
Abstract
Sirtuins are NAD+-dependent histone deacetylases (HDACs) that cleave off acetyl but also other acyl groups from the ϵ-amino group of lysines in histones and other substrate proteins. Five sirtuin isoforms are encoded in the genome of the parasitic pathogen Schistosoma mansoni. During its life cycle, S. mansoni undergoes drastic changes in phenotype that are associated with epigenetic modifications. Previous work showed strong effects of hSirt2 inhibitors on both worm life span and reproduction. Thus, we postulate smSirt2 as a new antiparasite target. We report both the optimization of a homogeneous fluorescence-based assay and the development of a new heterogeneous fluorescence-based assay to determine smSirt2 activity. The homogeneous assay uses a coumarin-labeled acetyl lysine derivative, and the heterogeneous version is using a biotinylated and fluorescence-labeled oligopeptide. Magnetic streptavidin-coated beads allow higher substrate loading per well than streptavidin-coated microtiter plates and make it possible to screen for inhibitors of either smSirt2 or its human isoform (hSirt2) for selectivity studies. We also present hits from a pilot screen with inhibitors showing an IC50 lower than 50 µM. Binding of the hits to their targets is rationalized by docking studies using a homology model of smSirt2.
Introduction
Epigenetics is the study of inheritable changes in gene expression without any changes in underlying DNA sequence. Major epigenetic mechanisms are alterations of the methylation state of the DNA base cytosine, the interaction of chromatin with small noncoding RNAs (ncRNAs), and posttranslational histone modifications. The latter mainly occur at the basic amino acids lysine and arginine of unstructured N-terminal histone tails that protrude out of the histone octamer. 1 Threonine or tyrosine residues and amino acids of the nucleosomal core, however, can be modified as well. 2 The different modifications are diverse, and in their manifold combinations they generate the epigenetic code. The epigenetic code is much more complex than the genetic code, and it is far from being deciphered completely. Nevertheless, specific modifications, or a combination of single modifications, can be associated with a well-defined change in gene expression. Chemical groups that can be transferred to the above-mentioned sites are the methyl, acetyl, and phosphoryl groups, as well as bigger molecules [e.g., ubiquitin, fatty acids, and adenosine diphosphate (ADP)-ribose]. 3 Enzymes that catalyze the transfer of these groups to the N-terminal histone tails are called writer enzymes. They take part in a dynamic interplay with eraser enzymes, which are able to restore the unmodified state.
Sirtuins belong to the class of eraser enzymes and represent a specific NAD+-dependent class of histone deacetylases (HDACs), also referred to as class III HDACs. These enzymes catalyze the deacetylation of acetylated ϵ-amino groups of lysines in histones and other proteins such as p53, tubulin, FOXO proteins, and p300. 4 Because an increasing number of nonhistone substrates have been discovered in recent years, it is more accurate to term HDACs as protein deacetylases, and maybe the sirtuins are more signal transduction targets than epigenetic modifiers, at least in the human context. Sirtuins have been found in species from bacteria to humans in a highly conserved manner. The hallmark of the family is a domain of approximately 260 amino acids with a high degree of sequence similarity in all sirtuins. 5 Sirtuins can be grouped into 5 classes. The 7 mammalian sirtuins belong to classes I–IV, whereas sirtuins of the fifth class, termed class U, are present in only bacteria and archaea. 6 Human sirtuins (hSirt1–7) differ in their subcellular localization and their catalytic activity. Sirtuins can act as amidohydrolases and/or mono-ADP-ribosyltransferases. hSirt1 shows a nuclear and cytosolic localization, and deacetylates histones as well as transcription factors (e.g., p53 and FoxO). 4 hSirt2 is a cytosolic protein 7 with a major impact on cell cycle regulation and apoptosis by deacetylating substrates like α-tubulin, 8 p53, 9 and BubR1. 10 The mitochondrial sirtuins are hSirt3–5. hSirt4, for example, downregulates glutamine dehydrogenases by mono-ADP-ribosylation. 11 Moreover, hSirt5 controls the activity of carbamoyl-phosphate synthase 1 (CPS-1) via desuccinylation. 12 This underlines the effect of mitochondrial sirtuins on cellular metabolism. hSirt6 and hSirt7 are located exclusively in the nucleus. hSirt6 has been reported to have only low deacetylase activity, whereas it efficiently hydrolyzes long-chain fatty acyl groups from ϵ-amino groups of lysine residues. 13 The sirtuin PfSir2A from Plasmodium falciparum also hydrolyzes long-chain acid amides. 14
Keeping the variety of sirtuin substrates and the effects of substrate conversion in mind, such as regulation of apoptosis, the cell cycle, metabolism, and aging, it is obvious that sirtuins have emerged as potential drug targets in recent years. Although a remarkable number of sirtuin inhibitors have been discovered, there is still a need for subtype-selective and drug-like sirtuin inhibitors. 15 To date, there is only one compound, Selisistat (or SN196), which is a hSirt1 inhibitor, in Phase II clinical trials for Huntington’s disease. 16 Nevertheless, the biological effects of sirtuin inhibitors highlight these inhibitors’ potential, especially in cancer therapy.15,17 The use of epigenetic drugs as anti-infectives is a rather new approach that is particularly usable on eukaryotic parasites due to their chromosomal genome organization. This anti-infective strategy is even more promising for metazoan parasites, which undergo drastic changes in phenotype during their life cycle. Changes in phenotype without an alteration of genotype are caused by epigenetic modifications. An interruption of the fine-tuned epigenetic interplay should have significant effects on parasites that are at the point of switching phenotype, whereas host cells may remain rather unaffected. The development of species-selective modulators of epigenetic regulators, however, should be the main goal of epigenetic drug discovery in the field of anti-infectives. Because of a high sequence homology of epigenetic regulators (e.g., HDACs from protozoa to humans), this is quite challenging. Nevertheless, Marek et al. identified small-molecule inhibitors of the Schistosoma mansoni HDAC8 (smHDAC8) with a reduced affinity for human HDACs by a structure-based approach. Crystal structures of smHDAC8 with generic HDAC inhibitors reveal specific structural changes in the active site on ligand binding, which cannot be accommodated by human HDACs. These species-selective changes were exploited to develop the smHDAC8 inhibitor J1075, which is capable of inducing apoptosis and mortality in schistosomes. 