
Abstract
Phosphatidylinositol 4-phosphate 5-kinases (PIP5Ks) regulate a variety of cellular processes, including signaling through G protein-coupled receptors (GPCRs), endocytosis, exocytosis, and cell migration. These lipid kinases synthesize phosphatidylinositol 4,5-bisphosphate (PIP2) from phosphatidylinositol 4-phosphate [PI(4)P]. Because small-molecule inhibitors of these lipid kinases did not exist, molecular and genetic approaches were predominantly used to study PIP5K1 regulation of these cellular processes. Moreover, standard radioisotope-based lipid kinase assays cannot be easily adapted for high-throughput screening. Here, we report a novel, high-throughput, microfluidic mobility shift assay to identify inhibitors of PIP5K1C. This assay uses fluorescently labeled phosphatidylinositol 4-phosphate as the substrate and recombinant human PIP5K1C. Our assay exhibited high reproducibility, had a calculated adenosine triphosphate Michaelis constant (Km) of 15 µM, performed with z’ values >0.7, and was used to screen a kinase-focused library of ~4700 compounds. From this screen, we identified several potent inhibitors of PIP5K1C, including UNC3230, a compound that we recently found can reduce nociceptive sensitization in animal models of chronic pain. This novel assay will allow continued drug discovery efforts for PIP5K1C and can be adapted easily to screen additional lipid kinases.
Keywords
Introduction
Phosphatidylinositol (PI) lipids tightly regulate many cellular processes, including cell migration, vesicle trafficking, endocytosis, surface receptor signaling, cell proliferation, and cell survival—all of which underlie normal physiology and disease mechanisms.1–3 The role of phosphatidylinositol 3-kinases (PI3Ks) in cancer proliferation and survival has been extensively characterized, explaining why the vast majority of lipid kinase drug discovery efforts during the past 20 years (starting with the discovery of the first PI3K inhibitor, wortmannin) have been directed toward PI3Ks. 4 There has been relatively little effort on identifying inhibitors of other lipid kinase family members, likely due to a lack of conclusive evidence for a direct link between specific diseases and individual lipids and/or lipid kinases. Recent studies connecting additional lipid kinases to disease-relevant cellular processes include sphingosine 1-kinase in cancer progression,5,6 phosphatidylinositol 5-phosphate 4-kinase (PIP4K2B) in insulin signaling,7,8 and phosphatidylinositol 4-phosphate 5-kinase (PIP5K1C) in nociceptive sensitization. 9 There is thus growing interest in using high-throughput screening (HTS) assays to identify inhibitors of these lipid kinases.8,10 Here, we describe an HTS assay developed during our studies that examined the role of PIP5K1C in nociceptive signaling and sensitization. 9
Type I PIP5 kinases (PIP5K1s) and type II PIP4 kinases (PIP4K2s) synthesize the predominant lipid second messenger, PI(4,5)P2, from phosphatidylinositol monophosphates (PIPs) phosphatidylinositol 4-phosphate [PI(4)P] and phosphatidylinositol 5-phosphate [PI(5)P], respectively. Although there have been two recent publications reporting the development of independent high-throughput assays to identify inhibitors of PIP4K2A 11 and PIP4K2B, 8 there were no known inhibitors of any of the three mammalian PIP5K1 isoforms (alpha, beta, or gamma). Our studies using genetic Pip5k1c knockout mice demonstrated the need for a pharmacological inhibitor that could be used to complement our genetic studies by acutely inhibiting PIP5K1C and as a tool for target validation.9,12
When we began our studies, available assays to monitor PIP5K1-dependent phosphatidylinositol 4,5-bisphosphate (PIP2) synthesis required the use of lengthy lipid extraction protocols, radiolabeled adenosine triphosphate (ATP), and/or thin-layer chromatography to separate substrate and product, none of which were amenable to an HTS assay.10,13,14 Here, we overcame these limitations by developing an HTS assay using fluorescently conjugated PI(4)P, the natural substrate for PIP5K1C, and full-length recombinant PIP5K1C.
