
Abstract
Translating existing and emerging knowledge of cancer biology into effective novel therapies remains a great challenge in drug discovery. A firm understanding of the target biology, confidence in the supporting preclinical research, and access to diverse chemical matter is required to lower attrition rates and prosecute targets effectively. Understanding past successes and failures will aid in refining this process to deliver further therapeutic benefit to patients. In this review, we suggest that early oncology drug discovery should focus on selection and prosecution of cancer targets with strong disease biology rather than on more chemically “druggable” targets with only modest disease-linkage. This approach offers higher potential benefit but also increases the need for innovative and alternative approaches. These include using different methods to validate novel targets and identify chemical matter, as well as raising the standards and our interpretation of the scientific literature. The combination of skills required for this emphasizes the need for broader early collaborations between academia and industry.
Keywords
Introduction
In recent years, major scientific and technical advances have promised to improve our understanding of disease biology, provide novel targets for translation, and catalyze a new era in the development of medicines. However, notwithstanding some notable successes, the number of new molecular entities (NMEs) approved by the U.S. Food and Drug Administration (FDA) in the recent past has remained relatively static, despite increased investment in research and development (R&D). This has led many to comment on the major productivity crisis faced by the pharmaceutical industry and to question the long-term sustainability of the model in its current form.1–8
Without doubt, one of the major reasons for the spiraling costs of R&D is the high level of attrition. The reasons for failure of a discovery project or drug are diverse. In the early stages, many projects will fail during the initial target validation phase because of a lack of compelling supportive data. Further attrition arises in hit-to-lead and lead optimization because of challenges in obtaining progressable chemical matter and/or poor efficacy in preclinical models. In the clinic, the majority of drug candidates between phase II and submission will fail because of lack of efficacy (56% between 2011 and 2012) or safety (28% in the same time period, including failures due to an insufficient therapeutic index). 9 The impact of this attrition is evident in recent analyses modeling the number of projects required (by development phase) to achieve a single successful NME approval.1,7 By combining R&D survival data from 14 large pharmaceutical companies over the period from 2005 to 2009, Bunnage et al. 1 reported that ~24 development candidates were required to achieve a single NME approval. Over this period, the greatest attrition (75%) is seen in phase II, in which the biological hypothesis of the project is directly tested in clinical proof-of-concept studies. 1 It is concerning that the proportion of phase II failures citing efficacy or safety reasons has increased in recent years.9,10 Although the costs of failure in the clinic are considerable, the cumulative costs of multiple failures at the early stages of discovery can also have a detrimental effect on industry productivity. These levels of failure question the effectiveness of the reductionist target-based paradigm that has become the mainstay within the pharmaceutical industry11–13 and implies that radical improvements are required in the selection of disease-relevant therapeutic targets.
To address this, many companies have recently reevaluated their target selection criteria in hope of improving their overall productivity. A good example of this is illustrated by a recent publication from AstraZeneca describing some of the lessons learned following a comprehensive longitudinal review of their small-molecule programs from 2005 to 2010. 14 This led to the implementation of an operating model described as the “5 R’s.” The framework seeks to ensure continual review of all targets against the following criteria—right target, right patient, right tissue, right safety, and right commercial potential—and marks a move away from the volume-based measures of success that many see as contributing, in part, to the decline in industry productivity.8,15 Although it is too early to determine the impact of this framework, it is encouraging to see a cultural shift toward truth seeking based on strong scientific reasoning rather than program progression to satisfy numerical-based objectives.
Within the area of cancer therapeutics, improving target selection is a considerable challenge. This review will focus on what we can learn from recent successes and failures in small-molecule oncology drug discovery. A review of potential oncology targets and key signaling pathways arising from recent cancer genomics initiatives has been extensively covered elsewhere and will not be addressed here (see, for example refs. 16–18). We will discuss ways in which we may be able to overcome some of the major challenges faced in oncology translational research through increased innovation, embracing alternative technologies, and creating new collaborative models for drug discovery. Specific focus will be given to the issue of data reproducibility, alternative approaches to finding hit matter against challenging oncology targets, and the recent growth of industry-academia collaborations, as these have not been covered extensively elsewhere.
Challenges Identified from Recent History
Targeted Therapies
Advances in our understanding of cancer biology have led to the development of targeted therapies for the treatment of several human cancers. To date in 2014, a number of molecularly targeted agents have been approved by the FDA, with examples including ceritinib (Zykadia, Novartis, Basel, Switzerland), ramucirumab (Cyramza, Eli Lilly and Company, Indianapolis, IN), and ibrutinib (Imbuvica, Pharmacyclics, Inc., Sunnyvale, CA). These agents target ALK, VEGFR-2, and Bruton’s tyrosine kinase (BTK), respectively.