18 Another example for a smHDAC8 selective inhibitor is the thiol-based inhibitor discovered by Stolfa et al. 19 Schistosomal histone acetyltransferases SmGCN5 and SmCBP1 have been identified as potential drug targets as well. 20 The blood fluke S. mansoni is the main pathogen of schistosomiasis, a disease affecting more than 200 million people in tropical and subtropical countries. Due to the absence of a vaccine, praziquantel is currently the only way of controlling schistosomiasis, and it is distributed mainly through mass administration programs to millions of people every year. Even though praziquantel shows a number of positive features, especially with regard to safety, efficacy, cost, and ease of distribution, a major problem is its lack of efficacy against the immature stages of the parasite. Also, in view of praziquantel’s massive and exclusive use on a large number of individuals, the development of drug resistance is a much-feared possibility. Furthermore, the mechanism of action is still unclear, which does not favor the development of derivatives or alternatives. A large number of compounds have been tested as potential antischistosomal drugs, some with promising results (e.g., oxamniquine, derivatives of antimalarial drug artemisinin, furoxan, and the kinase inhibitor imatinib), but none so far represents a suitable substitute for praziquantel.21,22 The current situation emphasizes the urgent need for novel therapeutic strategies to discover new antischistosomal agents. To evaluate schistosomal sirtuins and their potential as therapeutic drug targets, Lancelot et al. identified and characterized 5 sirtuins encoded in the S. mansoni genome and established their homology relationship to human isoforms. Phylogenetic analysis showed that they are orthologs of mammalian Sirt1, Sirt2, Sirt5, Sirt6, and Sirt7. Initial experiments with adult worms and schistosomula showed strong effects of hSirt2 inhibitors on both life span and reproduction. 23 In this study, we present two novel complementary in vitro assays to determine smSirt2 activity as well as hits from our screening campaign with IC50 values lower than 50 µM. The heterogeneous assay protocol was transferred to and optimized for hSirt2 as well.
Materials and Methods
Proteins
A truncated version of smSirt2 (smST1-N1C1; amino acid residues 21 to 322) was amplified using the forward primer smSirT2-N1 (5′-GGATATCCATATGGAACTTAAGTCTCTCAATATAGAG-3′) and the reverse primer smSirT2-C1 (5′-CGCGGATCCTCAATTTAAGCGAGAGTCCGT TTC-3′) inserted into the pnEA-tH vector.
24
Overexpression was carried out in Escherichia coli BL21(DE3) cells containing the pRARE vector (Novagen, Darmstadt, Germany) in lysogeny broth medium. Induction was done at 25 °C by adding isopropyl-1-thio-β-D-galactopyranoside (IPTG; Euromedex, Souffelweyersheim, France) in a final assay concentration of 0.5 mM, in the presence of 100 μM ZnCl2. Harvested bacteria were resuspended in lysis buffer (10 mM Tris-HCl, 400 mM NaCl, pH 8.0) and lysed by sonication. The lysate was clarified by centrifugation at 17,000 rpm for 1 h. The supernatant was loaded onto Talon Metal affinity resin (Clontech, Mountain View, CA) pre-equilibrated with the lysis buffer. The 3C Protease treatment was used to remove His-tags from the recombinant protein, which was subsequently loaded onto a HiLoad 16/60 Superdex 200 gel filtration column (Amersham Bioscience, Amersham, Little Chalfont, UK) equilibrated in 400 mM NaCl, 10 mM Tris-HCl pH 8.0, and 2 mM DTT. Finally, the protein was concentrated with Amicon Ultra centrifugal filter units (Millipore, Billerica, MA) to reach a final concentration of 1.5 mg/mL as assayed by the Bio-Rad Protein Assay reagent (Bio-Rad, Hercules, CA). The Coomassie-stained sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel of the purified smSirt2 protein and the chromatogram of the size-exclusion chromatography are depicted in
Inhibitors
The Sirt2 reference inhibitor nicotinamide and Tyrphostin AG-825 were purchased from Sigma-Aldrich, Vatalanib 2HCl was purchased from Selleckchem (Houston, TX), and resveratrol from TCI (Tokyo, Japan). PU92, 27 MS3, 28 and MS1028 were synthesized in our lab according to literature procedures.
Homogeneous Fluorescence-Based Assay for smSirt2
To determine smSirt2 activity, we optimized a homogeneous fluorescence-based deacetylase assay, which was developed in our group for human sirtuins. 29 The assay was performed in black 384-well nonbinding plates (Greiner Bio-One, Monroe, NC) with a reaction volume of 20 µL per well. All tests were performed at least in duplicate using Z-(Ac)Lys-AMC (ZMAL) as a substrate. ZMAL was synthesized according to published procedures. 29 14 µL SmSirt2 (86 ng/µL, final assay concentration 60 ng/µL) in assay buffer (50 mM Tris, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.015% Triton X-100, pH 8.0) was mixed with 2.5 µL of a ZMAL solution [prepared from ZMAL stock solution (12.6 mM) in DMSO diluted with assay buffer; final assay concentration, 10.5 µM], and the inhibitor dissolved in DMSO at various concentrations or DMSO as a control [final DMSO concentration, 5% (v/v)]. Deacetylation was started by addition of NAD+ (final assay concentration, 500 µM). After an incubation of 1 h at 37 °C and 140 rpm, deacetylation was stopped by adding 20 µL of a solution containing trypsin and nicotinamide [50 mM Tris, 100 mM NaCl, 6.7% (v/v) DMSO, trypsin 1 mg/mL, 8 mM nicotinamide, pH 8.0]. The microplate was further incubated for 20 min at 37 °C and 140 rpm. Finally, fluorescence intensity was measured in a microplate reader (BMG Polarstar; λEx = 390 nm, λEm = 460 nm; BMG Labtech, Ortenberg, Germany). Rates of inhibition were calculated using the controls, containing no inhibitor, as a reference for 0% inhibition. The value for 100% inhibition was generated by blank controls containing only ZMAL, NAD+, DMSO, and assay buffer. Graphpad Prism software (Graphpad Software, La Jolla, CA) was used to determine IC50 values.