Materials and Methods
Materials
Fluorescein-conjugated phosphatidylinositol 4-phosphate [PI(4)P; 9000655] and fluorescein-conjugated PIP2 (10010388) were purchased from Cayman Chemical (Ann Arbor, MI) and reconstituted in 100% DMSO to 1.5 mM. N-terminal His6-tagged full-length (90 kDa) recombinant human PIP5K1C was purchased from Millipore (14-845M; Millipore, Billerica, MA). ProfilerPro separation buffer (760367) and coating reagent 8 (CR-8; 760278) were purchased from PerkinElmer (Waltham, MA). PIP5K1C enzyme was used at a final concentration of 3 nM in assay buffer (
Library of Pharmacologically Active Compounds
The Library of Pharmacologically Active Compounds (the LOPAC library) was purchased from Sigma-Aldrich (St. Louis, MO) and was used as an assay validation library. The 1280 compounds were supplied as 1 µL samples (10 mM) in 384-well polypropylene microplates (Greiner, Monroe, NC). On the day of screening, plates were thawed and diluted (1:100) to 0.1 mM (10× the final assay concentration) with assay buffer (
Kinase-Focused Library
The 4727-compound kinase-focused library was prepared and generously provided by the UNC Center for Integrative Chemical Biology and Drug Discovery (CICBDD). 15 On the day of screening, plates were prepared as described for the LOPAC library.
Screening
A Multidrop Combi Reagent Dispenser was used for the addition of all reagents to assay plates. First, 10 µL of 90 mM EDTA (in assay buffer) was added to each well in columns 1 and 2 and served as positive control reactions (100% inhibition). Nine microliters of 2× enzyme solution were added to each well of the entire plate. Plates were incubated at room temperature for 10 min, then 9 µL of 2× substrate solution was added to each well of the entire plate. Assay plates were incubated in the dark for 40 min at room temperature. Ten microliters of 90 mM EDTA (in assay buffer) were then added to columns 3–24 to stop the reactions. Fluorescently conjugated substrate, PI(4)P, and product, PIP2, were detected using the LabChip EZ Reader II microfluidic mobility shift assay (MSA) platform from PerkinElmer and ProfilerPro separation buffer containing 1.5% CR-8.
For dose–response curves, compounds were plated as threefold serial dilutions starting with a top concentration of 10 mM. The lowest concentration prepared in the 10-point dose response was 0.0005 mM. Dose–response compound plates were prepared using a Tecan Genesis 200 (Research Triangle Park, NC). Dose–response plates were heat-sealed and stored at −20 °C until the day of use. On the day of use, plates were prepared as described from the LOPAC library (see above). The final top assay concentration was 10 µM, and the lowest assay concentration was 0.005 µM.
Data Analysis
Screening data were analyzed using Screenable software (Screenable Solutions, Chapel Hill, NC). Screenable was used to calculate the mid-mean of the positive and negative controls (positive controls in columns 1 and 2, and negative controls in columns 23 and 24), the percent inhibition (with respect to on-plate controls) for each reaction, and the common assay performance measure, z’, for each plate:
where p is the positive control (+90 mM EDTA; 100% inhibition); and n is the negative control (no compound addition; no inhibition). 16 A z’ >0.5 was considered acceptable for the plate to be included in the overall data analysis. The LabChip software calculated percent conversion for each reaction.
Compounds from the kinase library were considered hits if they inhibited PIP5K1C at ≥80%. The 80% threshold was determined as greater than three standard deviations from the mean percent inhibition for the entire screen. Mean inhibition was 14% with a standard deviation of 21%. Dose–response curves were calculated using Screenable Software by converting the percent conversion to percent inhibition with respect to on-plate controls and using a three- or four-parameter curve fit.
Results
Assay Design and Development
Our initial goal was to develop an assay that was compatible with high-throughput techniques. To avoid lengthy lipid extraction and preparation protocols required when using eukaryotic cell lysates, we used recombinant human PIP5K1C and a fluorescein-conjugated PI(4)P substrate in an in vitro biochemical reaction ( Fig. 1A ). Although there are no reported assays that monitor PIP2 from any of the three natural substrates, there was a recent assay developed that used the PerkinElmer LabChip microfluidic MSA platform to monitor the conversion of PIP2 to phosphatidylinositol 3,4,5-triphosphate (PIP3) by PI3K. 17 Using the reported PI3K assay buffer and the PerkinElmer LabChip microfluidic MSA platform, we observed baseline separation of 1 µM fluorescein-conjugated PIP2 and 1 µM fluorescein-conjugated PI(4)P using ProfilerPro separation buffer with 1.5% CR-8 ( Fig. 1B ). Notably, we found that the presence of sodium cholate in the assay buffer is imperative to achieve separation and prevent aggregation of lipids. The proprietary CR-8 promotes run-to-run separation consistency, prevents aggregation of substrate and kinase, and reduces adsorption of substrate and kinase to the microchannel surfaces. 18 Baseline separation of the substrate and product indicated that the LabChip MSA platform would be suitable for continued assay development.
