divisions. After a 150-min incubation, the cells were pelleted by centrifugation at room temperature and resuspended in M9 to OD600 between 0.8 and 1. Then, 110-µL aliquots of the cultures were distributed in the wells of black polystyrene 96-well plates, and 3 µL of either DMSO (in control samples) or the compounds to be tested in DMSO solution was added into the wells. The plates were incubated at 37 °C for 15 min before adding 10 µL M9 supplemented with either 2.4% arabinose or, for negative controls (not induced samples), 2.4% glycerol. The plates were then incubated at 37 °C in the dark with slow agitation. Irrespective of the C source, the cultures poorly grew in these conditions (they did only 0.5–1.5 divisions even upon overnight incubation). The fluorescence was detected by means of a Packard FluoroCount microplate reader (Packard Instrument Company, Meriden, Connecticut; excitation/emission wavelengths 485/530 nm). For the Prestwick library screening, the bacterial strains were grown as described above. The compounds were assayed at 50 µg/mL (average final concentration, ~0.1 mM) and their effect on fluorescence assessed by means of a VersaDoc Imaging System (Bio-Rad, Hercules, CA) after a 3- and 16-h incubation. The wells signals were quantified with ImageQuant software (Molecular Dynamics). For each strain, the molecules were considered inhibitors when they reduced fluorescence to 30% or less of the respective positive controls (i.e., the induced cultures expressing either the L- or the CI-GFP without any added compound). The screening cascade was as follows. First, the whole library was assayed with the strain AS19/pGM991. Candidate molecules not listed as antibacterials were retested with the same strain. The compounds that reproducibly inhibited fluorescence were assayed with both the AS19/pGM999 and the DH10B/pGM991 strains.
Z′ Factor and Signal-to-Background Determination
Cultures of E. coli strains carrying either pGM991 or pGM999 were grown and distributed in the wells of black polystyrene 96-well plates as described above. Then, 3 µL of either 600 µg/mL chloramphenicol resuspended in DMSO or DMSO was added in the wells of either positive (+c; inhibited) or negative (–c; not inhibited) controls, respectively. Forty-eight wells were used for each type of control. The plates were incubated at 37 °C 15 min before adding 10 µL M9 supplemented with 2.4% arabinose. The plates were then incubated at 37 °C in the dark with slow agitation. After 3 h, fluorescence was detected by means of a Packard FluoroCount microplate reader. The signal-to-background (S/B) parameter was calculated as µ–c/µ+c, and the Z′ factor was calculated according to the following formula: Z′= 1 – [(3σ–c + 3σ+c)/(µ–c – µ+c)], where µ and σ are the average and standard deviation, respectively, of the positive (+c) and negative (–c) controls. 24 For each bacterial strain, two experiments were done on different days.
Results
Rationale and Setup of a Whole-Cell Assay for S1-Dependent Translation Initiation Pathway
We devised a two-step whole-cell assay for inhibitors of S1-dependent translation initiation. The theoretical bases of the assay rest on differential requirements exhibited by leaderless and leadered transcripts in E. coli. In the first step, compounds of interest are screened for their effect on the expression of a reporter gene with a 5′-UTR (leadered reporter). Inhibitors are then analyzed for their effect on the expression of a leaderless variant of the same reporter. Compounds that selectively inhibit translation of the leadered (and not of the leaderless) reporter will be considered hits for further analysis ( Fig. 1 ).