Heterogeneous Fluorescence-Based Assay for smSirt2
The heterogeneous fluorescence-based assay for smSirt2 was performed at least in duplicate in Hard-Shell low-profile thin-wall 96-well skirted PCR plates (Bio-Rad) using Ac-Glu-Glu-Lys(biotin)-Gly-Gln-Ser-Thr-Ser-Ser-His-Ser-Lys(Ac)-Nle-Ser-Thr-Glu-Gly-Lys(5/6-carboxytetra methylrhodamine)-Glu-Glu-NH2, hereafter called Ac-p53-TAMRA, as a substrate (final assay concentration, 250 nM). 30 It was custom synthesized (PSL, Heidelberg, Germany). The reaction volume was adjusted to 50 µL per well. Twenty microliters of SmSirt2 (250 ng/µL, final assay concentration 100 ng/µL) in assay buffer (50 mM Tris, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, pH 8.0) were mixed with inhibitor dissolved in DMSO at various concentrations or DMSO as a control [final DMSO concentration, 5% (v/v)]. The enzymatic reaction was started by adding 10 µL of start solution (1.25 µM Ac-p53-TAMRA, 750 µM NAD+ in assay buffer). Then, the plate was covered with a TopSeal-A cover (PerkinElmer, Waltham, MA) and incubated for 2 h at 37 °C and 140 rpm. Deacetylation was stopped by adding 20 µL of a stop solution containing trypsin and nicotinamide [50 mM Tris, 100 mM NaCl, 11.7% (v/v) DMSO, trypsin 35 µg/mL, 14 mM nicotinamide, pH 8.0]. The reaction mixture was incubated for 20 min at 37 °C and 140 rpm. The next step consisted of the addition of streptavidin-coupled magnetic beads (Dynabeads MyOne™ Streptavidin T1, 10 mg/mL stock solution; Life Technologies, Carlsbad, CA), 10 µg per well (1 µL bead stock solution), suspended in 30 µL assay buffer, and a further incubation of 20 min at 37 °C and 140 rpm. Afterward, the microplate was placed on a DynaMag™-96 side magnet (Life Technologies, Carlsbad, CA) for 3 min to pull the magnetic beads to the side of the PCR tubes. The supernatant was discarded, and the beads were washed with washing buffer [80 mM Tris, 20 mM Tris-HCl, 150 mM NaCl, Tween-20 0.1% (v/v), pH 7.5, 80 µL per well, 4 times]. Subsequently, the magnet was removed, and the samples were resuspended in 80 µL washing buffer and transferred to a black 96-well plate (OptiPlate-96 F, PerkinElmer). Fluorescence intensity was measured in a microplate reader (EnVision; PerkinElmer; λEx = 531 nm, λEm = 591 nm). Rates of inhibition were determined by using controls, containing no inhibitor, as a reference for 0% inhibition. The top of the assay window was defined by samples containing 750 µM nicotinamide as a point of reference for 100% inhibition. Graphpad Prism software (Graphpad Software) was used to determine IC50 values.
Heterogeneous Fluorescence-Based Assay for hSirt2
To determine hSirt2 activity in a heterogeneous fluorescence-based assay, we used the same protocol as we used for smSirt2 with minor modifications. hSirt2 was adjusted to a final assay concentration of 38 ng/µL, and incubation time of deacetylation was prolonged to 2.5 h.
Counterscreening for Trypsin Inhibition
Because the presented assay protocols rely on trypsin cleavage of the converted substrate, a counterscreening for trypsin inhibition was performed (see Suppl. Material).
Km Determination
For the characterization of substrate affinity, Km values for both substrates were determined (see Suppl. Material).
Z-Factor (Z’-Factor) Measurement
Z-factors (Z’-factors) were determined by measurements of signals for 0% and 100% inhibition (n = 24). 31
Computational Studies
See supplementary material.
Results
Screening Procedure
For this initial screen, we preselected known sirtuin inhibitors of the human isoforms, nicotinamide analogs and kinase inhibitors from our internal libraries. Nicotinamide analogs were selected due to their similarity to the endogeneous sirtuin inhibitor nicotinamide. Kinase inhibitors were tested because adenosine triphosphate mimetics could fit in the NAD+-binding pocket of sirtuins as well, because both enzymes use cofactors that bear an adenosine moiety.
32
In the initial screen, all compounds were tested at concentrations of 20 and 100 µM. Because this is the first inhibitor screen with smSirt2, we initially chose a rather high threshold to definitely identify potential inhibitors. Hits of initial screening (inhibitor @ 100 µM > 50%) from the homogeneous fluorescence-based assay for smSirt2 were tested for trypsin inhibition (
Homogeneous Fluorescence-Based Assay for smSirt2
To determine smSirt2 activity, we optimized a homogeneous fluorescence-based deacetylase assay, which was developed in our group for human sirtuins.