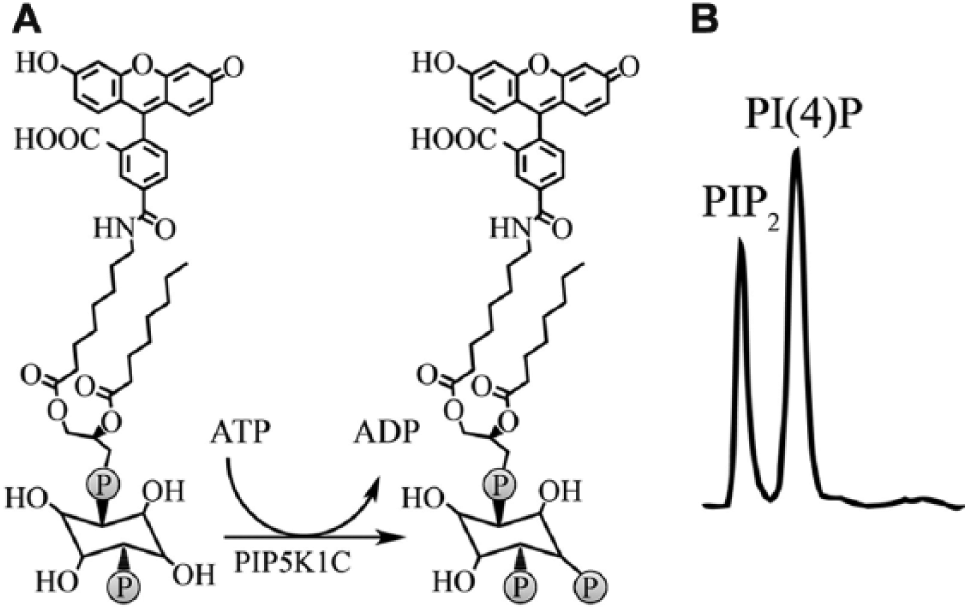
Recombinant PIP5K1C generates fluorescein-conjugated phosphatidylinositol 4,5-bisphosphate (PIP2) from fluorescein-conjugated phosphatidylinositol 4-phosphate [PI(4)P]. (
Fluorescein-conjugated PI(4)P (1 µM) was incubated with 100 nM N-terminal His6-tagged full-length (90 kDa) recombinant human PIP5K1C and varying concentrations of ATP (threefold serial dilution starting at 1000 µM; 11 concentrations total) to determine the ATP Km. The LabChip MSA platform was used to monitor the reactions kinetically (every 5 min for ~80 min). Michaelis–Menten analysis, using initial rate velocities, revealed an ATP Km of 15 µM (
Fig. 2A
and
2B
). Next, reactions containing 1 µM PI(4)P, 15 µM ATP, and varying concentrations (twofold serial dilution starting at 10 nM; seven concentrations total) of PIP5K1C were read kinetically (every 6 min for 45 min) to determine the concentration of enzyme that would give ~30% conversion of substrate to product at an endpoint within the linear range of the reaction (
Fig. 2C
). A final concentration of 3 nM PIP5K1C and a 40-min incubation time were chosen. Kinase titrations were performed for every vial of enzyme purchased during the course of assay development and screening to account for slight differences in kinase activity from lot to lot. Varying concentrations of PI(4)P (twofold serial dilution starting at 100 µM; 11 concentrations total) were incubated with 3 nM PIP5K1C and 15 µM ATP, and reactions were read kinetically (every 4 min for 45 min) using the LabChip platform to calculate substrate Km. Percent conversion of the reactions was converted to pmol product produced (
