
Abstract
The Bispecific T-cell Engager (BiTE®) antibody modality is a clinically validated immunotherapeutic approach for targeting tumors. Using T-cell dependent cellular cytotoxicity (TDCC) assays, we measure the percentage of specific cytotoxicity induced when a BiTE molecule engages T-cells, redirects T-cell mediated cytolysis, and ultimately kills target cells. We establish a novel luminescence-based TDCC assay quantified by measuring cell viability via constitutive expression of luciferase. The luciferase-based TDCC assay performance is valid and comparable to an adenosine triphosphate (ATP)-based detection method. We demonstrate that the luciferase-based TDCC assay is an efficient homogeneous assay format that is amenable to both suspension and adherent target cells. The luciferase-based TDCC assay eliminates the need for plate-washing protocols, allowing for higher-throughput screening of BiTE antibodies and better data quality. Assay capacity is also improved by performing serial dilutions of BiTE antibodies in 384-well format with an automated liquid handler. We describe here a robust, homogeneous TDCC assay platform with capacity for in vitro assessment of BiTE antibody potency and efficacy using multiple tumor cell lines and T-cell donors.
Keywords
Introduction
Bispecific T-cell Engager (BiTE®) Antibodies
Immunotherapeutic approaches for the treatment of cancer exploit the potent cytotoxic properties of T-cells. 1 The Bispecific T-cell Engager (BiTE) antibody modality is a breakthrough bispecific antibody platform in which various target-specific immunotherapeutics are engineered for the treatment of diverse hematological and solid-tumor oncology indications. 2 BiTE antibodies are derived from the variable domains of two distinct monoclonal antibodies that (1) bind to T-cells with one arm (anti-CD3ϵ) and (2) bind to a specific surface antigen expressed on the tumor cell with the other arm [i.e., anti-EGFR (epidermal growth factor receptor)]. The BiTE antibody molecule consists of two single-chain variable fragment (scFv) regions from the anti-CD3ϵ and the antitumor-associated antigen antibody connected by a short linker, producing a bispecific antibody with a molecular weight of approximately 55 kilodaltons. 3 Through this dual binding interaction, BiTE antibodies induce formation of a cytolytic synapse and thus direct specific lysis of target positive tumor cells by the T-cells.4,5 Activated T-cells induce a cascade of cytotoxic events, including release of perforins and granzymes, which compromise the cell membrane. This leads to the activation of caspases, fragmentation of DNA, morphological changes, and ultimately apoptosis of target cells.5–7 The potent cytotoxic effects of T-cells are also magnified by the release of cytokines that further stimulate T-cells to kill proximal target cells serially and proliferate. 7
The identification and characterization of BiTE antibodies require an in vitro cellular-based bioassay to screen for functional activity against multiple target-positive tumor cell lines and with multiple T-cell donors. The T-cell dependent cellular cytotoxicity (TDCC) assay is used to measure the cytotoxicity that is specifically induced when a BiTE antibody molecule engages effector T-cells and redirects cytolysis toward target-positive tumor cells. Given that there are multiple techniques to determine cytotoxic activity, our primary focus was to develop a robust method that is rapid, scalable, and reproducible. Ideally, it is desirable to have a cell-based assay that can be performed in a homogeneous format, in which reagents are simply added, and signal is detected without the need to remove supernatant or to wash the assay plate. Here, we describe a successful TDCC screening assay that is validated for precise and accurate in vitro assessment of BiTE antibody molecule potency and efficacy, and also has the capacity to evaluate multiple tumor cell lines and with multiple T-cell donors. Furthermore, characterization of TDCC activity will support the rapid identification of discovery research BiTE antibodies against various targets and drive lead selection for additional characterization and further preclinical development.
Cytotoxicity Assays
A spectrum of cytotoxic cell fates exists, including decreased cell viability, apoptosis, and necrosis, each of which can be quantified by various biochemical and cell-based assay methods. 8 As cells stop actively growing or dividing, a proportional decrease in cell viability may be observed through metabolic readouts such as adenosine triphosphate (ATP) levels measured via luminescence detection, for example with CellTiter-Glo. Cell membrane integrity, another surrogate of cell viability, can also be measured via a fluorometric lactate dehydrogenase (LDH) release assay. Apoptosis is a more defined stage of cell death that can be measured via caspase enzyme activation.
The chromium (51Cr) release assay is a long-standing TDCC method, in which target tumor cells are pre-loaded with 51Cr and cell death is quantified by measuring release of the radiometric marker into the supernatant during T-cell mediated cytolysis.3,9 Although this is a sensitive method, it is not desirable due to the costs and risks associated with using radioactive reagents. There are analogous methods to detect the release of nonradiometric compounds from target cells, such as DELFIA, although this method is not recommended for assay durations greater than 4 h. Spontaneous release of pre-loaded detection reagents into the supernatant can confound data when measuring cytotoxicity after 24 or 48 h of incubation by increasing the background signal and thus decreasing the ability to measure a robust response.
Morphological changes in dying cells can be observed and measured using vital dyes and can be measured via cellular imaging and flow cytometry. Flow cytometry and high-content imaging methods allow simultaneous quantification of both live and dead cell populations with multiple fluorescent reagents (e.g., propidium iodide, DIO, and YoPro-1).9,10 Although imaging and flow cytometry are data rich and can be high-throughput approaches, they are not easy to use without proper instrument training. Typically, they also require significantly more resources to ensure optimal gating, correct data analysis algorithms, as well as informatics overhead compared with biochemical endpoint measurements on a luminescence/fluorescence plate reader.