One of the most successful targeted agents is the tyrosine kinase inhibitor imatinib mesylate (Gleevec, Novartis, Basel, Switzerland), which is the standard of care for patients with chronic myelogenous leukemia (CML) and gastrointestinal stromal tumors. The success of imatinib in CML is in part due to the relative genetic homogeneity of the disease. More than 95% of CML arises through a single driving fusion protein (BCR-ABL) related to a recurrent chromosomal translocation (t(9;22)(q32:q11)), thereby providing a clear disease-linkage rationale for targeting a single protein via small-molecule intervention. 19 However, in contrast to CML, most other cancers are driven by multiple factors that are being increasingly used to divide cancers into defined molecular subtypes. 16 This knowledge of the genetic heterogeneity of tumors, previously classed as similar diseases, can decrease confidence in a specific target being the main driver of tumor growth and survival (right target). It also potentially narrows the range of patients available for therapy (right patient). Although in many cases, the disease subdivisions are of sufficient size to encourage commercial investment, there is a major challenge in enabling the prosecution of targeted therapies for rare cancers with small patient populations (right commercial potential). For example, ceritinib is the most recent approval for the treatment of metastatic non–small-cell lung cancer (NSCLC) only in patients with ALK-positive tumors who have previously been treated with crizotinib (Xalkori, Pfizer, New York, NY). Although NSCLC represents approximately 85% to 90% of lung cancers, only 3% to 5% of these contain the driving EML4-ALK fusion gene. 20
In NSCLC, genetic profiling has defined subtypes based on mutation of known oncogenes including AKT1, ALK, BRAF, EGFR, ERBB2, KRAS, MEK1, MET, NRAS, PIK3CA, RET, and ROS1 (reviewed in ref. 21). Of this long list, EGFR and KRAS mutations are the most prevalent (10–35% and 15–25% of NSCLC, respectively), tend to occur mutually exclusively, and are attractive targets with strong disease-linkage.22–26 The development of small-molecule inhibitors against these two well-characterized targets has posed quite different challenges and provides interesting lessons for future drug discovery efforts.
ERBB receptor tyrosine kinases are well-established drivers of human cancer, including lung, head and neck, colon, and pancreatic cancers.27,28 The first generation of reversible EGFR inhibitors, gefitinib (Iressa, AstraZeneca, London, UK) and erlotinib (Tarceva, Roche, Basel, Switzerland), were designed to inhibit the wild-type receptor, known at the time to be overexpressed in many epithelial tumors including NSCLC. 29 Initial clinical trials recruited NSCLC patients with undefined genetic backgrounds and showed a poor overall response rate (reviewed in ref. 30). Following the discovery that kinase domain mutations in EGFR were linked to patients demonstrating tumor sensitivity to gefitinib or erlotinib,23–25 a retrospective analysis of data from the IPASS phase III gefitinib versus chemotherapy trial demonstrated that a response rate of 71% was achieved when data from patients with EGFR-mutant tumors were analyzed versus 43% for the whole cohort. 31 Further clinical trials confirmed this correlation and led to the approval of these agents for the treatment of advanced EGFR-mutant NSCLC. 30 Erlotinib is now a first-line treatment for metastatic NSCLC with exon 19 deletions or exon 21 (L858R) substitution mutations. 32 The realigning of anti-EGFR therapies to the appropriate NSCLC patients highlights the importance of informed selection of the right clinical trial populations for success. The in-depth genomic profiling of cancers to identify tumor-specific aberrations must therefore not only inform initial target selection but also be used to develop strong disease-positioning hypotheses that can be developed throughout the life of a program and tested much earlier in preclinical development.
In contrast to anti-EGFR therapies, targeting RAS proteins with small-molecule inhibitors has proven to be much more challenging.33–35 The family of RAS GTPases, particularly KRAS, have activating mutations not only in NSCLC but also in ~20% of all human cancers, including in approximately 90% of pancreatic cancers.33,36 Mutant RAS proteins are very well-validated targets, with exceptionally strong linkage to disease pathology, but they have historically been considered “undruggable” by nucleotide analogs.33–35 A number of approaches to indirectly target the RAS family have been attempted over the years, including inhibition of farnesyl transferase, 37 the enzyme that activates RAS via membrane localization, or of downstream effector kinases such as RAF, MEK, ERK, AKT, and PI3K (reviewed in refs. 33–35). Although inhibitors of these key signaling molecules have been successful in numerous preclinical models and a few clinical settings (advanced melanoma in the case of the BRAF inhibitors vemurafenib and dabrafenib 38 ), limited efficacy in a monotherapy setting has been observed in mutant RAS-driven cancers to date. However, as the RAS family remains one of the most important groups of proto-oncogenes in human cancer, researchers have and will be forced to consider alternative methods for targeting these clearly validated target proteins. These include, but are not limited to, combination approaches to overcome potential compensatory mechanisms associated with inhibiting a single downstream effector pathway of RAS, as well as synthetic lethal approaches.
Synthetic Lethality and RNA Interference Studies
Synthetic lethality is defined as an interaction between genetic perturbations in which the loss of either gene alone is compatible with cellular viability, but loss of both genes results in cell death. Thus, inhibiting the product of a gene that is synthetic lethal with a cancer driver mutation should lead to a unique vulnerability in the cancer that can be exploited by therapeutic intervention.39,40 Following on from the early promise of PARP inhibitors in cancers lacking the BRCA1 and/or BRCA2 genes involved in DNA homologous recombination,41–44 several groups took a similar approach to find synthetic lethal partners for common oncogenic drivers. Unsurprisingly, KRAS was a prime target for such studies, and many groups performed genetic screens looking for protein targets that interact with KRAS in a synthetic lethal manner.45–53 Although technically distinct, each of these studies followed a similar premise by screening sh/siRNA libraries in genetically distinct or isogenic cancer cell lines with defined wild-type or mutant KRAS status. These screens have resulted in a wide, but largely nonoverlapping, range of interesting synthetic lethal partners for KRAS being proposed. Many of these (in particular those considered highly tractable via a small-molecule approach) have been the subject of a substantial drug discovery effort. One such gene, STK33, a calcium/calmodulin serine-threonine kinase, was shown to be critical for the survival of cancers with mutant KRAS in multiple tumor models. 50 Based on these results, several translational groups explored this target further. Unfortunately, the dependence of KRAS for STK33 activity was not confirmed in these follow-up studies using RNA interference (RNAi), dominant mutant overexpression, or small-molecule inhibitors of STK33.54–56 Moreover, none of the identified synthetic lethal partners have, to our knowledge, given rise to successful new drug discovery programs to date. Despite the disappointment of the STK33/KRAS synthetic lethality story, it does represent an encouraging, but still all too rare, example of researchers publishing target devalidation data. This is an ethos that must be cultivated within academia and industry if we are to improve productivity.