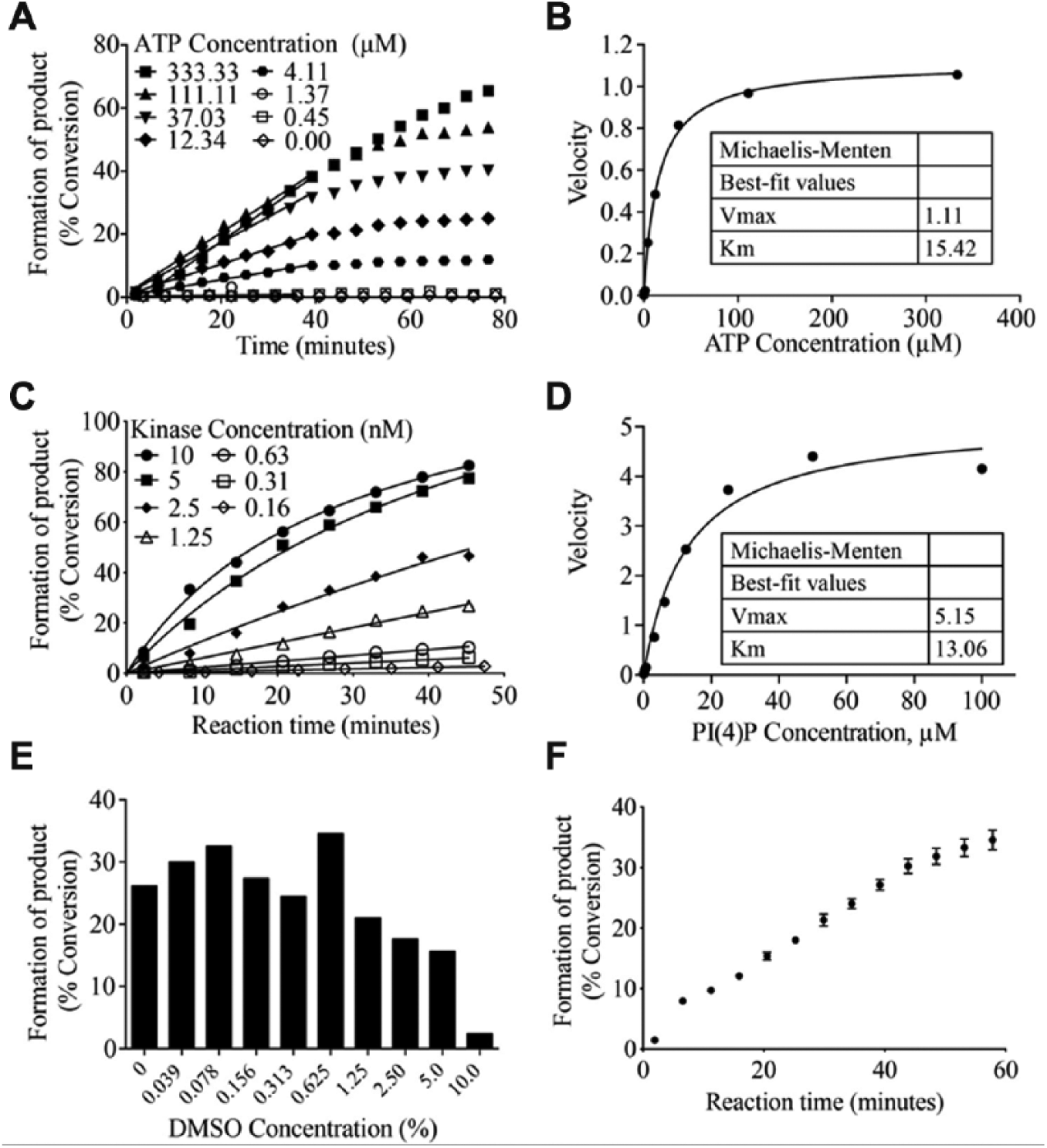
Assay development to determine kinase concentration, adenosine triphosphate (ATP) and substrate Michaelis constant (Km), and DMSO sensitivity. (
Given that all UNC compound libraries are dissolved in 100% DMSO, reactions containing 1 µM PI(4)P, 15 µM ATP, 3 nM PIP5K1C, and varying concentrations of DMSO (twofold serial dilutions starting at 10%; nine concentrations total) were monitored using the LabChip MSA platform to determine DMSO tolerance ( Fig. 2E ). There was decreased activity in reactions containing >1% DMSO, indicating that the final concentration of DMSO within the reaction must be lower than 1%. Compounds are prepared as 10 mM stocks in 100% DMSO. Compounds are then diluted in the compound plate to 0.1 mM with assay buffer (DMSO concentration of 1%). Compounds are then transferred to the assay plate, where the final concentration after enzyme and substrate addition is 10 µM in ~0.125% DMSO (0.1% from compound addition and 0.125% from substrate solution), which is well within the DMSO tolerability of the reaction. Finalized reactions containing 1 µM PI(4)P, 15 µM ATP, 3 nM PIP5K1C, and 0.15% DMSO result in 30% conversion of substrate to product following a 40-min incubation ( Fig. 2F ), an ideal percent conversion (within the linear range of the reaction) for detecting inhibitors.