Cell viability can be detected via surrogates for metabolic activity such as cellular ATP levels, alkaline phosphatase (AP) activity, and LDH release. 8 These are simple and cost-effective approaches with numerous commercial kits available. These approaches eliminate the use of vital dyes to label the different populations of target cells or T-cells, and they can be adapted to homogeneous formats; however, detection can be complicated by the signals from the T-cells. Although an ATP-based luminescent cell viability method is compatible with adherent target cell lines, it is less compatible with suspension and semi-adherent cell lines that could be washed away, so there is a clear need for a homogeneous TDCC method that could be used with various target cell types.
Here, we describe a luminescence-based TDCC assay platform that quantifies live target cells through the detection of constitutively expressed luciferase.11,12 Luminescence readout is a common approach for quantitative cell-based assays that exhibits good sensitivity and robustness. 13 Firefly luciferase has a short half-life of a few hours in cells and rapidly decays in assay media.11–13 Target tumor cell lines are stably transduced to express firefly luciferase. 14 T-cells are not modified and do not provide luminescence to the assay, so the remaining live target cells can be quantified specifically and robustly. We show that this homogeneous/no-wash assay format further increases screening throughput and data quality, and is compatible with suspension, semi-adherent, and adherent target cell lines.
Materials and Methods
BiTE Antibody Generation
BiTE antibodies were constructed by recombinant DNA technology with the sequence of the variable heavy- and light-chain domains of anti-EGFR (cetuximab), anti-MEC14 (hapten), and anti-CD3 antibody sequences as previously described. 4 BiTE antibodies were purified from supernatants generated by stably transfected Chinese hamster ovary (CHO) cells. Target and CD3 binding of BiTE antibody constructs were confirmed by flow cytometry.
Human Pan T-Cells
Normal peripheral blood (NPB) was collected via leukapheresis from normal healthy human donors who were nonsmokers and were not taking any allergy medication (AllCells, Alameda, CA). NPB pan T-cells were isolated by negative selection from mononuclear cells using indirect immunomagnetic methods and characterized by flow cytometry for viability and activation markers. Donor T-cells were pre-screened and validated in the TDCC assay using the SW480 cell line and EGFR and MEC14 positive and negative control BiTE antibodies to determine efficiency of cell killing. Freshly isolated primary human T-cells were evaluated in comparison with previously frozen T-cells in TDCC assays. Aliquots were banked and frozen in liquid nitrogen for subsequent use in TDCC assays to reduce lot variability and to maximize cost efficiency during screening.
Human Cancer Cell Lines
SW480, OVCAR-8, A2058, and additional acute myeloid leukemia (AML) human cancer cell lines were received from the Amgen research cell bank. Cells were cultured at 37°C, 5% CO2 in RPMI medium with non-essential amino acids, 100 U/ml penicillin, 100 µg/ml streptomycin, and 10% heat-inactivated fetal bovine serum (Invitrogen, Waltham, MA). Cell viability was monitored with a Vi-CELL automated cell counter (Beckman Coulter, Pasadena, CA). Target cell surface expression was characterized by flow cytometry with a BD LSRII (BD Biosciences, East Rutherford, NJ).
Generation of Firefly Luciferase–Expressing Stable Pools
Target cells were transduced with a reporter to constitutively express firefly luciferase. A constitutive luciferase reporter plasmid was generated with the typical firefly luciferase [ffLuciferase (1-398), ~60 kDa] and cloned into pLV417 lentiviral vector (huEF1α promoter). 14 Lentivirus was produced in supernatant of 293-6E (serum-free suspension) cells with a separate lentivirus envelop (VSV-G pseudotyped) and packaging plasmid (Gag-Pol). Human tumor cell lines were transduced with lentivirus and selected with 5–10 µg/mL of blasticidin to generate stable pools constitutively expressing the luciferase reporter. Luciferase expression was confirmed with the Steady-Glo Luciferase Assay System (Promega, Fitchburg, WI).
T-cell Dependent Cellular Cytotoxicity (TDCC) Assay
Tumor cells expressing various cell surface target antigens were co-cultured with non-activated human pan T-cells at an effector-to-target (E–T) ratio of 10:1 in the presence of the BiTE antibodies. Twofold or threefold dilution series for BiTE antibodies were performed on the Bravo (Agilent, Santa Clara, CA) liquid handling platform in TDCC assay media. TDCC assays were performed in the following media: RPMI Medium with non-essential amino acids, 10 mM HEPES, 50 µM 2-β-mercaptoethanol, 1 mM sodium pyruvate, 100 U/mL penicillin, 100 µg/mL streptomycin, and 5% heat-inactivated fetal bovine serum (Invitrogen). Using a Multidrop Combi (Thermo Fisher, Waltham, MA) bulk liquid dispenser, 2500 cells per well of target cells (20 µL per well) and 25,000 cells per well of T-cells (25 µL/well) were plated sequentially into 384-well, low-flange, white, flat-bottom, polystyrene TC-treated microplates (Corning, Corning, NY). BiTE antibody dilutions as well as positive and negative controls were added (5 µL/well) and mixed three times to achieve uniformity within each well (total volume, 50 µL/well). Plates were incubated for 48 h at 37°C, 5% CO2 before cell viability quantification.
ATP-Based Cell Viability Determination
Target cells’ viability was determined via ATP quantification and was performed with the CellTiter-Glo Luminescent Cell Viability Assay System (Promega). Cell supernatant was aspirated and washed twice with phosphate buffered saline (PBS) (50 µL/well), aspirated, and followed by addition of PBS (20 µL/well). Automated plate washing was carried out using an EL406 microplate washer dispenser (BioTek, Winooski, VT). Per Promega’s protocol, reconstituted CellTiter-Glo reagent was added (20 µL/well), and assay plates were incubated for 30 min at room temperature. Luminescence was quantified with an EnVision multilabel reader (Perkin Elmer, Waltham, MA) with an ultrasensitive luminescence detector.