Although it is still too early to make firm conclusions about the success of these and similar RNAi-based screening approaches, in our experience a major challenge exists in independently confirming the basic research findings even when the candidate target(s) were further validated in the literature through RNAi deconvolution, genetic rescue, and model expansion. A significant challenge also lies in the building of a credible disease-positioning hypothesis for novel targets derived from RNAi screens that lack clear genetic linkage to human pathology.12,57 This is in contrast to the outputs of recent genetic profiling studies that provide clear evidence of where the most disease-relevant targets can be found.16 –18 These considerations have led us and others to reassess the value of this technique in its current form to deliver robust targets for drug discovery.58 –62
Data Reproducibility and Target Validation
The drug discovery industry relies heavily on the preclinical scientific literature, large-scale genomic data sets, and seminar and conference disclosures as a rich source of candidate drug targets. In addition, this knowledge helps shape disease-positioning and patient stratification hypotheses as discovery targets progress toward the clinic. However, it is increasingly acknowledged, both within industry and academia, that a large proportion of the published scientific literature cannot be extended to other similar disease models or, worse, even be reproduced when assessed by independent laboratories (see Nature Focus on this topic at http://www.nature.com/nature/focus/reproducibility/). This inability to reliably exploit findings from the primary literature is a particularly acute problem and is a major contributing factor to project attrition throughout preclinical discovery. 63
The issue of poor data reproducibility has received a considerable amount of attention in recent years within the scientific press and increasingly in mainstream media. A catalyst for this was a report by Bayer Healthcare in 2011 disclosing that the majority of their in-house target validation projects (predominantly within oncology) showed significant inconsistencies when compared with the original literature claims. Their analysis revealed data generated in-house to be wholly consistent with the literature in just 21% of cases, resulting in a high level of project terminations, often after a substantially resourced and protracted evaluation phase. 63 A year later, Amgen reported similar concerning findings. In this instance, just 11% of projects (6 of 57 projects) led to confirmation of the original landmark scientific findings. 64 Despite these and other recent disclosures, there is still a lack of widespread recognition of the problem and the negative impact it has on academic and translational research.
Our contribution to this debate is based on the assessment of more than 350 oncology targets over the past 5 y. Targets were assessed by a multidisciplinary team according to a number of criteria including biological rationale (strength of the disease-positioning hypothesis and validation data, biomarker considerations, safety concerns), targeting strategy (feasibility of protein and assay development, discovery cascade build), and commercial fit. This led to the prioritization of 50 targets that were experimentally assessed with a view to repeating and extending the original observations and testing the hypothesis that the target in question is sufficiently compelling to initiate hit finding activities. We are confident that many of the targets assessed over this period were also of significant interest to other pharmaceutical and biotech companies at the time. Consistent with others,63,64 we have experienced high attrition rates in this target validation phase because of a lack of data reproducibility (unpublished findings). Consequently, most projects (78%, 39 of 50) were discontinued after an initial period of assessment in the lab. There are many valid reasons why reported findings may not be reproducible between independent laboratories, including subtle differences in models and reagents, an incomplete understanding of the biology, variability in the magnitude of effect, and technical difficulties. However, all too often, we have experienced difficulties in confirming the original observations. In line with previous reports,64,65 our experience suggests that two major reasons underpinning the low level of reproducibility are the misinterpretation of poorly controlled RNAi studies and the use of weakly validated reagents. To be a good drug target, the original observation must ideally be robust enough to bear examination in multiple related systems using multiple well-validated reagents and a variety of orthogonal techniques that lead to supportive conclusions. 66
A key lesson from the workflow described above is the need to account for the well-recognized off-target effects of RNAi. 61 Probes against the target should be employed from multiple independent suppliers with a full provision of positive (e.g., death-inducing RNAi probes such as PLK1) and negative control probes in all experiments. Typically, studies are performed in-house with a minimum of four control and four target-specific probes in several appropriate cell models using low concentrations of siRNA (<20 nM) optimized with multiple transfection reagents to mitigate off-target effects. This is essential as off-target phenotypic effects in siRNA experiments can differentially manifest depending on a number of factors such as probe manufacturer, strand modification, transfection method, and cell line (unpublished observations). In the case of shRNA, it is equally important to build in appropriate extensive controls and not rely on a low number of stable clones that demonstrate the desired effect. To build further confidence, it is necessary to correlate knockdown efficiency with both pharmacodynamic biomarker and phenotypic readouts. Alongside this, it is important to invest the time to thoroughly test and characterize multiple relevant reagents to decrease experimental uncertainty and to provide additional supportive evidence through the use of tool compounds where available, mutant proteins, or modulation of additional components of the target pathway to maximize chances of success. We typically perform this work in collaboration with the originating laboratories, which aids collective early “go/no go” decisions on the validity of a target with high confidence. Decisions are usually reached within a relatively short period of 3 to 6 mo, allowing, in the case of a “no go,” valuable resources to be refocused on further early-stage targets or elsewhere in the portfolio. For projects in which the target biology has been sufficiently “de-risked” through this process, we will consider taking risks elsewhere during its prosecution (such as chemical tractability or screening strategy).