Next, we transitioned this assay into an HTS format beginning with automation validation using a Multidrop Combi Reagent Dispenser for delivery of the enzyme and substrate solutions. We initially observed inconsistent delivery of the lipid substrate and kinase to the plate, likely because of lipid adsorption to the silicone tubing of the Multidrop delivery cassette. 19 To overcome adsorption problems, final concentrations of 0.01% BSA and 0.05% DMSO were included in the enzyme and substrate solutions, respectively, to serve as carriers and mitigate adsorption. 20 There was a slight reduction in PIP5K1C activity upon addition of 0.01% BSA to the enzyme solution (23% conversion in the presence of BSA compared to 30% conversion in the absence of BSA). Regardless, reactions in the presence of BSA remained in the linear range of detection and displayed the desired 20–40% conversion of substrate to product after a 40-min incubation, indicating that no adjustments were needed to the previously determined PIP5K1C concentration of 3 nM. To further minimize adsorption problems, the silicone lines of the Multidrop delivery cassette were primed with 5 mL of 0.2% BSA prior to kinase dispensing and 5 mL of 1% DMSO prior to substrate dispensing. The addition of BSA and DMSO to the enzyme and substrate solutions, respectively, allowed for consistent delivery of both solutions to multiple plates (data not shown). By incorporating these changes, all plates had coefficients of variation (CVs) less than 10% (CVs >20% were observed prior to making these changes) and no discernible, systematic patterns, thus completing automation validation.
Assay Validation
Following automation validation, the assay was revalidated in an HTS format using the Nanoscreen MultiMek to transfer 1% DMSO (to mimic compound delivery) to each assay plate, followed by Multidrop delivery of the enzyme and substrate solutions. During this stage, we confirmed the plate layout for the assay in which positive controls (+90 mM EDTA in assay buffer; 100% inhibition) would occupy columns 1 and 2, and negative controls (no compound; no inhibition) would occupy columns 23 and 24; all plates were run in this manner for HTS validation. HTS validation was carried out over 3 days, two plates per day. HTS validation revealed excellent assay performance with z’ values higher than 0.9 on all 3 days ( Fig. 3A ). We then used the LOPAC library as a validation library to examine reproducibility of our assay. The 1280 compounds were run in duplicate for 2 days (four plates per day). The assay was deemed excellent with respect to z’, which ranged from 0.7 to 0.9 ( Fig. 3A ) for all plates. The data fitted with a linear regression line revealed an r2 and slope of 0.966, indicating a highly reproducible assay ( Fig. 3B ).
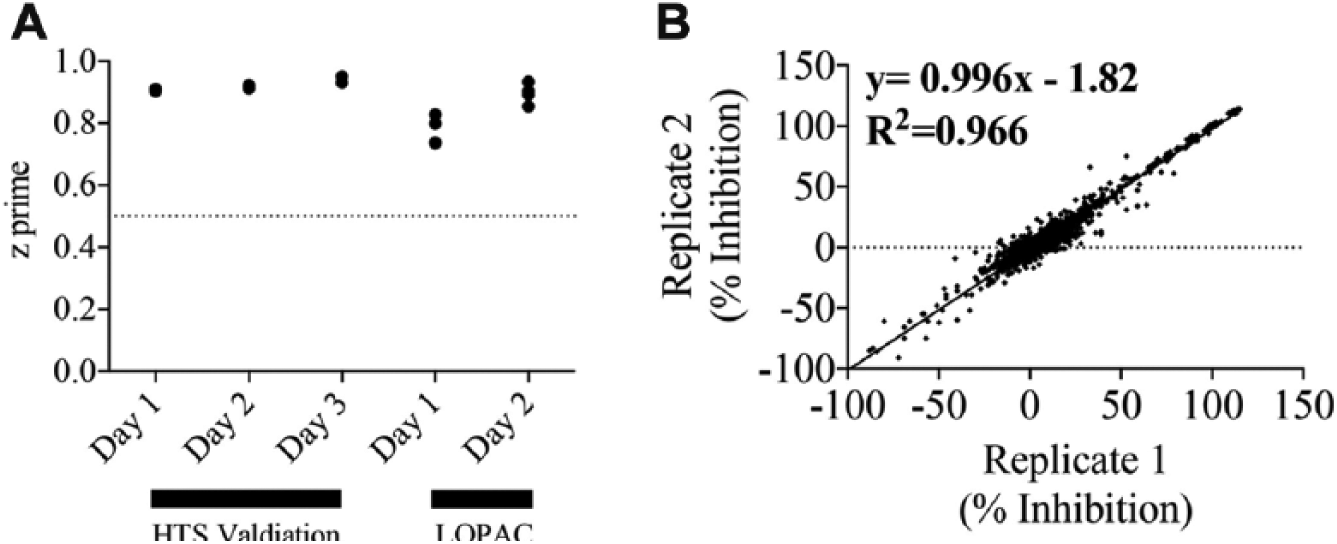
Assay validation using the Library of Pharmacologically Active Compounds (the LOPAC library). (
Screening
A kinase-focused library of 4727 compounds that was designed and made available by the UNC CICBDD was screened at 10 µM in 0.1% DMSO.