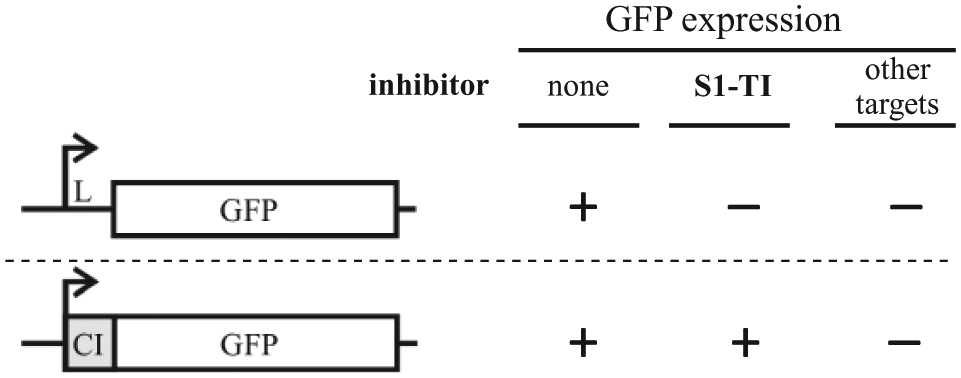
Rationale of the whole-cell reporter assay for translation initiation inhibitors. Fluorescence by bacterial cells carrying green fluorescent protein (GFP) fusions transcribed as either a leadered (upper part) or a leaderless (lower part) messenger RNA should be differentially inhibited by compounds affecting the canonical, S1-dependent pathway of translation initiation. Conversely, both reporter variants should equally respond to generically toxic molecules (i.e., detergents) and inhibitors targeting other cellular pathways. L, leader region; S1-TI, S1-dependent translation initiation; CI, λ cI gene 5′-end. Box, open reading frame; bent arrow, promoter.
As reporter gene, we exploited the eGFP gene, whose expression is readily monitored and amenable to high-throughput analysis. 25 Two constructs were assembled in the multicopy vector pGM931—namely, L-GFP, which carries the leader region of the E. coli recA gene fused with the eGFP gene, and CI-GFP, in which the eGFP gene is cloned in frame with the first 63 codons of λ cI. Transcription from the araBp vector promoter produces either a leadered (L-GFP; expressed by plasmid pGM991) or a leaderless (CI-GFP; expressed by pGM999) variant of green fluorescent protein (GFP) mRNA. The plasmids were constructed in the DH10B strain and transferred into AS19, an E. coli B derivative highly permeable to different antibiotics.19,26
We preliminarily tested the kinetics of fluorescence induction by the two constructs. The assay was set up in 96-well plates inoculated with AS19 cultures expressing either L-GFP or CI-GFP or with DH10B cells expressing the L-GFP. The fluorescence was measured at different times after the induction of transcription with arabinose ( Fig. 2 ). For all tested strains, the fluorescence steadily increased in the first 5 h after induction. Further incubation up to 20 h did not augment the signal. We decided to perform