Much has been written about the multitude of factors that can contribute to poor data reproducibility,63,64,67 and many potential solutions have been proposed.64,68,69 These include increasing opportunities to publish and present target devalidation data and linking such studies to the original findings within PubMed, mentoring of students and postdocs to recognize potential warning signs of irreproducibility, 70 increasing methodological transparency in journals, 68 and improving the characterization of commonly used research reagents. In addition, understanding and improving the predictive value of preclinical models is also undoubtedly a great challenge (reviewed in ref. 71). To raise the standards of preclinical research in this way will require a concerted and synchronized effort from all stakeholders, but it is essential if preclinical research is to remain the engine that drives translational progress. Such improvements will take time to manifest, but in our view, even making stepwise progress along the lines described above will likely deliver a significant impact on industry productivity and ultimately on the benefit realized for patients.
Taken together, there appears to be no simple formula to identify promising oncology targets. Although we should continue to explore the cancer genome for novel tractable targets through genetic screening and profiling approaches, we should also place emphasis back on the question of tackling valuable but traditionally “undruggable” targets directly. In the next section, we highlight collaborative approaches that bring the strengths of industry and academia together to develop new cancer therapeutics. Such approaches, which seek to integrate previously disparate sectors to a common goal, are becoming increasingly common and are opening up new opportunities in drug discovery.
Collaborative Approaches in Oncology Drug Discovery
New models of industry-academia collaboration are redefining drug discovery and have the potential to reduce the attrition of novel therapies in the clinic by directly tackling some of the challenges outlined in this review. The precise drivers for each partner within a collaboration vary but are typically aligned to overlapping research interests and are often associated with gaining access to enabling technologies and platforms as well as target biology, bioinformatic, and clinical expertise. These collaborations look to couple the drug discovery engines of industry with cutting-edge academic scientific breakthroughs and the rapidly expanding wealth of patient/tumor data from clinical centers.72,73 Although the overarching aim for these models is clear, several different approaches are emerging ( Table 1 ).
Examples of Collaborative Models within and between Academic and Industry Partners.
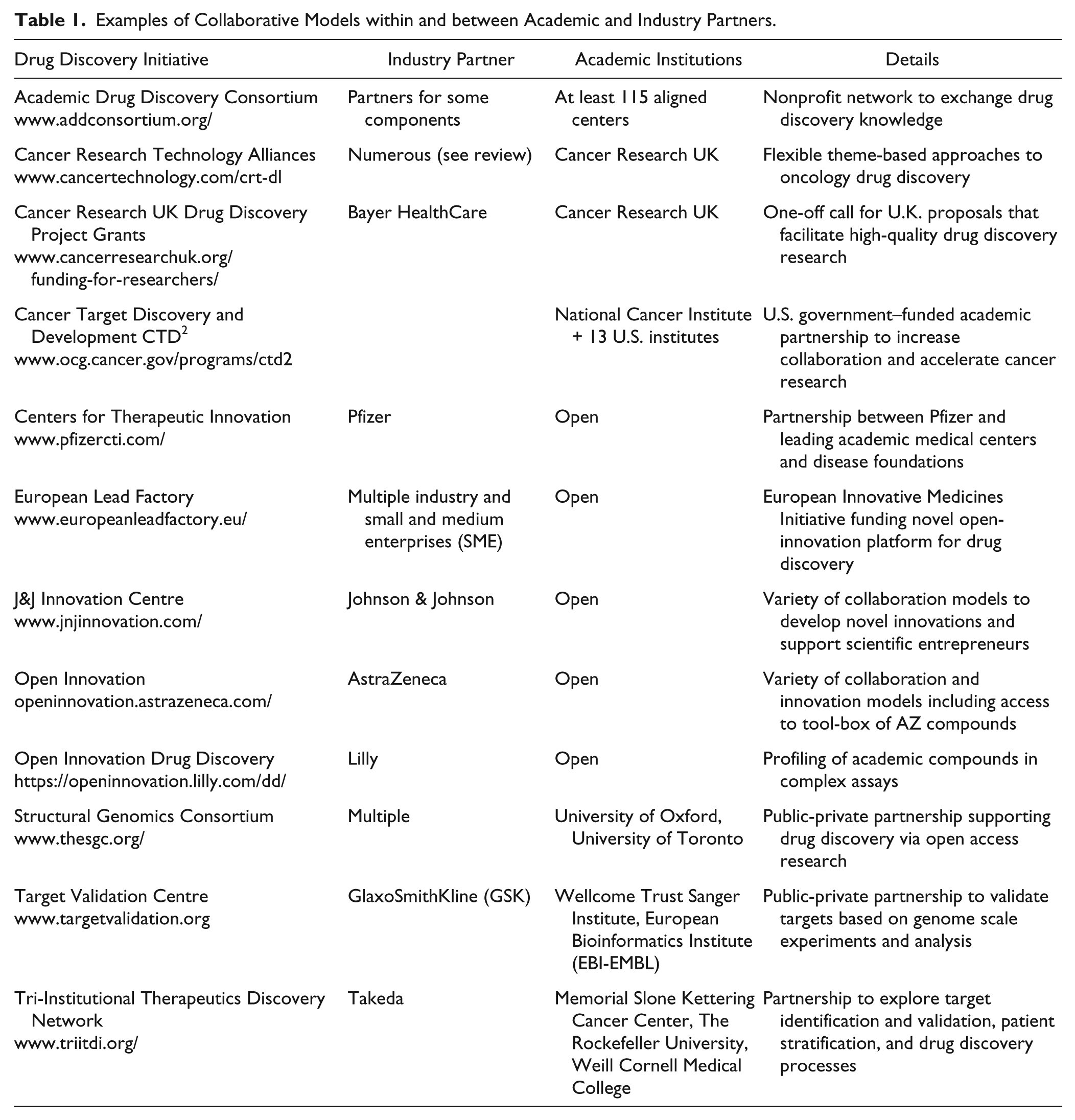
Institute-wide funding from a major partner has become a method to strengthen collaborations between academia and industry and allow industry greater access to high-quality science and early-stage targets/validation. Gilead Sciences, for example, established a long-term collaboration (10 y/$100 million) with the Yale School of Medicine to fund interdisciplinary research to understand the fundamental mechanisms of cancer. 74 Similarly, the Broad Institute linked up its oncogenomic expertise with Bayer HealthCare’s drug discovery capabilities to “jointly discover and develop therapeutic agents that selectively target cancer genome alterations.” 75
Industry are also looking to access novel drug discovery opportunities by opening up their tool compound and screening capabilities to academic institutes at no upfront cost via open innovation initiatives. For example, Eli Lilly and Company’s open innovation drug discovery program, which combines their Phenotypic Drug Discovery (PD2) and in vitro target-based assays (TargetD2) initiatives, allows external partners to test compound-based hypotheses, access Lilly’s design tools, and develop further drug discovery collaborations. 76
Pfizer’s Centers for Therapeutic Innovation is another model designed to bridge the gap between academic scientific discovery and its translation into therapies. 77 Pfizer has established four innovation centers in Boston, New York, San Diego, and San Francisco, where Pfizer scientists work alongside academic teams to create a synergistic biomedical research environment and to “share their understanding of target biology and translational medicine expertise.” In return, Pfizer provides funding for programs and access to select compound libraries, screening technologies, and a wealth of other resources for drug discovery.