Luciferase-Based Cell Viability Determination
Target cells’ viability was determined via quantification of constitutively expressed firefly luciferase and was performed with the Steady-Glo Luciferase Assay System (Promega). Per Promega’s protocol, reconstituted Steady-Glo reagent was added (25 µL per well), and assay plates were incubated for 30 min at room temperature. Luminescence was quantified with an EnVision multilabel reader (Perkin Elmer) with an ultrasensitive luminescence detector.
Specific Cytotoxicity, EC50 Calculation, and Data Analysis
For analysis of bioactivity, we calculated percent (%) specific cytotoxicity from relative luminescence units (RLUs).
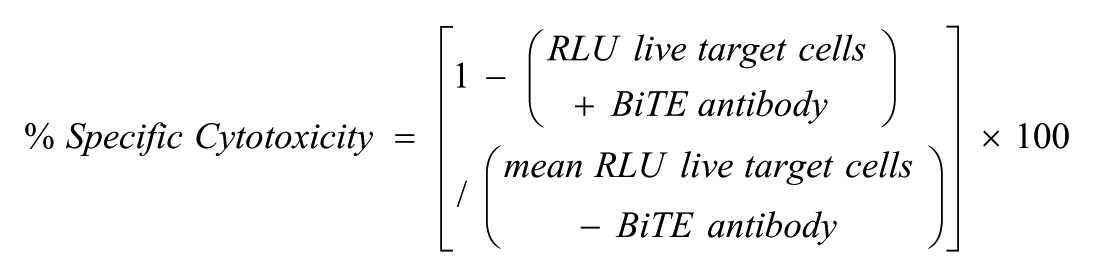
Results were reported as the mean EC50 inflection point (IP) value ± standard error of the mean. For EC50 determination, data were transformed in Microsoft Excel and analyzed with GraphPad Prism 6 software “log(agonist) vs. response—variable slope (four parameters)” or directly with Genedata Screener data analysis software with robust regression and outlier removal (ROUT fit method). The TDCC assay 384-well plate layout included 2 to 8 rows of replicates per plate and/or with duplicates on separate plates for each of the positive and negative control BiTE molecules. TDCC assays were performed with two different human T-cell donors and/or repeated on three independent days (n = 3). Signal-to-background (S/B) and Z’-factor (Z’) calculations were used to evaluate assay data quality and performance. 15
Results and Discussion
TDCC Assay Development
Developing a TDCC cell-based assay requires a number of considerations and criteria. Assay media selection is critical for ensuring proper cell viability, especially because two different cell types are involved (primary T-cells and target cell lines). The TDCC assay medium is suitable to maintain cell health and growth at an E–T ratio of 10:1 after being co-cultured for 48 h. We also evaluated pre-plating the targets cells overnight before addition of T-cells as well as plating the two cell types simultaneously on the same day, and we observed comparable results (
Figs. 1A
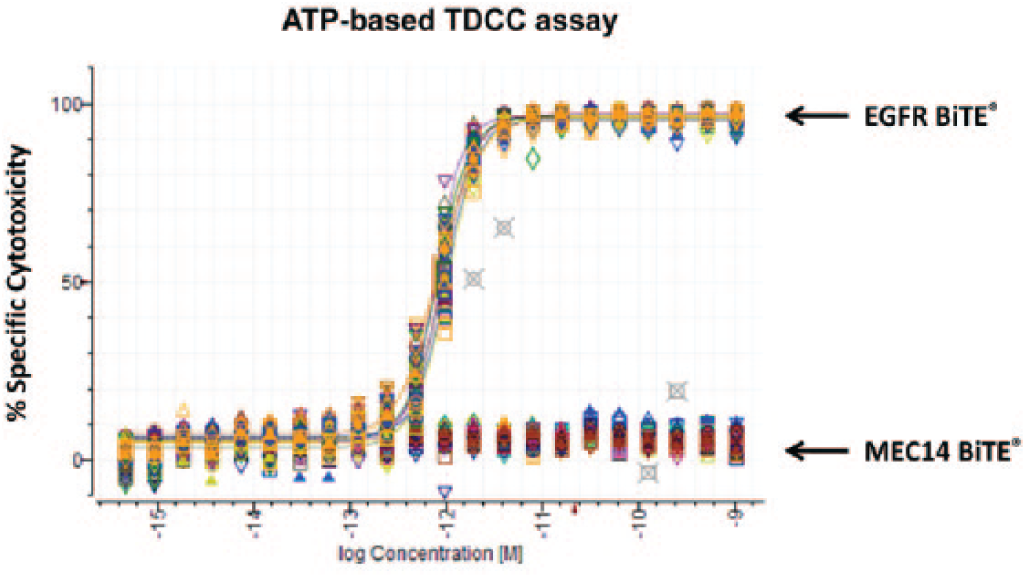
T-cell dependent cellular cytotoxicity (TDCC) assay process automation and performance in 384-well format. Adenosine triphosphate (ATP)-based TDCC assay performed with pre-plating of SW480 cells, followed by plating Donor 3809 unstimulated human T-cells [effector-to-target (E–T) ratio 10:1] the next day and Bispecific T-cell Engager (BiTE) antibody dilutions added without mixing. After 48 h incubation at 37°C, plates were washed with phosphate buffered saline to remove T-cells and dead target cells, and luminescence signal was detected with Cell Titer-Glo. (
TDCC Assay Process Automation and Performance in a 384-Well Format
Appropriate cell densities were determined (1200–3600 target cells/well) for 384-well plating, typically using 2500 target cells per well and 25,000 T-cells per well for a constant E–T ratio of 10:1. We confirmed that we could use either an automated liquid handler or bulk liquid dispenser to achieve uniform cell plating in 384-well plates, as well as automating plate washing for the ATP-based TDCC assay method. We improved assay capacity and precision by using a liquid handler for automating BiTE antibody serial dilutions in a 384-well format. Also, this was critical to improve ergonomics because tip changing between dilution steps is necessary to eliminate well-to-well carry-over of high-potency BiTE molecules. BiTE antibodies were added into assay-ready plates pre-loaded with cells. We compared the luminescence readout on multiple plate readers and selected the instrument that provided the sensitivity to measure a robust assay window with minimal cross-talk between wells. Luminescence detection requires less than 1 min per 384-well plate, which greatly improved data acquisition times compared with flow cytometry or imaging platforms.