29
Under assay conditions, which were used for the human isoform, smSirt2 showed less conversion of the substrate ZMAL. In the course of a buffer optimization, we identified Triton X-100 to have beneficial effects on smSirt2 activity (
Fig. 1A
). Triton X-100 is a nonionic surfactant that prevents adsorption of proteins to the assay plate surface. We adjusted a final assay concentration of 0.015% (v/v) to take full advantage of the surface-blocking capability without exceeding the critical micellar concentration of 0.0155%. By adding Triton X-100 to the assay buffer and a miniaturization of assay volume, we were able to reduce enzyme consumption per measurement by a factor of 50. A Km value of 59 µM was determined, and the ZMAL concentration was adjusted to 10.5 µM to be sensitive to substrate competitive inhibitors (
Fig. 1B
). Measuring enzyme kinetics enabled us to reduce the incubation time from initially 4 h to 1 h (
Fig. 1C
). With 1 h of incubation, we are already at the upper limit of linearity. We use these conditions to maximize the signal window, however. In
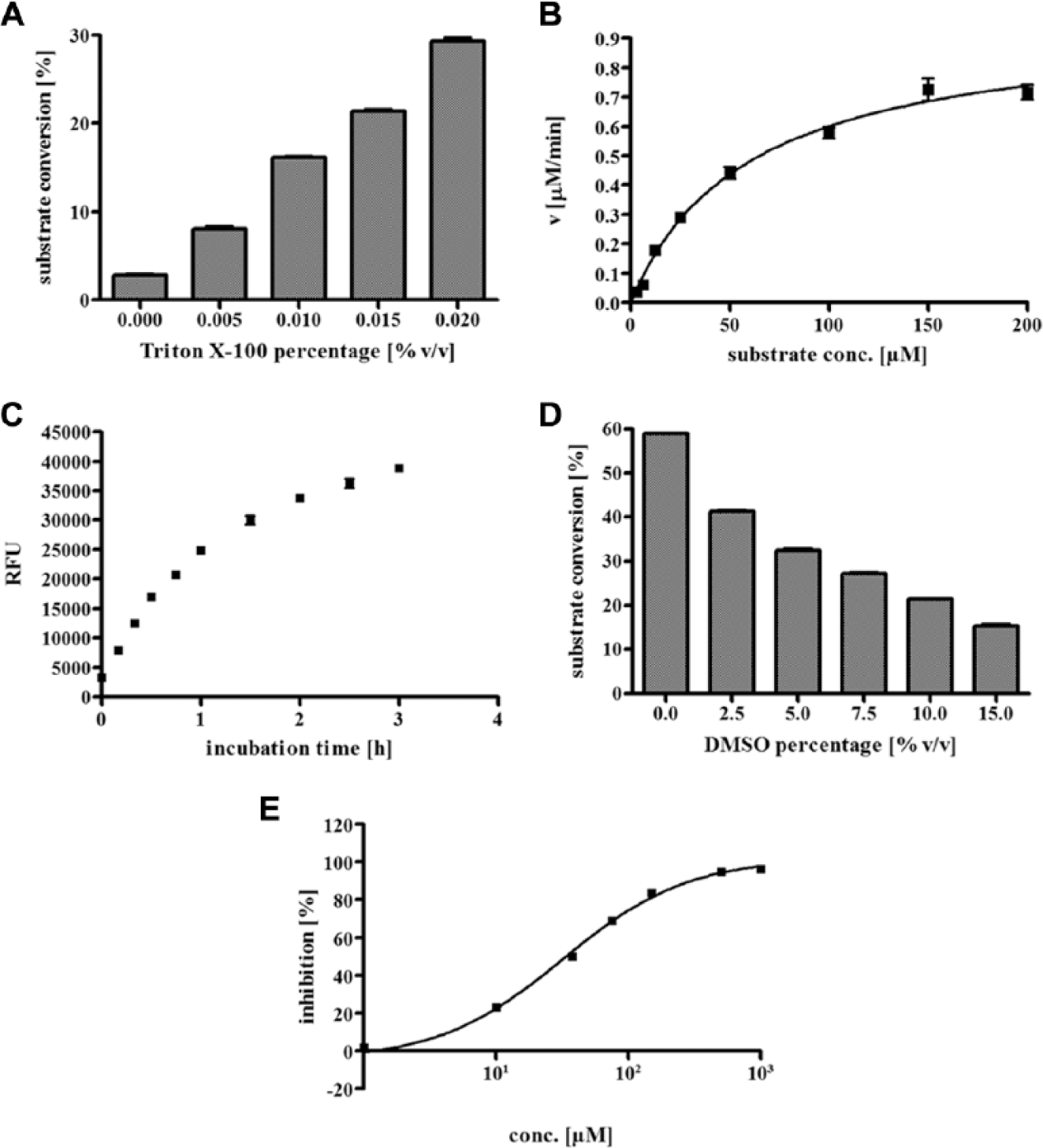
Assay development of homogeneous fluorescence-based assay for smSirt2. (
Inhibition Results (IC50 Values ± Standard Deviation and Corresponding Hill Slope) in Relation to Compound Fluorescence.