GlaxoSmithKline (GSK), the European Bioinformatics Institute (EMBL-EBI), and the Wellcome Trust Sanger Institute have recently joined forces to create the Centre for Therapeutic Target Validation, aiming to “harness the power of big data and genome sequencing to improve the success rate for developing new medicines.” 78 To achieve this objective, the center will use the biological expertise of the EBI and the Sanger Institute’s genomic data sets, pathway analysis, cell screening technologies, and animal models and couple this with the drug discovery expertise of GSK. These organizations will co-fund up to 50 scientists based at the Wellcome Trust Sanger Institute as part of this public-private initiative. This constitutes a nonexclusive, precompetitive model aimed to attract external members with a cornerstone principle to “share its data openly in the interests of accelerating drug discovery.”
The European Lead Factory (ELF) is another variation of the partnerships model. 79 This public-private partnership uses Innovative Medicine Initiative funding, crowd sourcing, and open innovation approaches to provide resources and funding in kind to academics and small and medium enterprises (SMEs) to promote drug discovery. Within this framework, the ELF provides access to substantial screening libraries and compound logistics to assist collaborating partners in developing drug candidates or high-quality pharmacological tools. This initiative has brought together several major pharmaceutical companies with a number of SMEs and academic institutions in a “collaborative approach to finding small molecule candidates” and provides an excellent example of how a multi-partner collaboration can effectively function in drug discovery.
Finally, the Structural Genomics Consortium represents another public-private partnership supporting the discovery of new medicines through an open access research model. This consortium of academic and industry scientists aims to advance research in new areas of biology and drug discovery within an altruistic framework that is less influenced by financial gain. An excellent example of this can be seen in their epigenetics programme which has generated well characterized, potent, and selective chemical probes for the community that have opened up new insights and therapeutic opportunities in cancer. 80
In summary, a common feature of these approaches is that they extend beyond collaborations on a single target toward partnerships around a biological theme or technology. Furthermore, these interactions are developing across multiple partners and are embracing a variety of models including open innovation, precompetitive interactions, and less restricted access to the industry’s compound collections and tools.
Establishing the right environment for industry-academia partnerships to flourish is crucial to the future of oncology drug discovery. To this end, we have developed an industry-academia multiproject alliance model that aligns Cancer Research UK–funded academic and clinical scientists and other key opinion leaders with industry partners to progress novel targets. We find that industry partners are keen to engage from the earliest stages of drug discovery if they see that novel target risk is shared and mitigated by creating multiple project opportunities and by the involvement of world-leading researchers. The attraction for the scientific community is the substantial translational capability that can be brought to bear through CRT-DL and their industry partners. Alliances can be built around a biological theme, as demonstrated by our cancer metabolism alliance with AstraZeneca 81 and our DNA damage response alliance with Teva Pharmaceuticals. 82 Similarly, an alliance can be centered on a specific target class, such as our alliance with Forma Therapeutics to target deubiquitinating enzymes involved in cancer. 83 By focusing alliances around a common subject in this way, we find that it creates opportunities for increased cross-learning and experimental synergy (e.g., biological and screening assays) across the portfolio. Such alliances aim to align the biological strength of academic and clinical labs, CRT-DL’s own drug discovery engine and alliance management expertise, and the platforms and capacity that industry partners can offer.
In our experience, a clearly defined set of outputs for the partnership and an understanding of the different drivers of each party, which are reflected in the shared objectives, are vital for success. Strong communication, honesty, flexibility, and a joint governance process that is able to maintain alignment to the agreed strategy, as well as to make concessions where needed, is crucial. Partnerships will work best where each party’s opinions are respected and where each party plays to their strengths in helping advance the collaboration toward its goal(s).