ATP-Based TDCC Assay Performance
The performance of the 384-well TDCC assay with ATP-based readout was evaluated with the adherent SW480 colorectal cancer cell line with non-activated human pan T-cells (E–T of 10:1) and a BiTE antibody that targets EGFR and CD3 (positive control is expressed on cells) or a nontargeting irrelevant antibody MEC14 and CD3 (negative control is not expressed on cells). The target cells were pre-plated overnight with the T-cells, and BiTE antibody was added the following day. After preparing the TDCC assay with the pre-plating method, plates were then incubated for 48 h to allow for cytotoxic activity to reach steady-state kinetics and to achieve stable efficacy levels for dose–response determination. ATP-based TDCC assay results showed that the EGFR BiTE antibody potently kills SW480 target cells with non-activated pan human T-cells (EC50 = 0.886 ± 0.097 pM; n = 6, two replicate plates per day repeated on 3 independent days) and also demonstrated nearly complete efficacy of greater than 99% specific cytotoxicity at 1 nM, the highest BiTE antibody concentration tested ( Fig. 1A ). The MEC14 BiTE did not show any activity because this hapten target antigen is not expressed on SW480 cells. Good intraplate and interplate reproducibility was observed, and no significant variability was seen from different locations on the plate. Kinetics of cytotoxicity may vary with different target cells and especially at early time points before T-cells are activated; however, similar TDCC data quality and results were obtained when cytotoxicity was quantified after 48 h or after 72 h (data not shown). The automated ATP-based TDCC assay process offered reproducible potency and efficacy with adherent target cells.
Luciferase-Based TDCC Assay Development
Although the ATP-based TDCC assay was validated for adherent target cells, we also required a homogeneous TDCC assay protocol for suspension and semi-adherent target cell lines, which can be more difficult to work with for cell-based assays. Here, we describe the luciferase-based TDCC assay development using target cells with constitutively expressed luciferase to measure specific cytotoxic activity.11–14 The luciferase-based TDCC assay was prepared with simultaneous, same-day plating, and, more significantly, plate detection did not require a wash step.