all successive analyses by incubating the samples 3 to 4 h. In these experimental conditions, the signal-to-noise ratio was 4- to 5-fold for AS19/pGM999 and higher for the two strains carrying pGM991 (14- to 19-fold for AS19 and 7- to 9-fold for DH10B), and no appreciable growth of the cultures was detected (data not shown). Thus, in these conditions, fluorescence should not depend on differential growth of the cultures in the plate wells but only on the expression efficiency of the constructs. This implies that the assay should be more sensitive to inhibitors of mRNA or protein synthesis than to compounds that interfere with the growth by targeting other cellular pathways.
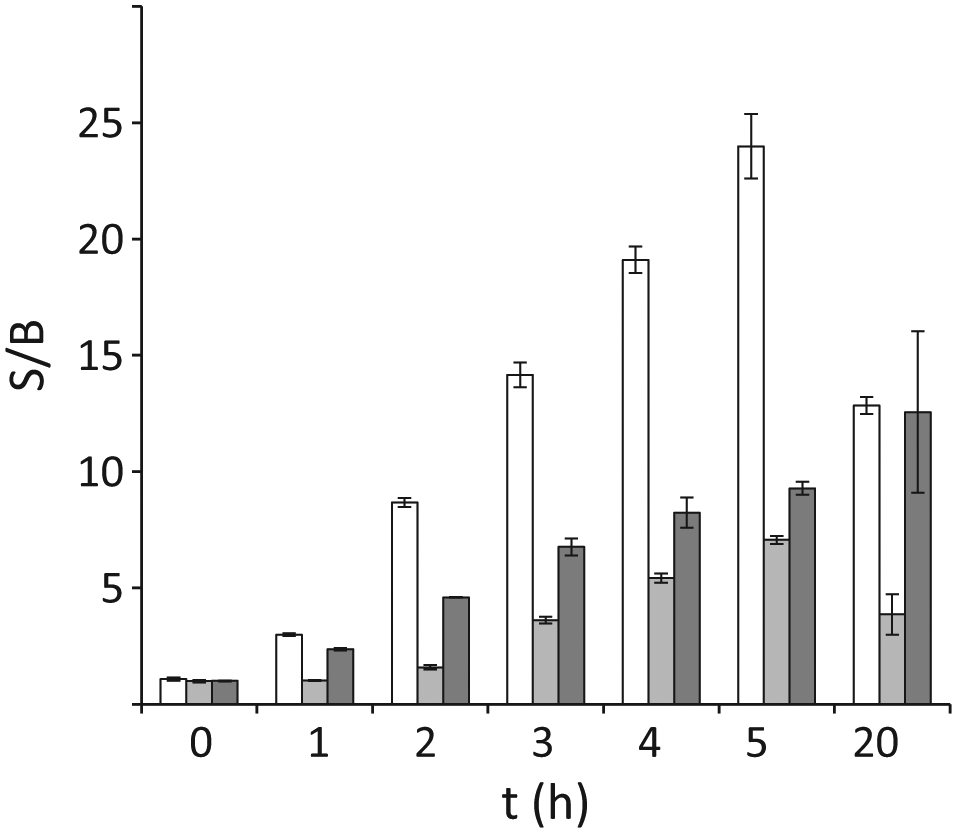
Kinetics of fluorescence induction in different reporter strains. Cultures of AS19/pGM991 (white bars), AS19/pGM999 (light gray), and DH10B/pGM991 (dark gray) were diluted at OD600 = 0.8 in M9+2.5% DMSO and induced with 0.2% arabinose (final concentration). In control samples, 0.2% glycerol was added instead of arabinose. The cultures were incubated at 37 °C, and the fluorescence of the wells was detected by means of a Packard FluoroCount microplate reader at the indicated time points. Three cultures for each strains and conditions were analyzed. The bars represent the average fluorescence of the induced cultures (signal, S) normalized for the average fluorescence of the control samples (background, B). The standard deviation is reported on top of the bars.
To verify this prediction, the effect on fluorescence of antibiotics inhibiting different macromolecular syntheses was assayed in the permeable strain. As expected, the screen was sensitive to molecules that target transcription and translation, whereas most inhibitors of folate (i.e., sulfa drugs) or cell wall biosynthesis did not affect fluorescence with few exceptions that, at least in some cases, could be explained by an indirect effect on gene expression (