Collectively, the partnerships across the oncology field highlight the steady but relentless move toward broader academic and industrial collaboration. The increasing two-way communication has allowed the breakdown of some of the previous barriers to collaboration by addressing issues such as intellectual property rights, publication embargos, and loss of research control and has enabled a fresh view of how academia and the pharmaceutical industry can work more openly together. As more industry-academia partnerships develop, it will be important for the models to evolve to fit the changing needs of partners and the prevailing drug discovery landscape. Equally, it will be important for the outputs of these collaborations to be reviewed and compared with other previous and existing models of translational research.
Alternate Hit-Finding Strategies for Validated Oncology Targets
Although strong disease-linkage and convincing target validation define good cancer targets, the identification of a pharmacological agent for any given target that can be optimized into a drug is a considerable challenge. It is estimated that only 10% to 14% of the total human proteome possess suitable molecular properties to bind small molecules with high affinity and specificity.84–86 These “druggability” models have defined molecular features for chemical tractability, 87 and proteins displaying these features are highly suited to the reductionist target-based screening paradigm. However, an objective assessment of oncology drug targets reported a tension between their importance in the molecular pathology of cancer and their chemical tractability. 17 Thus, as we have described above for KRAS, some of the most attractive targets in oncology are also the most difficult to develop drugs against. Innovative strategies are emerging toward pharmacological inhibition of such targets, 88 and the following sections discuss ways in which we can maximize our chances of identifying chemical hit matter.
Improving Compound-Screening Libraries
In any high-throughput screen (HTS), the size and diversity of the chemical library are related to the success of hit identification and the ability to initiate lead discovery. A potential strategy to maximize the output is to increase the molecular complexity and diversity of compound libraries by populating new areas of chemical space. 89 The ultimate goal would be to populate screening libraries with compounds that can give excellent potency and selectivity in both biochemical and cell-based assays, providing a set of experimental compounds that (1) validates pharmacological modulation of the target as a viable therapeutic strategy, (2) provides substrate for the generation of high-quality tool compounds for use as biological probes, 90 and (3) identifies suitable chemistry to develop into lead molecules.
In a recent evaluation of the sources of novel structures for anticancer agents approved since the 1940s, nearly half were found to be either natural products or directly derived from them. 91 However, high cost, chemical purity, accessibility, and synthetic challenges in chemistry remain significant hurdles to optimizing natural product hits through the drug discovery process. Advanced chemical synthesis approaches toward expanding chemical diversity include function- and diversity-orientated synthesis (FOS and DOS). These attempt to recapitulate the biological function of complex natural products with simpler scaffolds (FOS 92 ) and create collections of compounds with diverse scaffolds and stereochemistry (DOS 93 ).
Another approach to sample a larger theoretical diversity space than is practical through standard HTS is through fragment-based screening. This technique requires a robust structural biology platform for optimization of weakly binding hits. 86 Further diversity can be achieved with cyclic peptides. Their large size, conformational flexibility, and cell-permeable properties enable high-affinity interactions with the relatively flat protein surfaces that can categorize challenging targets. 94 Although macrocyclic peptide synthesis is far from trivial, novel methodologies have been developed, enabling the creation of large screening collections of highly diverse structures.95,96 Finally, DNA-encoded chemical libraries have the potential for the synthesis and interrogation of hundreds of millions of compounds through affinity selection followed by deconvolution of the chemical display library to identify discrete screening hits. 97
Collaborative models, such as the one recently described by AstraZeneca and Bayer Pharma AG,98,99 are also forging new ways to improve the chemical diversity of screening libraries. In this instance, the sharing of compound collections between the two companies, with suitable measures in place to protect intellectual property, was shown to be a valid strategy to quickly enhance the coverage of chemical space in respective screening collections. This may allow new sources of chemical leads to be identified for targets that have been traditionally viewed as poorly tractable.
Developing Irreversible Inhibitors
There are many examples of irreversible inhibitors in the clinic; however, safety considerations are often cited as a reason to avoid such compounds in the development of new therapeutics. Safety certainly can be a concern in the case of covalent nonselective inhibitors and when sustained inhibition of the target in question is not desirable. However, with high-value oncology targets, advances in the development of irreversible inhibitors may lead to improved efficacy in the clinic by providing a sustained duration of target inhibition even when pharmacokinetics are suboptimal.100,101 There is also emerging evidence that irreversible inhibitors may circumvent some types of acquired resistance.102,103
An example approach has come from the use of irreversible modifiers for targets amenable to specific covalent inhibition, such as targets containing a cysteine residue that is either inside or near to a functionally relevant binding site. 101 This has been used to identify irreversible inhibitors to target KRAS directly, exploiting the oncogenic glycine to cysteine mutation at position 12 (G12C) in the KRAS protein present in many cancers, in particular lung adenocarcinoma. 33 Inhibitors containing an electrophilic functionality positioned to react with the cysteine residue were shown to irreversibly bind and impair only the mutant KRAS.104,105 These molecules alter the affinity of KRAS for GTP, converting it to the GDP form, thus allowing it to accumulate in an inactive state. Decreased cellular viability and apoptosis in response to these inhibitors were observed in a small panel of lung cancer cell lines containing the G12C mutation, whereas cell lines lacking the mutation were unaffected. 105 Although the magnitude of effect observed was moderate in this study, at this stage it provides a starting point for further development of the chemistry and represents an exciting outlook for targeting KRAS in the future.
A similar approach has recently gained momentum in the kinase inhibitor field. 106 Several targeted covalent inhibitors have recently been developed to treat patients who have acquired mutant forms of EGFR (activating mutations and T790M resistance mutation) including Afatinib 107 (Gilotrif, Boehringer Ingelheim, Ingelheim, Germany), AZD9291 108 (AstraZeneca, London, UK), and rociletinib/CO-1686 109 (Clovis Oncology, Boulder, CO). Afatinib has recently been approved by the FDA as a first-line treatment of patients with metastatic NSCLC whose tumors have exon 19 deletions or exon 21 (L858R) substitution mutations in EGFR.