Before performing TDCC assays, each target cell line was transduced with a constitutively expressed firefly luciferase reporter gene, and stable pools were selected based on luciferase expression. We determined that the luciferase signal was linear with cell number for each stable luciferase-expressing target cell pool and established that luciferase is a reliable method for quantifying cell viability. For example, OVCAR-8 and A2058 luciferase transduced stable pool were plated at various cell densities and demonstrated good linear correlation between luciferase signal and cell number (
Figs. 2A
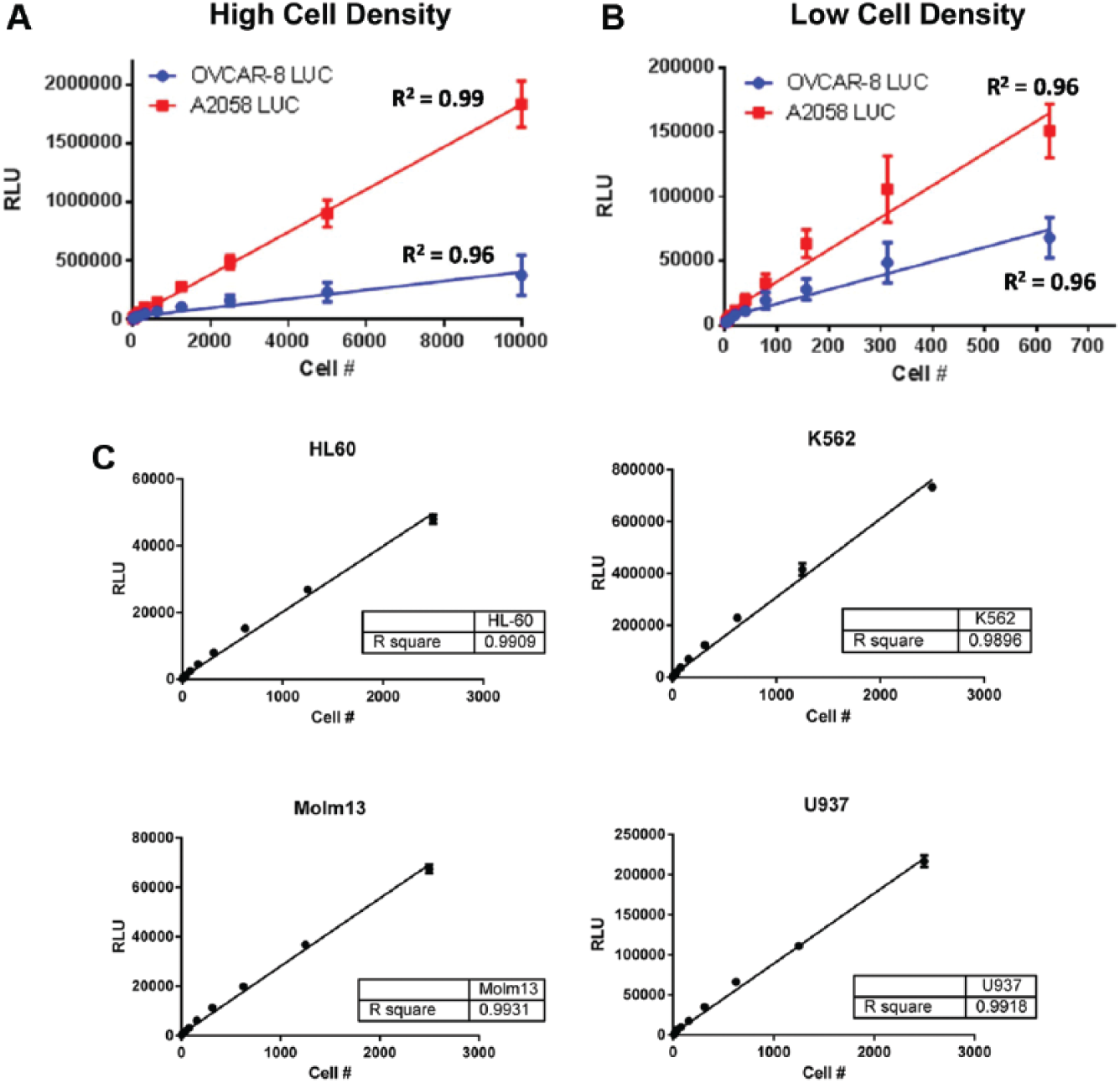
Luminescence quantitation has linear correlation with target cell number. Adherent OVCAR-8 luciferase (LUC) and A2058 LUC cells expressing firefly luciferase were (
Comparison of ATP-Based and Luciferase-Based TDCC Assay Methods
To compare assay performance between the ATP-based and luciferase-based detection methods side by side, the TDCC assays were prepared with the same assay components in duplicate 384-well plates. Specifically, the adherent SW480 luciferase-expressing stable pool cells were plated with unstimulated human T-cells (simultaneous plating, E–T 10:1). BiTE antibodies were added to the cells, and assay plates were incubated for 48 h at 37°C, 5% CO2. The key difference between methods was that the ATP-based (CellTiter-Glo) detection required that assay plates be washed beforehand to remove T-cells, whereas for the luciferase-based (Steady-Glo) detection assay plates were not washed. The TDCC assay results derived from the ATP-based or luciferase-based detection methods are shown to be highly comparable (
Figs. 3A
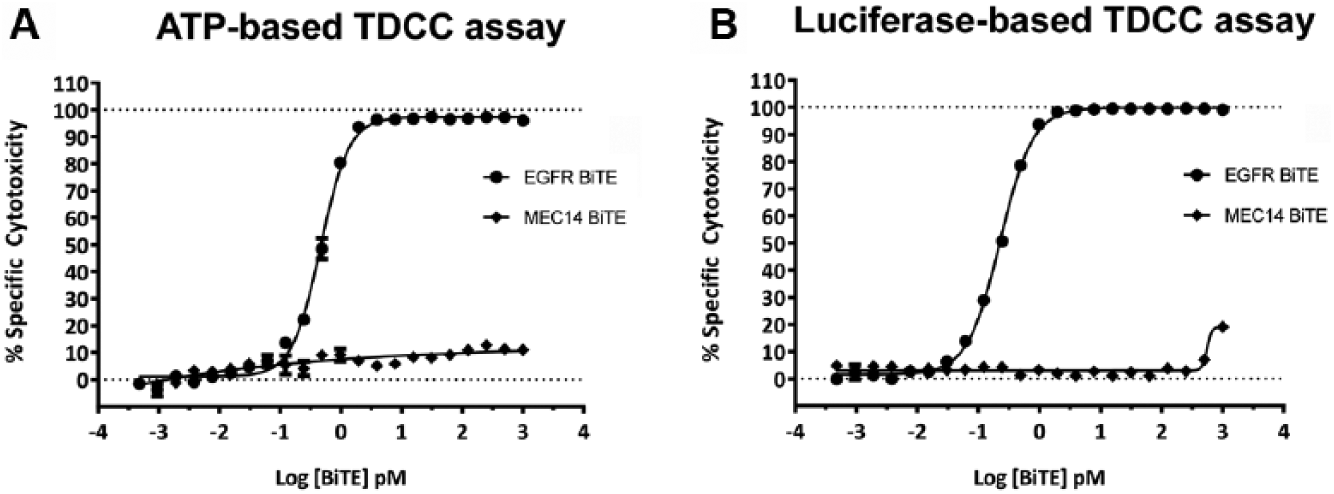
Comparison of T-cell dependent cellular cytotoxicity (TDCC) assay by various luminescence detection methods. Adenosine triphosphate (ATP)-based TDCC assay performed with same-day plating of SW480 cells expressing constitutive firefly luciferase with Donor 4217 unstimulated human T-cells [effector-to-target (E–T) ratio 10:1] and Bispecific T-cell Engager (BiTE) antibody dilutions added with mixing. After 48 h incubation at 37°C, plates were washed with phosphate buffered saline to remove T-cells and dead target cells, and luminescence signal was detected with Cell Titer-Glo. (
Luciferase-Based TDCC Assay Is Homogeneous and Luciferase Is Not in the Cell Supernatant