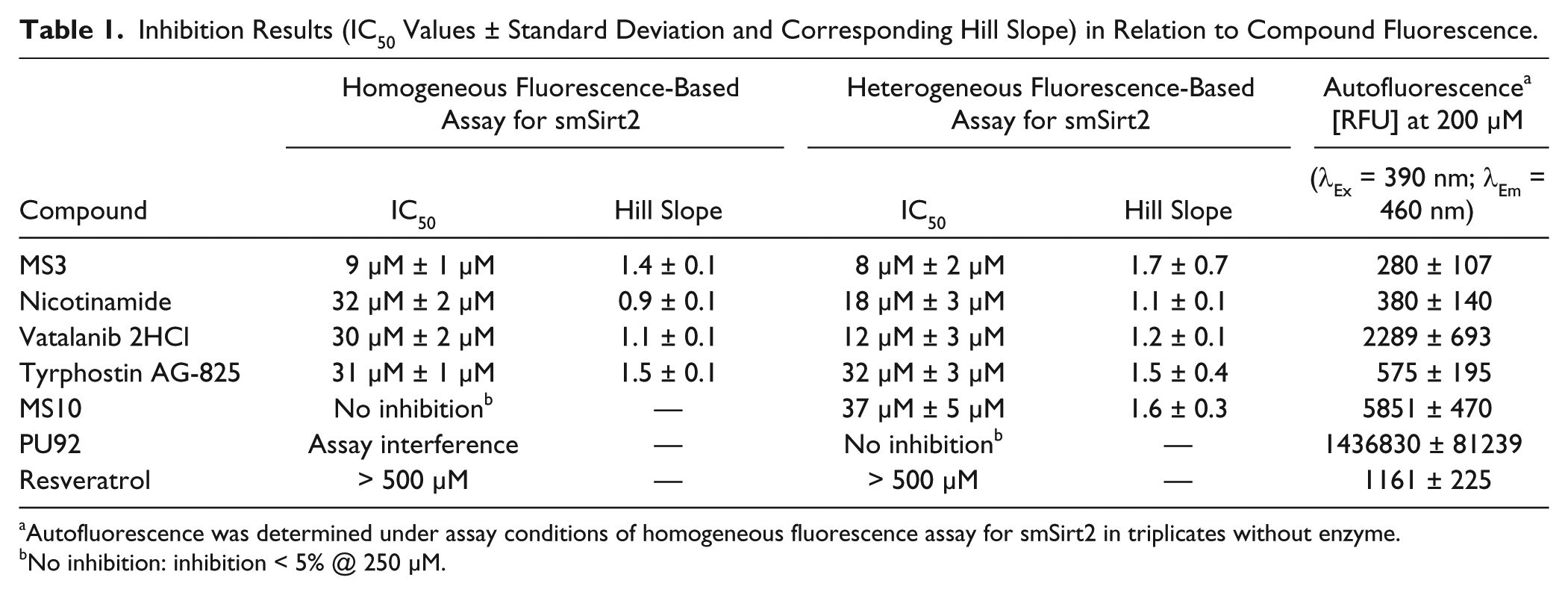
Autofluorescence was determined under assay conditions of homogeneous fluorescence assay for smSirt2 in triplicates without enzyme.
No inhibition: inhibition < 5% @ 250 µM.
Heterogeneous Fluorescence-Based Assay for smSirt2
By means of our novel homogeneous fluorescence-based assay, we are not able to evaluate the inhibitory effects on smSirt2 activity of library compounds showing high autofluorescence or quenching effects under given assay conditions. Therefore, we developed a heterogeneous fluorescence-based assay for smSirt2 that is not prone to assay interference by compound properties. As a substrate, we choose Ac-p53-TAMRA, a 20–amino acid substrate derived from the sequence of p53, which is a substrate for both hSirt1 and hSirt2. 30 The peptide is N-terminally linked to biotin and C-terminally modified with a fluorescent rhodamine-derived tag. These modifications allow immobilization on streptavidin-coated surfaces via biotin and fluorescence detection due to the fluorescence label TAMRA. The reaction for monitoring enzyme activity is a coupled enzymatic reaction. The first reaction, the deacetylation reaction of Lys(Ac)-12, catalyzed by smSirt2, is detected by coupling to a cleavage at the newly exposed lysine residue by the endopeptidase trypsin. 