15
Results from the kinase-focused library were found to follow a normal distribution (
Fig. 4A
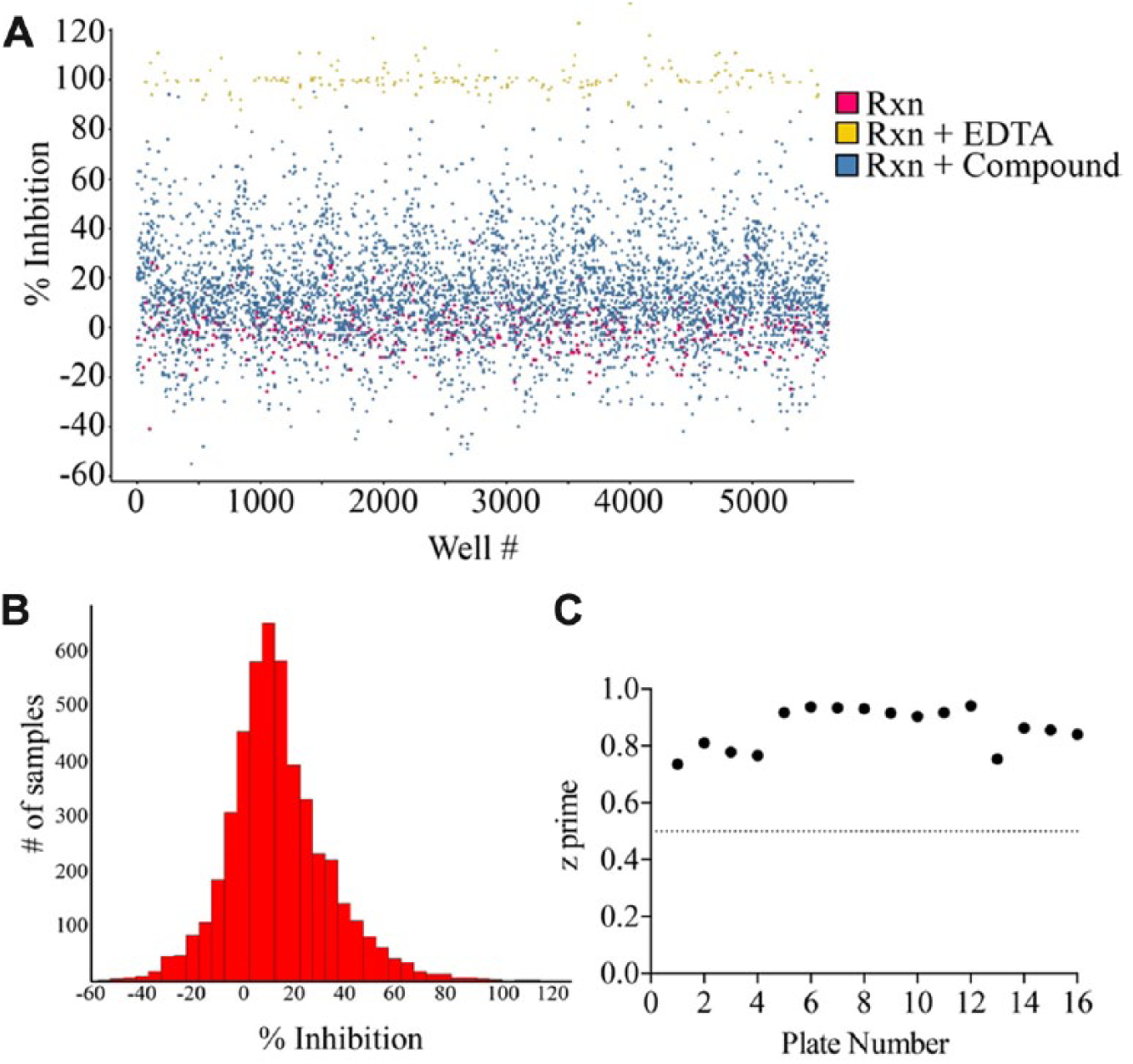
High-throughput screening of a 4727-compound kinase-focused library. (
Hit Confirmation and Follow-Up of Select Inhibitors
All available active compounds were retested in triplicate in a 10-point dose–response assay to confirm activity and provide potency information (
Of the 18 active kinase-focused library compounds, compound
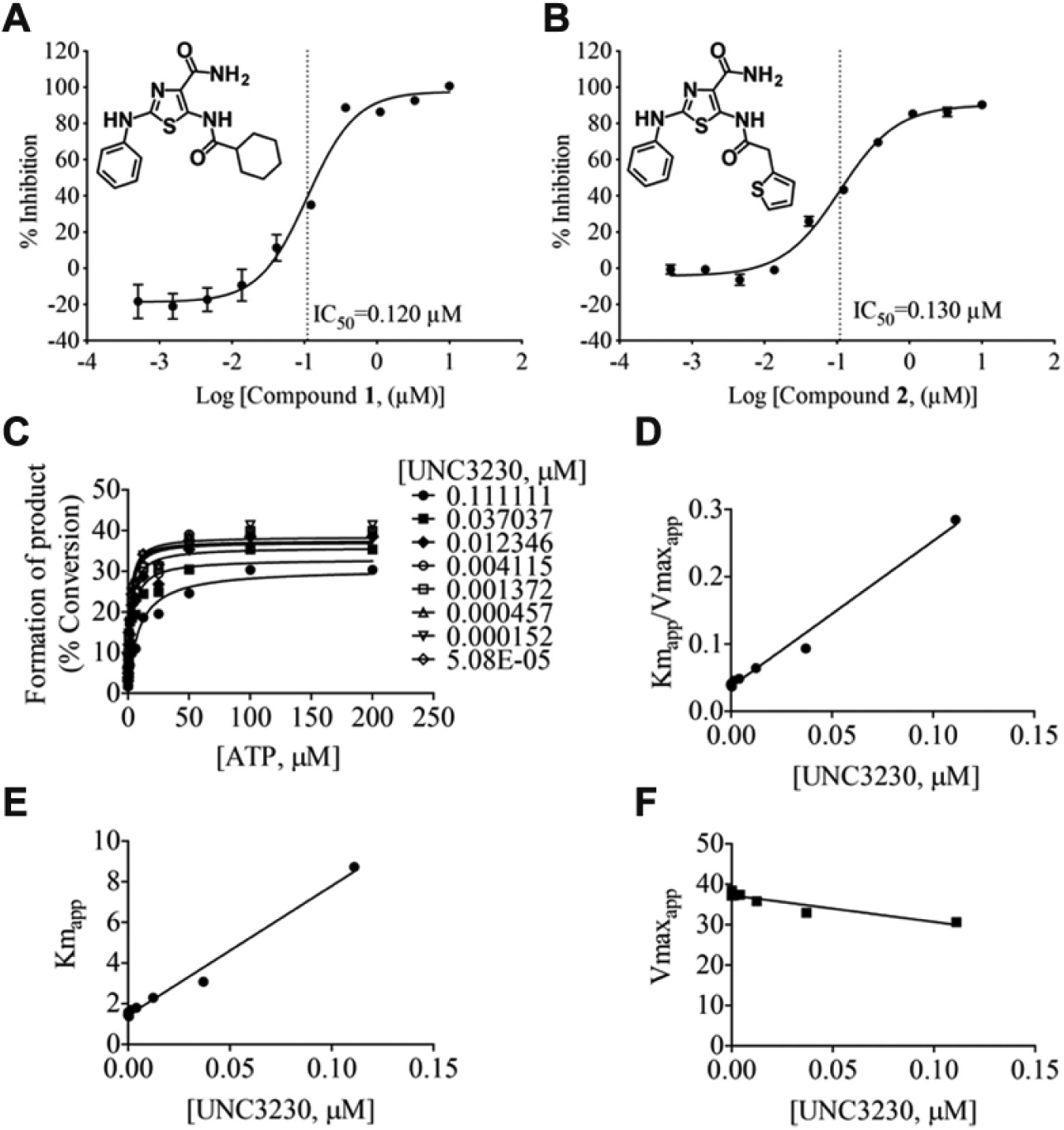
The two most potent inhibitors of PIP5K1C have the same core structure, and UNC3230 exhibits an adenosine triphosphate (ATP)-competitive mode of inhibition. (
In additional follow-up studies, the mode of inhibition of our most potent compound, UNC3230, was evaluated in two independent ATP competition studies using our mobility shift assay ( Fig. 5C–5F ). These studies demonstrated that UNC3230 was competitive against ATP with an average Ki of 23 nM ( Fig. 5C–5F ). The slight decrease in Vmaxapp is not statistically significant; thus, we conclude that UNC3230 is an ATP-competitive inhibitor of PIP5K1C.