Fig. 3
and
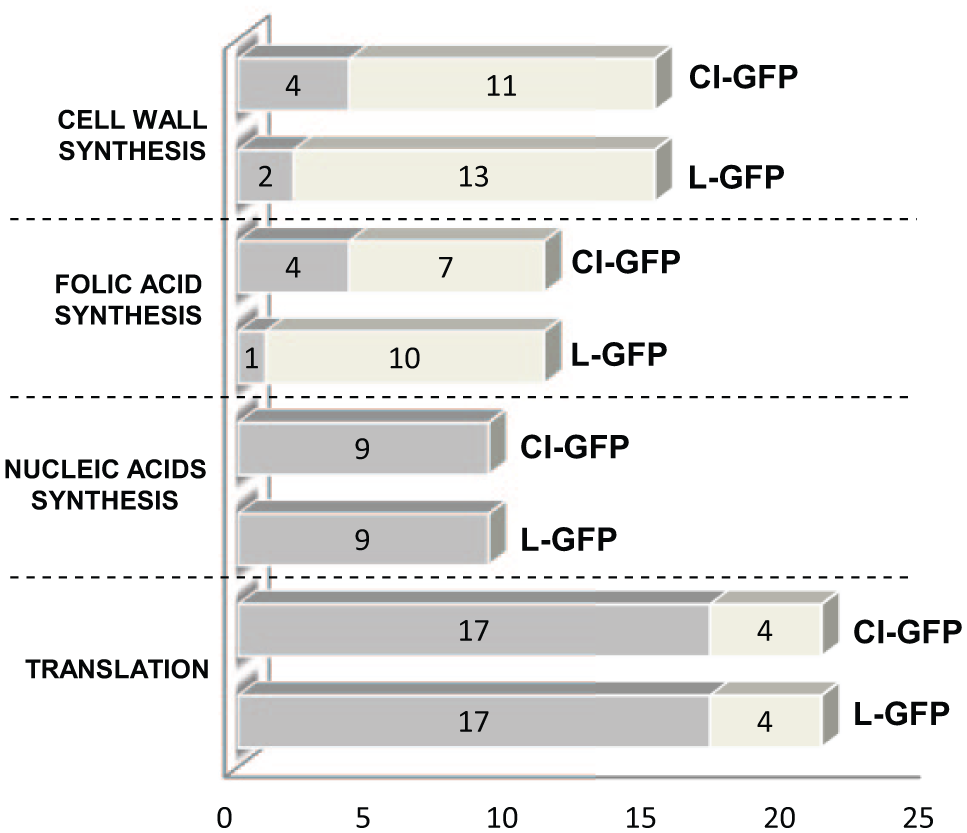
Antibiotics’ effect on the fluorescence of the reporter strains. Cultures of AS19/pGM991 (L-GFP) and AS19/pGM999 (CI-GFP) induced with 0.2% arabinose were incubated with different antibiotics (final concentration, 50 µg/mL; listed in
Evaluation of the Assay Specificity
To validate the assay outlined above, we exploited the aminoglycoside antibiotic kasugamycin (Kas), which specifically inhibits canonical translation initiation.17,18 Several E. coli strains have been reported to be basically resistant to kasugamycin (minimal inhibitory concentration [MIC], ≥500 µg/mL), 28 probably because of poor cell entry, but the AS19 strain is sensitive to such an antibiotic (MIC between 75 and 100 µg/mL; data not shown).
As shown in Figure 4 , fluorescence by the L-GFP construct was progressively inhibited in the presence of increasing concentrations of Kas. Conversely, the expression of the CI-GFP variant did not significantly change at Kas ≥200 µg/mL (ca. 0.5 mM) and increased at lower Kas concentration, probably because the partial inhibition of bulk leadered mRNA translation may boost the number of ribosomes available for the interaction with rare leaderless mRNAs. Interestingly, enhanced leaderless mRNA translation was observed also in conditions that affect S1 assembly into the ribosome, such as upon S1 depletion or in rpsB mutants with S1/S2-deficient ribosomes.16,29
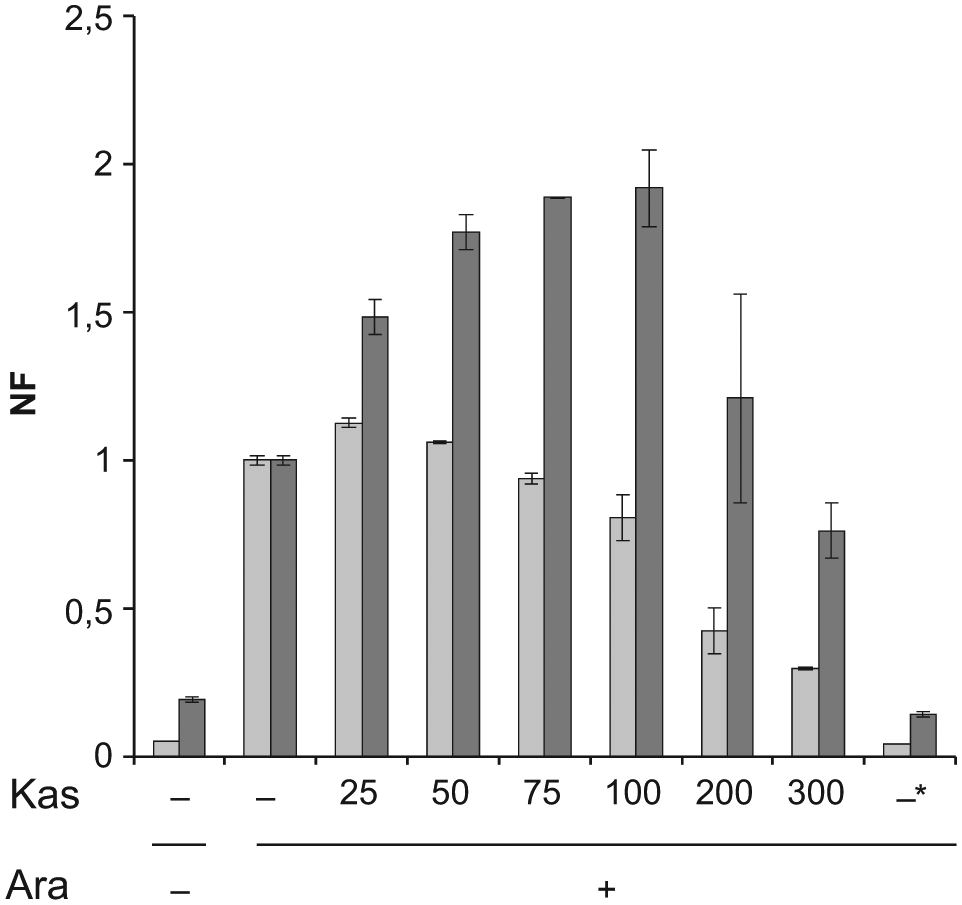
Kasugamycin effect on the fluorescence of the reporter strains. Cultures of AS19/pGM991 (L-GFP; light gray bars) and AS19/pGM999 (CI-GFP; dark gray bars) induced with 0.2% arabinose (Ara +) were incubated with kasugamycin at the concentrations indicated in µg/mL (Kas –, no kasugamycin added) or with 30 µg/mL chloramphenicol (Kas –*) in 96-well plates, as detailed in Materials and Methods. After 3 h, the fluorescence was measured by means of a Packard