30 Finally, after washing away all unbound assay components, the amount of remaining uncleaved but still biotinylated and TAMRA-labeled substrate is determined by measuring fluorescence intensity ( Fig. 2 ).
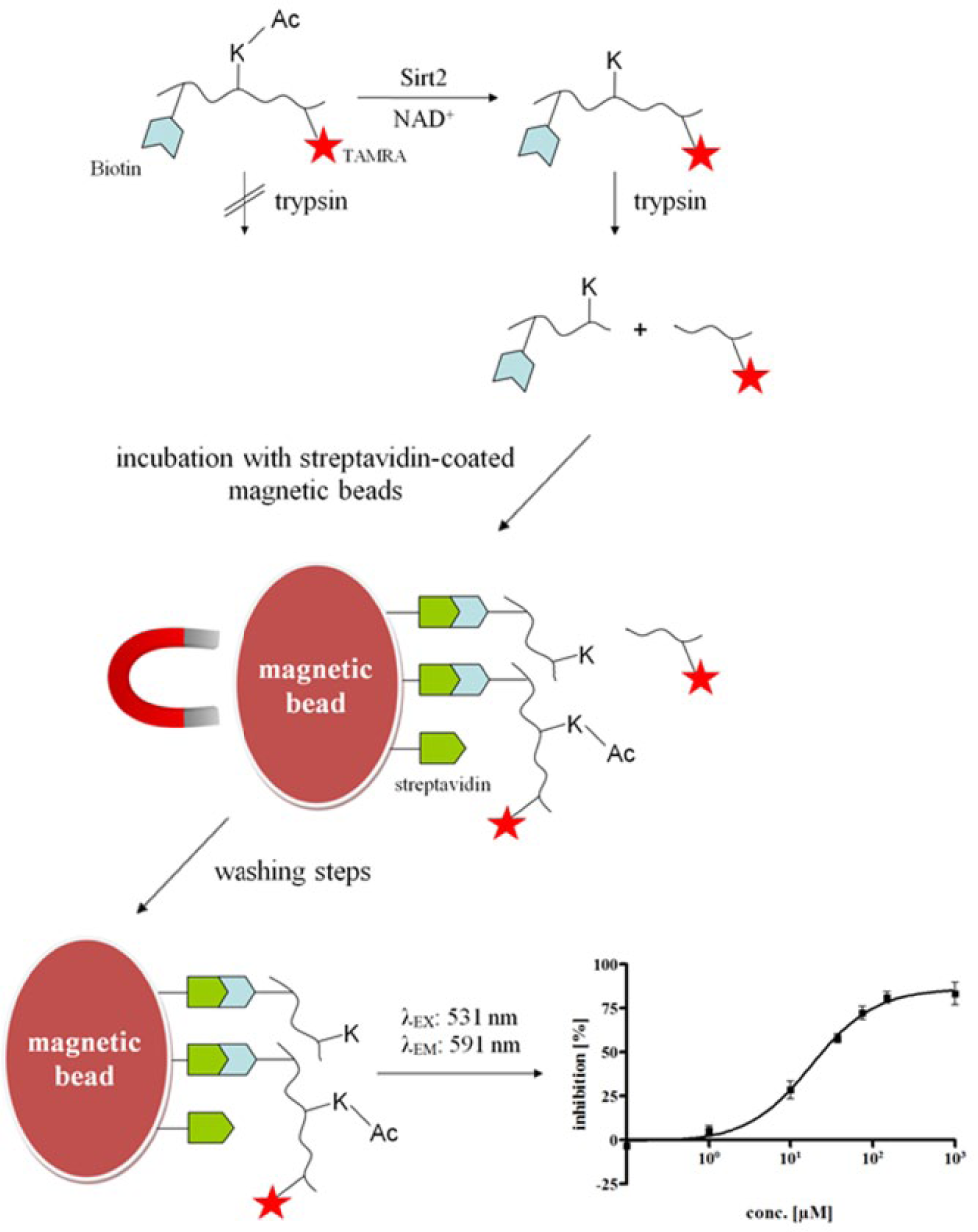
Schematic illustration of the heterogeneous fluorescence-based assay for smSirt2, respectively hSirt2. The sirtuin of interest is incubated with substrate and cofactor and inhibitor, or DMSO for controls. Substrate Ac-p53-TAMRA is deacetylated by active sirtuin. In a second step, the endopeptidase trypsin is added, which can only cleave deacetylated substrate molecules. Then, the streptavidin-coated magnetic beads are added to bind the biotinylated peptides. The samples are placed in a magnetic field to pull the magnetic beads to the sides of the reaction vessel. The supernatant, including all unbound assay components, gets discarded, and the beads are washed with washing buffer. Afterward, the magnet is removed, and the beads are resuspended in washing buffer and transferred to a 96-well plate. Finally, the amount of remaining uncleaved but still biotinylated and TAMRA-labeled substrate is determined by fluorescence readout (λEx = 531 nm; λEm = 591 nm).