Discussion
Previous studies of cellular processes that are regulated by type I PIP5Ks have been limited to genetic and molecular biology techniques, including the use of genetic knockout mice and targeted RNA interference, due to the lack of available pharmacological inhibitors.3,13,14,23 The use of genetic knockout mice is time- and resource-intensive, and it limits studies to irreversible deletion of the enzyme, thwarting information on acute inhibition of the enzyme. 24 Although PIP5K1 is thought to regulate cellular processes via its enzymatic activity (production of PIP2), PIP5K1s contain several protein binding motifs and hence might also exert some degree of regulation independent of catalytic activity.25–27 With the use of our inhibitors, studies of acute inhibition of PIP5K1C are now possible. In addition, these inhibitors could provide a necessary tool to differentiate between scaffolding and/or enzymatic regulation of each PIP5K1C-dependent cellular process,25,27,28 assuming that inhibitor binding reduces only the catalytic activity and does not change the protein conformation substantially. To make this assumption, additional studies are needed on inhibitor binding to the protein, which include acquisition of a crystal structure. As noted previously, our assay identified UNC3230, which was used successfully as a complementary pharmacological approach to validate PIP5K1C regulation of nociceptive signaling that was shown to be dependent on PIP5K1C catalytic activity using genetic-based experiments. 9
Furthermore, current techniques in the field of PIP5K1C-regulated cellular processes depend on overexpression of phosphatases and kinases that transiently eliminate or synthesize PIP2, respectively.3,29,30 These techniques perturb the complex, interconnected phosphatidylinositol signaling cascade at several critical enzymatic points in a single experiment. The identified inhibitors provide an attractive alternative to reduce PIP2 by specifically inhibiting PIP5K1C and PIP5K2C without disturbing other critical enzymes in the cascade.
Unlike the PIP4K2s, there are no crystal structures of any of the PIP5K1s, making structure-guided drug design impractical; 11 however, standard structure–activity relationship (SAR) studies are feasible and required. As we previously reported, 9 UNC3230 has limited aqueous solubility, highlighting the need for medicinal chemistry optimization to increase solubility while maintaining potency and selectivity. The assay described herein will be used for high-throughput evaluation of analogs derived from the identified thiazole carboxamide core structure that would be more feasible for further preclinical studies.
Importantly, our HTS assay can be extended to the study of other lipid kinases that regulate cellular signaling and disease physiology. The only limitation to extending our assay to other lipid kinases is the availability of fluorescently conjugated substrates and recombinant proteins. However, many fluorescently labeled lipid substrates are available for purchase, and recombinant proteins can be expressed and purified from bacculovirus or bacterial systems. Furthermore, this new assay can be expanded to screen larger libraries to provide additional scaffolds for continued development of PIP5K1C inhibitors for analgesic drug development.
Footnotes
Acknowledgements
We thank Chatura Jayakody for compound management, Melissa Porter for help with assay development, and Drs. Stephen Frye and Jian Jin for medicinal chemistry expertise and guidance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants to MJZ from the National Institute of Neurological Disorders and Stroke (R01NS081127 and R01NS067688).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.