FluoroCount microplate reader, and the values were normalized to those of the positive controls (induced sample of the same strain with no antibiotics). The average and standard deviation estimated from the results of duplicate experiments are shown. Ara –, 0.2% glycerol was added instead of arabinose; NF, normalized fluorescence.
These results show that the assay is suitable for the identification of specific inhibitors of canonical mRNA translation.
Screening of the Prestwick Chemical Library with the Whole-Cell Fluorescence Assay
We applied the whole-cell assay to screen the Prestwick library, a collection of 1120 chemically heterogeneous compounds approved by the Food and Drug Administration (FDA) for clinical use. The library was first assayed with the permeable AS19 strain expressing the L-GFP. We found that 108 compounds inhibited the fluorescence of the AS19 strain expressing the L-GFP (listed in
Assay Amenability to High-Throughput Analysis
A statistical parameter for evaluating the intrinsic quality of a screening is the Z′ factor, which keeps into account both the dynamic range of the signal (the signal-to-noise ratio) and the variability associated with measurements. Screens with Z′ ≥ 0.6 are considered amenable to the high-throughput analysis. 24 We measured such a parameter for the AS19 strains expressing the leadered or the leaderless GFP and the DH10B with the leadered fusion by comparing the fluorescence of induced cultures in the presence or absence of chloramphenicol, a translation inhibitor (see Materials and Methods). As already seen in the kinetics experiment ( Fig. 2 ), we found that in terms of S/B ratio, the assay gave the best performance with the AS19/pGM991. However, we found Z′ values >0.7 for the DH10B and >0.8 for the two AS19 derivatives, suggesting that the screen could be suitable for high-throughput analysis with all the strains ( Table 1 ).
Statistical Parameters of the Screen.