Cell-Based HTS and Target Deconvolution
An analysis of the origins of the 113 first-in-class NMEs approved by the FDA across all therapeutic areas between 1999 and 2013 showed that 30% (33 of 113) originated from target-agnostic approaches, including chemocentric (i.e., using compounds with known pharmacology as a starting point, 23%) and cell-based screening approaches (7%). 110 A similar analysis performed in 2011 using data from 1999–2008 claims that 56% (28 of 50) of first-in-class small-molecule approvals were derived from phenotypic screening. 111 Although there are many advantages to screening in a more physiologically relevant setting, there are also significant obstacles associated with this approach, such as chemistry optimization and molecular target deconvolution (see Table 2 for comparison between target-based and target-agnostic approaches). The lack of precise knowledge of the molecular target(s) and mechanism(s) through which a small-molecule inhibitor is functioning is often perceived as the most significant obstacle associated with a cell-based screening paradigm. Despite this, there are many examples of approved cancer drugs that were derived through cell-based screening approaches (see ref. 112 for analysis of the origins of NMEs approved between 1999 and 2013 and an excellent review of the different approaches to cell-based screening). Furthermore, the FDA guidance is unambiguous in that their focus is on safety and efficacy rather than knowledge of target or mechanism, which is borne out by the fact that many recently approved NMEs do not have a known target or molecular mechanism of action.110,111
Different Approaches to Primary Assay Design for Hit Identification.
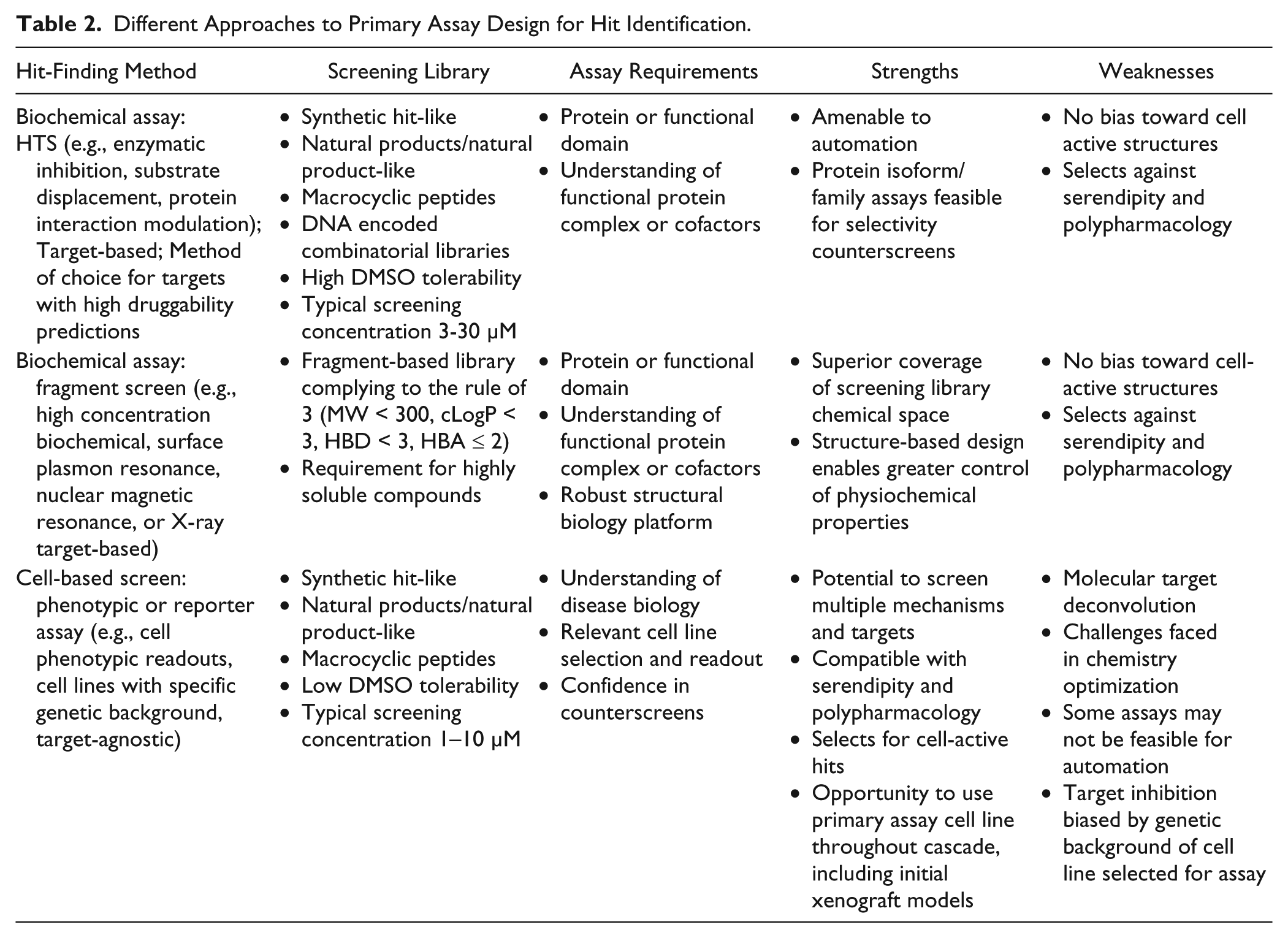
Selecting the optimal approach for cell-based screening in oncology is key for success. For “black box” screens, as opposed to more mechanism-informed strategies, a significant challenge lies in the interpretation of output and optimization as all polypharmacology relevant to the desired phenotype will be identified. Although challenges exist around the predictive value of preclinical cellular models, recent advances in high-content screening technologies and the use of models that better reflect human disease such as 3D cell culture, 113 patient-derived primary cells, 114 cancer stem cells,115,116 and co-culture systems 117 offer much promise for the future. 118 Providing there is a clear understanding of the cancer biology, including relevant pharmacodynamic biomarker(s) and/or phenotype(s), there may be a good rationale for using a well-designed cell-based screening cascade to run a drug discovery program. Indeed, a relevant pharmacodynamic biomarker often makes an effective primary assay for a mechanism-informed cell-based screen, allowing correlations between biomarker readout and phenotype to be demonstrated at an early stage. A further benefit of cell-based screens is realized through potency gains, in that validated hits from such screens are, by definition, “cell-active,” potentially bypassing months of chemical optimization to improve potency and permeability.