To ensure that the luciferase signal being detected is solely from within intact living target cells, we conducted experiments to demonstrate that luciferase is not present in the assay supernatant, perhaps as a result of being released from dead or dying target cells. The luciferase-based TDCC assay was prepared with adherent OVCAR-8 luciferase expressing stable pool cells and were plated with unstimulated human T-cells (simultaneous plating, E–T ratio 10:1). After 48 h, 50 µL of assay supernatant was removed from each well and was assayed for luciferase signal with Steady-Glo luminescence detection. Importantly, there were no significant levels of luciferase signal detected from the assay cell supernatant in wells treated either in the presence or in the absence of 1 nM EGFR BiTE (
Figs. 4A
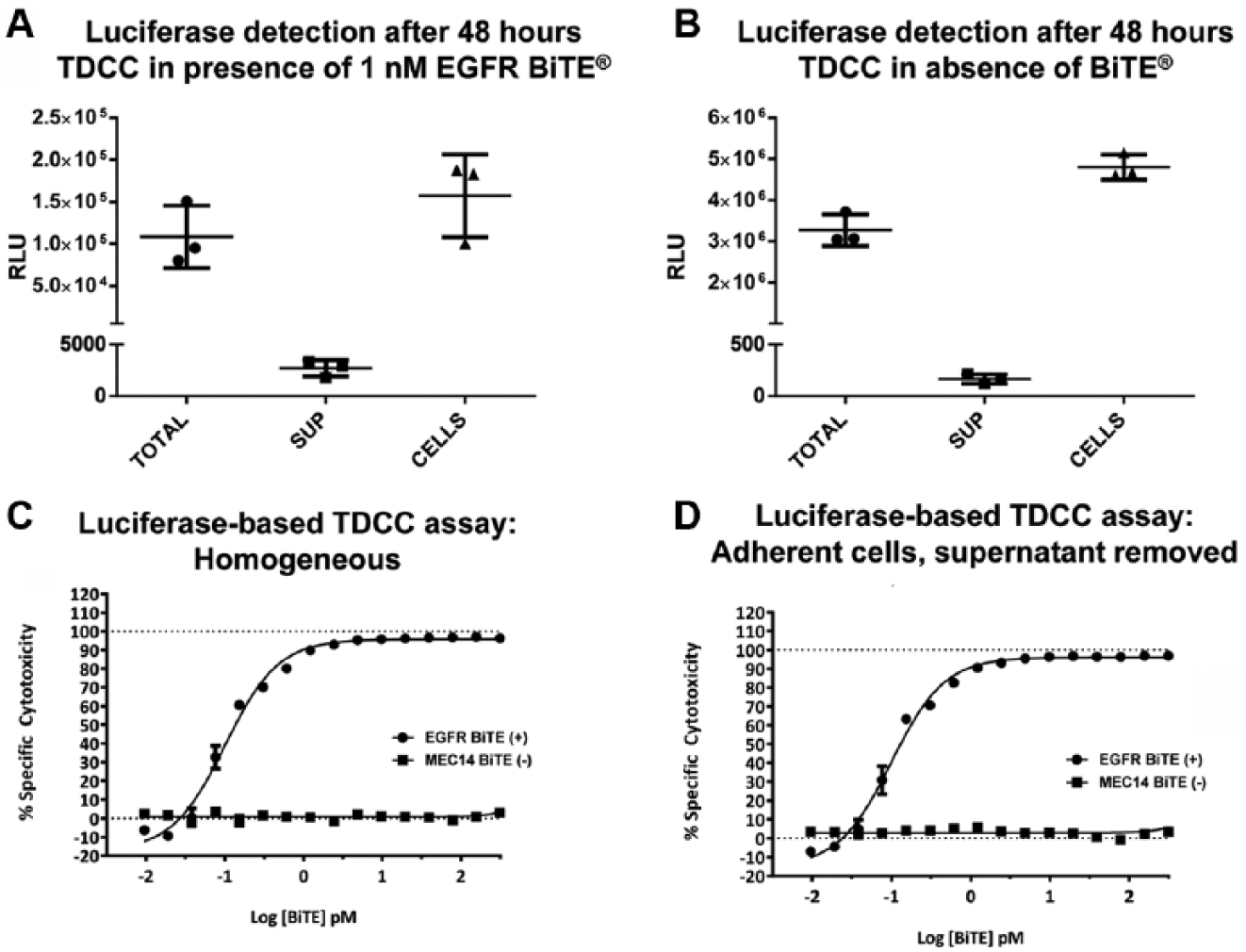
Firefly luciferase is not present in assay cell supernatant; luciferase-based T-cell dependent cellular cytotoxicity (TDCC) assay is homogeneous. Homogeneous luciferase-based TDCC assay performed with same-day plating of OVCAR-8 cells expressing constitutive firefly luciferase with Donor 5514 unstimulated human T-cells [effector-to-target (E–T) ratio 10:1] and Bispecific T-cell Engager (BiTE) antibody dilutions added with mixing. After 48 h incubation at 37°C, luminescence signal was detected with Steady-Glo. Mean results presented are from (
Luciferase-Based TDCC Screening Assays Identified Potent and Efficacious BiTE Antibodies against Human AML Cell Lines Expressing an Endogenous Tumor Target X
To characterize BiTE antibody molecules selectively targeting AML tumor cells, the luciferase-based TDCC assay method was applied to seven different human AML-derived suspension cell lines (i.e., U937, HL-60, PL-21, EOL-1, NOMO-1, HNT-34, and MOLM-13) and a target-negative Burkitt’s lymphoma cell line (Namalwa), and luciferase-expressing stable pools were generated. Target cell lines were evaluated by flow cytometry, and cell surface expression of target X was verified before and after luciferase stable pools were generated. The AML targeting BiTE antibody TDCC screening assays were performed with unstimulated human pan T-cells isolated from two donors. T-cell donors were characterized for off-target BiTE antibody independent cytolytic activity to avoid confounding TDCC assay screening results. Overall, unstimulated human donor T-cells did not show significant 37°C inhibitory effects with human target cells after 48 h of incubation at 37°C. To validate each particular target cell line with a particular T-cell donor, we evaluated target cell growth in the presence or absence of T-cells in the absence of BiTE antibody. Specifically, donor 4724 and donor 5514 T-cells demonstrated little to no significant effect on the survival and/or growth of most human tumor cell lines that were used for screening; target + effector T-cell / target only (T + E / T only) = 77–100%.