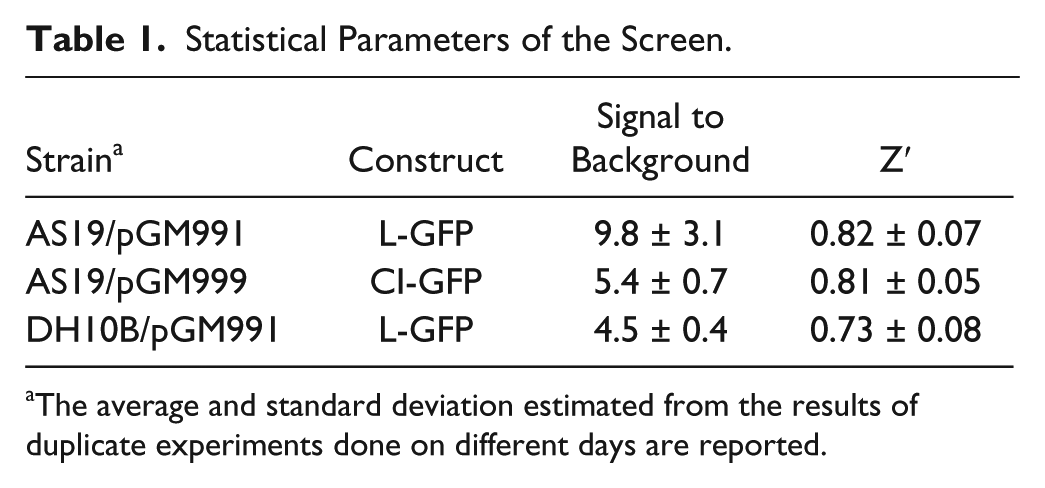
The average and standard deviation estimated from the results of duplicate experiments done on different days are reported.
Discussion
The discovery of new antibiotics is a very challenging task. Essentially, most of the currently in use antibiotic classes were originally discovered because of their inhibitory effect on the bacterial growth in whole-cell assays. However, recent high-throughput screening (HTS) campaigns run by pharmaceutical companies based on whole-cell assays have substantially failed to generate hits and leads. Major drawbacks in the application of this approach are the difficulty of generating hits because of poor cell permeability (this is especially true for Gram-negative bacteria), the generic toxicity of the hits identified in the screening, and the challenge posed by the identification of the hit’s molecular target(s), which is important for clarifying not only the mechanism of action of the active compounds but also for their chemical optimization.30,31
The two-step assay presented in this work addresses these problems as it combines the high sensitivity conferred by the usage of a permeable E. coli strain, which facilitates cell penetration of the chemicals, with the high specificity of the mechanism-based assay for inhibitors of the S1-dependent pathway of translation initiation. These properties may allow identifying compounds interfering with an essential and bacteria-specific step of gene expression, discriminating them from molecules with nonspecific cell toxicity. Moreover, as S1 is widely conserved and plays a role in translation initiation in distantly related bacteria,8–10 it seems sound that molecules targeting the S1-dependent mechanism identified in E. coli may inhibit translation and thus the growth of a large spectrum of bacteria. Another advantage of our assay is that, since only a few cellular factors seem to be specifically involved in leadered versus leaderless translation initiation,12,32 the determination of the molecular target(s) of the hits should be easier than with other phenotypic screenings.
A collection of 25,000 Actinomycete secondary metabolites was screened by Brandi et al. 33 by means of a cell-free in vitro assay specific for inhibitors of translation steps different from elongation. An active molecule was identified, which turned out to have poor antibacterial activity, probably because of problems of scarce penetration into the cells. 33 In the light of these results, and given the high specificity of our screen, it is not surprising that we have not identified any hits by analyzing a relatively small collection of compounds, as the Prestwick chemical library, with our whole-cell assay. The screening of a larger collection of compounds would be needed to find molecules with the sought specific properties. In fact, since the screen with the permeable strains is technically very simple and appears to be robust, as witnessed by the high Z′ values obtained with both reporters, it could potentially be adapted to HTS campaigns. Moreover, as the expression of the L-GFP can be monitored also in E. coli strains with normal outer membrane permeability (as shown by our results with the DH10B), cell penetration properties of the active compounds identified in the primary screening could be easily assessed.
Footnotes
Acknowledgements
We thank Pierfausto Seneci (Università degli Studi di Milano) for the generous gift of the Prestwick chemical library, Daniele Daffonchio and Aurora Rizzi (Università degli Studi di Milano) for pZR80-2 aadA::GFP, and Francesco Delvillani (Università degli Studi di Milano) for pGM963. We thank Gianni Dehò (Università degli Studi di Milano) for critical reading of the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The contributors are government employees of the Italy-Università degli Studi di Milano and provided and prepared this article as part of their official duties.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Italian Cystic Fibrosis Research Foundation (grant FFC#8/2013 to FB, sponsored by the FFC Delegation of Montebelluna “La bottega delle donne”).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.