Initial experiments with streptavidin-coated microplates showed that Ac-p53-TAMRA is accepted by smSirt2 as a substrate. Because of the low binding capacity of streptavidin-coated microplates, the assay window was too small to allow reproducible IC50 determinations (data not shown). Therefore, we switched to streptavidin-coated magnetic beads that can be suspended in the wells to increase substrate loading of the wells. A Km value of 4.5 µM was determined, and the Ac-p53-TAMRA concentration was adjusted to 250 nM to be sensitive to substrate competitive inhibitors (
Fig. 3A
). After optimization of substrate (
Fig. 3B
) and bead concentration, we were able to increase substrate concentration in the wells by a factor of 15. Enzyme kinetics indicated an incubation time of 2 h to be adequate (
Fig. 3C
). With 2 h of incubation, we are already at the upper limit of linearity. We use these conditions, however, to maximize the signal window. In
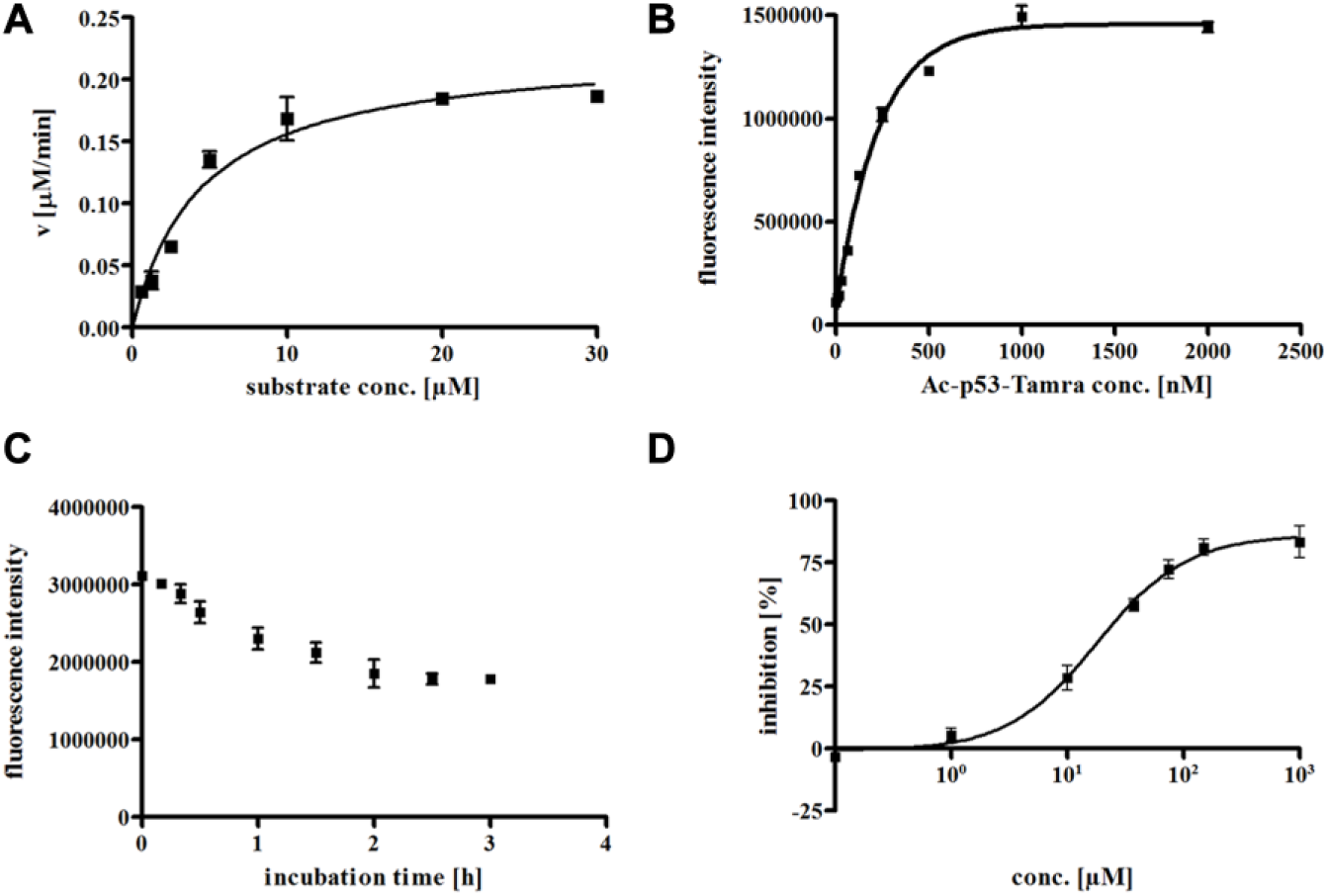
Assay development of heterogeneous fluorescence-based assay for smSirt2. (
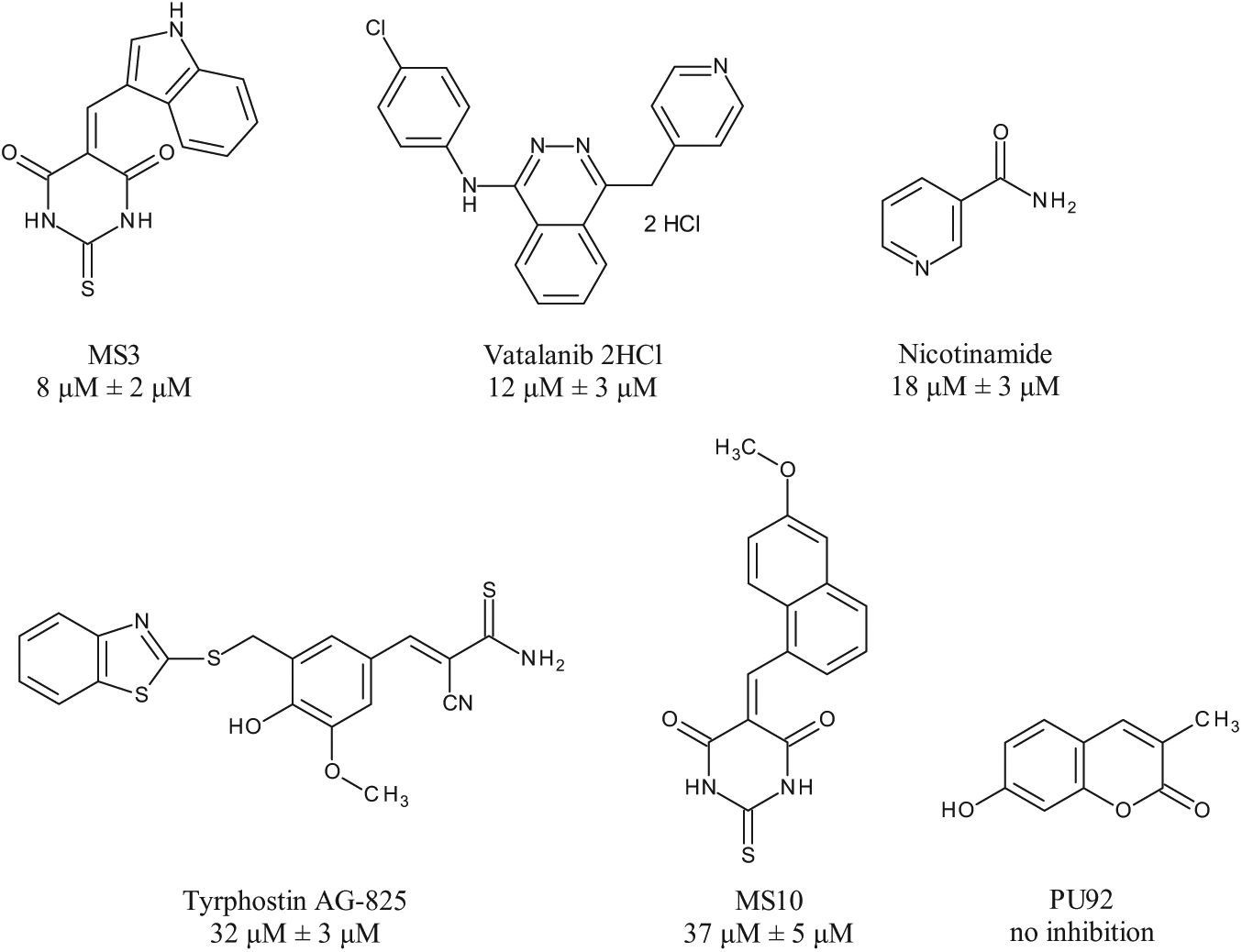
Inhibitors of smSirt2 identified by focused library screening and IC50 values determined with heterogeneous fluorescence-based assay for smSirt2.