The AML target X TDCC screening assays demonstrated reproducible and robust data quality with large assay windows (average S/B = 465-fold, Z’ = 0.821) for 20 total 384-well plates that were assayed. We identified four BiTE antibodies against an undisclosed target X for the AML indication with potent low picomolar TDCC activity and high maximum specific cytotoxicity (EC50 = 1 − 10 pM; 99.9% maximum activity) across a panel of multiple human AML cell lines expressing an endogenous tumor target with multiple T-cell donors ( Fig. 5A ). Conversely, there was no specific TDCC activity determined (EC50 undefined) against a target-negative Namalwa cell line, which does not express the endogenous tumor target ( Fig. 5B ). Data were consistent with a positive control CD33 and CD3 targeting BiTE antibody and an irrelevant control EGFR and CD3 targeting BiTE antibody. We later observed additional confirmation of the luciferase-based TDCC assays’ precision; EC50 values for new protein lots of lead BiTE antibodies were determined to be nearly equivalent to the original protein lots several months after initial screening. Luciferase-based TDCC assay results were considered valid and equivalent to data acquired using a flow cytometry–based TDCC assay, which was the alternative detection method available for TDCC assessment with suspension target cell lines. The development of a homogeneous and high-capacity luciferase-based TDCC assay platform allowed for the more rapid assessment of the AML targeting BiTE molecules than would have occurred with the flow cytometry–based method.
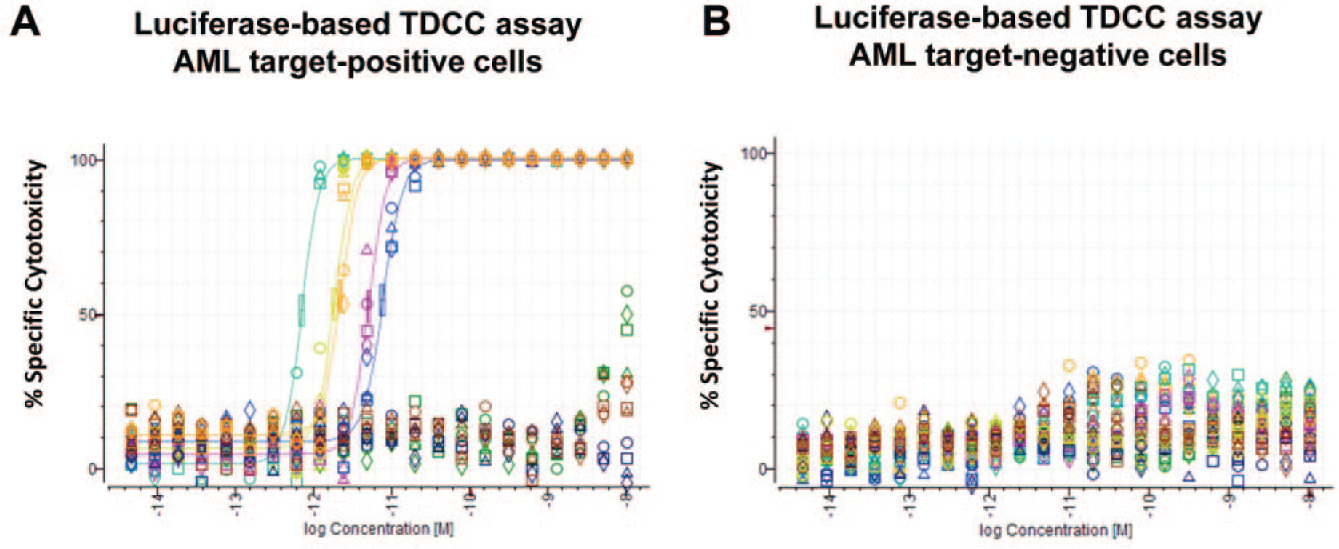
Luciferase-based T-cell dependent cellular cytotoxicity (TDCC) assays identified highly potent and fully efficacious Bispecific T-cell Engager (BiTE) antibodies against human acute myeloid leukemia (AML) cell lines expressing endogenous tumor target X. Homogeneous luciferase-based TDCC assays performed with same-day plating of PL-21 and Namalwa cells expressing constitutive firefly luciferase with Donor 4274 unstimulated human T-cells [effector-to-target (E–T) ratio 10:1] and BiTE antibody dilutions added with mixing. After 48 h incubation at 37°C, luminescence signal was detected with Steady-Glo. Representative dose–response curves for (
High-Capacity Luciferase-Based TDCC Assay Platform Established for Screening of BiTE Antibodies
The luciferase-based TDCC assay method using target cells constitutively expressing luciferase was developed to ensure precise and accurate measurements of in vitro cellular potency and efficacy of BiTE antibodies. The established luciferase-based TDCC process is being used for the rapid screening of novel immunotherapeutic BiTE antibody candidates targeting various indications. One potential limitation of this method is that it requires additional upstream time to generate stable luciferase-expressing target cells, although time is recovered later when performing homogeneous detection without the plate washing.
Typically, BiTE antibody TDCC screens consist of evaluating the activity of BiTE molecules against at least five different target cell lines and T-cells from at least two different donors. A summary of the luciferase-based TDCC assays performed to screen BiTE antibody molecules for various cancer target indications suggests a very robust cell-based assay platform with a large average assay window (S/B = 121-fold) ( Table 1 ). Moreover, the average Z’-factor during screening (Z’ = 0.738) was excellent and even more so considering that the TDCC assay involves two different cell types being co-cultured for 48 h. Observed potencies for the various tool BiTE antibodies were in the subpicomolar to high-picomolar range for each target indication. Overall, this BiTE antibody characterization workflow described here allowed for a broad evaluation of BiTE antibody activity before lead molecule selection for in vivo assessment. Molecules were rapidly assessed with the luciferase-based TDCC assay platform and reduced to a more manageable number before additional characterization in cytokine release assays or confocal imaging studies to determine cytolytic synapse formation. Additional experiments to determine the BiTE antibody mechanism of action as well as the kinetics of BiTE antibody activity can also be assessed with label-free and imaging-based methods.