We have assessed many new target opportunities with a strong biological rationale that fall into the classically “undruggable” category. For a number of these targets, we have adopted a cell-based screening approach and have found that the design of the primary screening assay and downstream cascade particularly benefits from collaborative input from academic partners. Within this strategy, there is a bias toward identification of high-potency screening hits, which will increase the chances of generating successful structure-activity relationships (SARs) and molecular target deconvolution. We therefore perform these screens at significantly lower concentration than is typical for standard biochemical target-based approaches (usually 3 µM). We find this helps to mitigate wasted effort on pursuing weakly active, typically nonspecific, hits. A high degree of confidence in the hits is required to progress the project and invest in target deconvolution, with demonstration of submicromolar cellular potency and emerging SAR being mandatory. More importantly, the SAR must not correlate with compound lipophilicity or reactivity. Finally, molecular target deconvolution is performed in parallel with medicinal chemistry optimization with the aim to identify new “on pathway” targets through the use of biologically active small molecules.
Significant progress has been made in the application of technologies for successfully identifying the molecular target(s) of lead molecules.119,120 In particular, photoaffinity labeling, via bio-orthogonal click chemistry reactions, negates the requirement for expensive and difficult-to-use radiolabeled probes. 121 Once targets are labeled, a combination of affinity enrichment and quantitative proteomics (e.g., stable isotope labeling by amino acids in cell culture, SILAC) is a powerful method for protein identification. 122 For instances in which chemical modification into a suitable affinity probe is not readily available, drug affinity responsive target stability (DARTS) has been developed as a label-free method that takes advantage of a reduction in protease susceptibility of the target protein(s) upon ligand binding. 123
As these and other target identification methodologies improve, they could also be incorporated into ongoing drug discovery programs to gain a clearer understanding around the molecular targets of tool, candidate, and clinical compounds. Identifying additional beneficial (or detrimental) polypharmacology could help to improve therapies or reveal potential opportunities for repurposing. Indeed, the clinical repurposing of crizotinib from a c-MET inhibitor to an ALK inhibitor for the treatment of ALK-fusion gene-positive NSCLC clearly highlights the power of combining an understanding of pharmacological mechanism with targets that have compelling disease-linkage. 124 Moreover, many kinase inhibitors, including some undergoing clinical evaluation, have recently been observed to inhibit epigenetic bromodomains with therapeutically relevant potencies.125,126 These examples illustrate the value that may be gained in understanding the principal molecular targets of pharmacological agents.
Selecting the right target and the patient population for any associated therapeutic is critical for success in drug discovery. Although large-scale genomic initiatives and preclinical studies are revealing potential drug targets at an increasing pace, significant challenges remain in translating these into viable drug discovery projects and ultimately to patient benefit. The vast majority of molecularly-targeted projects will fail during target validation, and so an important challenge is to improve the reliability of translating the literature into robust drug discovery projects. The roots of this problem are multifaceted and include not only a need to increase the reproducibility of reported data but also to use in vitro and in vivo oncology models that give better predictions of efficacy in the clinic. The poor characterization of many of the tools and reagents routinely used in preclinical research is a related confounding problem that also needs addressing with urgency. To enhance productivity, we must incentivize and increase the frequency of our reporting of target devalidation data where they are available. These should not be perceived as negative publications but a valuable part of the scientific discourse, which, in some instances, will help to accelerate scientific self-correction through exposure within the public domain.
In the prevailing reductionist target-based drug discovery paradigm, an emphasis is often placed on what can be done (process), rather than answering the question of why it should be done (disease and biology). To effectively translate targets with strong disease-linkage, many of which are poorly tractable by conventional approaches, it will be necessary to explore alternative strategies for hit finding and to embrace increased biological complexity within the drug discovery process.
The need for a creative view in oncology drug discovery is very apparent, demanding flexibility and innovation coupled with the expertise and capacity to drive projects forward. New partnerships between industry and academia have the potential to raise success rates by opening up new disease-based models and by harnessing the wealth of “omics” data to help select the most relevant targets and strengthen their disease-positioning. Such collaborative efforts can allow the creation of synergies within the drug discovery process to reduce the risk associated with following a single target. They have the potential to tackle some of the biggest challenges we face in oncology drug discovery and enable the common goal of identifying the right patients, targets, and models to develop and deliver more effective therapies.
Footnotes
Acknowledgements
We would like to thank Hamish Ryder and Martin Swarbrick for critically reviewing the manuscript.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of Cancer Research Technology and are involved in drug discovery alliances between Cancer Research Technology and a number of pharmaceutical/biotech industry partners.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.