Heterogeneous Fluorescence-Based Assay for hSirt2
To expand the utility of our new heterogeneous assay format, we transferred the assay to the human isoform hSirt2. Incubation time was adjusted to 2.5 h due to kinetic measurements (
Computational Studies
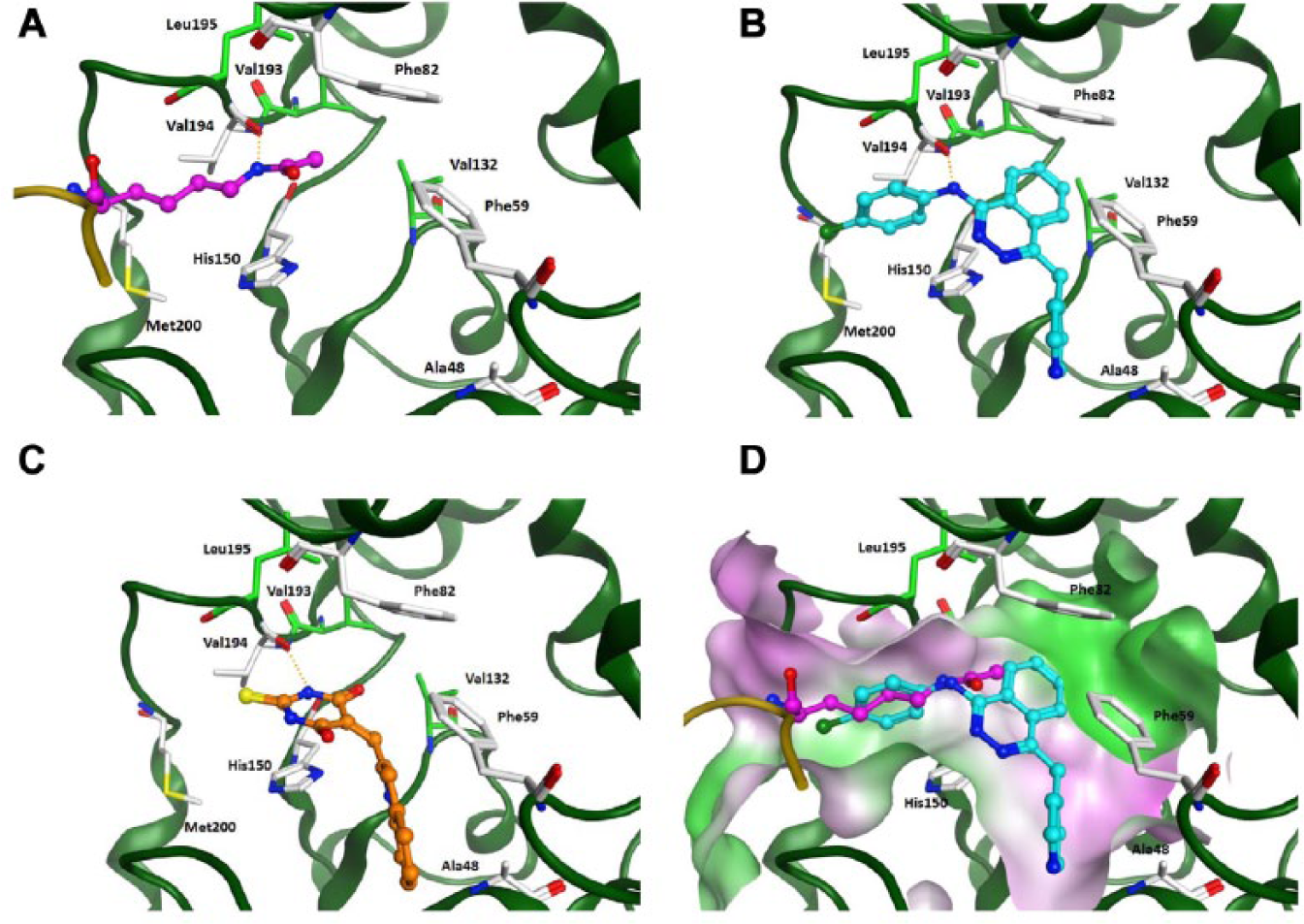
To rationalize the interaction of the identified inhibitors with smSirt2, we docked MS3 and Vatalanib into a generated homology model of smSirt2 (smSirt2-HM). Comparison of smSirt2-HM with the human Sirt2 crystal structure (PDB ID 3ZGV) reveals that smSirt2-HM shows the classical α–β scaffold folding of human sirtuins, and all major secondary structure elements are well matched. The largest deviation can be observed for the additional α helix, α13 (human Sirt2 numbering), that packs against the outside of the large domain containing the catalytic site (
(
Both docked inhibitors show favorable hydrogen bond interactions with the conserved residue Val194 in the peptide binding pocket ( Fig. 5B and 5C ), which is the interaction site for the amide group of the substrate. The NH group of the thiobarbiturate ring of MS3 as well as the amino group present in Vatalanib show favorable H-bonds to the backbone carbonyl of Val194. The aromatic rings of both inhibitors are making van der Waals interactions with the hydrophobic residues and fit nicely to the substrate pocket (Phe59, Phe82, Val132, His150, Val193, and Phe196) ( Fig. 5D ).
Discussion
The development of biochemical in vitro assays is one of the most important steps in the identification and characterization of novel modifiers of epigenetic targets, not only for drug discovery but also to elucidate biochemical functions of a specific enzyme. Here, we present a convergent assay platform, including the first two assays for smSirt2, a schistosomal sirtuin that is crucial for reproduction and survival of the parasite. This target-based approach will not guarantee to furnish compounds with in vivo activity on Schistosoma mansoni due to several reasons (e.g., pharmacokinetics). Nevertheless, the presented assay platform allows a rational pre-selection of the compounds showing in vitro activity, which will be further characterized in vivo. The homogeneous assay enables us, by using the small-molecule substrate ZMAL, to screen large compound libraries in a cost-efficient, high-throughput manner. IC50 values are highly reproducible and display minor standard deviations. This assay, however, is vulnerable with regard to compounds with strong fluorescence or quenching properties. Thus, the heterogeneous assay protocol is an ideal extension of the assay platform, because its detection is not influenced by compound interference. This allows us to characterize compounds exhibiting very high autofluorescence and helps us to identify inhibitors, which would be registered as false negatives in the homogeneous assay. We were able to show that the presented homogeneous and heterogeneous assays are not prone to a prototypical activator that produces artifacts with fluorescently labeled substrates (i.e., resveratrol). Furthermore, the heterogeneous protocol facilitates the confirmation of data of initial screening, using a peptide substrate derived from the physiological sirtuin substrate p53. The assay procedure of the heterogeneous assay is more complex and more costly due to the streptavidin-coated beads. Nevertheless, the assay is in principle suitable for high-throughput screening, and compared to other heterogeneous assays, which are often antibody based, the costs are rather low. In addition, we are not dependent on the availability of a highly selective antibody. We have shown that it might also be expanded to other sirtuins, as exemplified with human Sirt2. Our initial hits either show affinity to the hSirt2 (
Footnotes
Acknowledgements
We thank K. Schmidtkunz, University of Freiburg, for technical assistance.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work and the authors of this manuscript received funding from the European Union’s Seventh Framework Programme for research, technological development, and demonstration under grant agreements nos. 241865 (SEtTReND) and 602080 (A-ParaDDisE). Research on human sirtuins was funded by the Deutsche Forschungsgemeinschaft (DFG, Ju295/8-1, Si868/6-1).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.