Screening Performed with the Luciferase-Based TDCC Assay Identified Potent and Efficacious BiTE Antibodies against Various Cancer Targets.
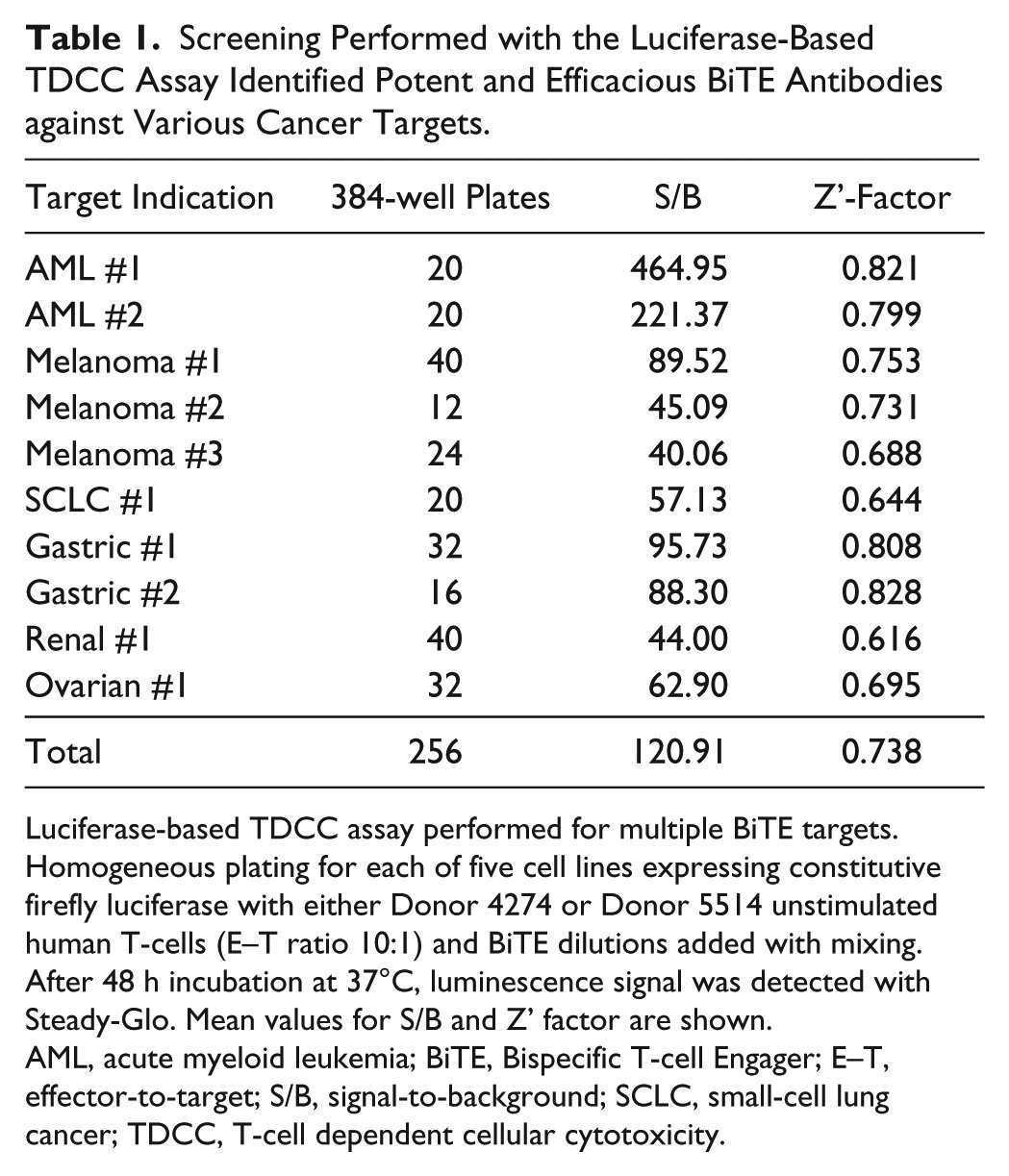
Luciferase-based TDCC assay performed for multiple BiTE targets. Homogeneous plating for each of five cell lines expressing constitutive firefly luciferase with either Donor 4274 or Donor 5514 unstimulated human T-cells (E–T ratio 10:1) and BiTE dilutions added with mixing. After 48 h incubation at 37°C, luminescence signal was detected with Steady-Glo. Mean values for S/B and Z’ factor are shown.
AML, acute myeloid leukemia; BiTE, Bispecific T-cell Engager; E–T, effector-to-target; S/B, signal-to-background; SCLC, small-cell lung cancer; TDCC, T-cell dependent cellular cytotoxicity.
We have established a high-capacity, homogeneous, luminescence-based TDCC assay that quantifies live target cells by measuring constitutively expressed luciferase. A robust TDCC assay platform with high data quality and high capacity for biologics assessment is a key driver of rapid drug discovery. A homogeneous, 384-well assay format allows for less time in the lab performing experiments as well as improving data quality. We demonstrated the value of incorporating laboratory automation to enable serial dilutions of biologics samples in a 384-well format. We enabled biologics high-throughput screening (HTS) further by using Genedata Screener data analysis informatics tools as with small-molecule HTS drug discovery. The luciferase-based TDCC assay platform will continue to drive forward high-quality immunotherapeutic molecules into preclinical development.
Footnotes
Acknowledgements
We thank members of Amgen’s Therapeutic Discovery, Oncology Research, Amgen Research Munich, Discovery Technologies, and particularly the BiTE assay working group for contributions to the discovery research BiTE platform. We thank Hong Ma and Cherylene Plewa for providing luciferase transduced human cancer cell lines. We thank Nilo Tayag for cell culture support. We thank Joe McGivern and Mark Sandberg for careful review of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.