
Abstract
Mycobacterium tuberculosis (Mtb) DNA gyrase ATPase was the target of a tuberculosis drug discovery program. The low specific activity of the Mtb ATPase prompted the use of Mycobacterium smegmatis (Msm) enzyme as a surrogate for lead generation, since it had 20-fold higher activity. Addition of GyrA or DNA did not significantly increase the activity of the Msm GyrB ATPase, and an assay was developed using GyrB alone. Inhibition of the Msm ATPase correlated well with inhibition of Mtb DNA gyrase supercoiling across three chemical scaffolds, justifying its use. As the IC50 of compounds approached the enzyme concentration, surrogate assays were used to estimate potencies (e.g., the shift in thermal melt of Mtb GyrB, which correlated well with IC50s >10 nM). Analysis using the Morrison equation enabled determination of
Introduction
History has taught us that emergence of resistance to antimicrobial agents is inevitable, with the consequence that there will generally be a need for new anti-infective agents. In addition, since the current therapies for tuberculosis were discovered several years ago, there is a need for better and more effective treatments to save the over 1.4 million patients who die of this disease every year. 1
The pharmaceutical industry is continuously on the search for new targets and starting points for drug discovery programs. Recently, emphasis has shifted from high-throughput screening (HTS) of genomics-derived targets to the use of clinically validated targets while searching for novel inhibitors or novel mechanisms of inhibition of these. One such target is DNA gyrase, which offers a mechanism that has not been fully exploited, that of its ATPase activity. DNA gyrase is an enzyme with two subunits, GyrA and GyrB, which introduces negative supercoils into DNA in an adenosine triphosphate (ATP)–dependent reaction, thus controlling the topological state of DNA in the bacterial cell. Classic inhibitors of the supercoiling activity are the fluoroquinolones, molecules that bind primarily the GyrA subunit and are widely used to treat antibacterial infections, including drug-resistant tuberculosis. However, resistance to the fluoroquinolones is slowly eroding the power of this class of antibiotics. 2 The ATPase activity of DNA gyrase can be uncoupled from its supercoiling activity and measured in isolation. Inhibition of the ATPase inhibits supercoiling; hence, inhibitors of this activity would be expected to be effective antibacterial agents. Targeting this activity offers an opportunity to develop novel anti-TB or anti-infective agents.
Assays have been developed to screen for inhibitors of GyrB ATPase. Novobiocin targets the GyrB ATPase; in addition, other chemical classes of GyrB ATPase inhibitors3–7 and co-crystal structures of many of these binding to GyrB are also reported. Mycobacterial DNA gyrase subunits have been purified from Mycobacterium smegmatis (Msm) or as recombinant proteins expressed in Escherichia coli.8,9 Virtual screening of the GyrB subunit 10 as well as a high-throughput screen 5 have been used to identify GyrB ATPase inhibitors. In addition, compounds with cellular activity against Mycobacterium tuberculosis (Mtb) have been developed without measuring the activity against the enzyme. 11 However, no reports have characterized the residence time or energetics of binding of compounds to GyrB. Compounds with a long residence time or t1/2 are expected to have an advantage in vivo, since compound effects can last beyond the time when compounds are cleared from the body. 12 The energetics of binding is useful in lead optimization, since inhibitors that are more specific to the target and also best in class compounds have an enhanced enthalpic binding component. 13
An accurate estimation of target potency (i.e., Kd or IC50) is critical in drug discovery programs to be able to drive the chemistry toward improvement of the affinity of inhibitors for the target. This is coupled with a need to use assays that are tractable and relatively high throughput. Quite often, a compromise has to be made and a surrogate used instead of the ideal assay. Described here is the development of mycobacterial DNA gyrase as a drug target and a toolkit of surrogate enzyme assays or analytical methods, beyond the primary assay, that was necessary to determine the potency of enzyme inhibition and guide the medicinal chemistry for progression of hits for the treatment of tuberculosis.
Materials and Methods
Materials
ColorLock malachite green reagent for the detection of inorganic phosphate (Pi) was from Innova Biosciences (Cambridge, UK). All other chemicals, unless otherwise specified, were from Sigma-Aldrich (St. Louis, MO).
Measurement of Cellular Activity against Mtb or Msm
Inhibition of the growth of Mtb or Msm was measured as described in the supplementary file. The minimum inhibitory concentration (MIC) was used as a measure of cellular activity of the compounds.
Cloning, Expression, and Purification of Mtb and Msm DNA Gyrase
Full-length Mtb gyrA and gyrB were cloned into pET20b and pET19b, respectively, as His-tagged proteins, with GyrA having a C-terminal His tag and GyrB an N-terminal His tag. Mtb DNA gyrase holoenzyme was purified from recombinant GyrB and GyrA subunit proteins expressed in E. coli as described. 14
A ~26-kDa fragment of Mtb gyrB (Gln20 to Gly266) was cloned into the pET20b vector and expressed in E. coli. Cells were lysed by freeze-thaw cycles or sonication. An ammonium sulfate precipitate (30%–50% saturation) was purified by novobiocin affinity chromatography; novobiocin was coupled to epoxy-activated sepharose in 200 mM sodium carbonate buffer (pH 9.5) overnight, followed by blocking of unreacted groups using ethanolamine. Mtb GyrB bound to the column and was eluted with 6 M urea, dialyzed, and concentrated. The concentrated eluate was further purified by gel filtration on a Superdex 200 (GE Healthcare, Piscataway, NJ, USA) column (320 mL). The protein was stored in 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM EDTA, and 10% glycerol in aliquots at −70 °C.
Msm gyrB was expressed and purified as described earlier from E. coli BL21 star (DE3) pLysS cells. 14 Briefly, E. coli cell pellets were lysed in a pH 7.5 Tris-HCl buffer containing glycerol, EDTA, dithiothreitol (DTT [TGED]), and protease inhibitors. Nucleic acids were precipitated by addition of streptomycin sulfate followed by ultracentrifugation. A 35% to 55% ammonium sulfate fraction of the supernatant was dissolved in and dialyzed against TGED buffer with 100 mM NaCl; this was loaded onto an anion exchange column (MonoQ, GE Healthcare, Piscataway, NJ, USA). The flow-through of the MonoQ column was loaded onto a heparin sepharose column. GyrB was eluted with a linear 0.1 to 1 M NaCl gradient; active fractions were pooled; concentrated; dialyzed against TGED, 100 mM NaCl, and 1 mM DTT; and stored in aliquots at −70 °C.
Both mycobacterial GyrB preparations contained a novobiocin-resistant ATPase, which was reduced to ≤15% of total ATPase by the use of heparin sepharose during purification.
Msm gyrA was cloned, expressed, and purified from E. coli as described (supplementary information). Msm holoenzyme was reconstituted prior to the start of the reaction by incubating GyrB and GyrA at a ratio of 1:3 and in the presence (or absence) of 12 µg/mL DNA on ice, in a buffer containing 50 mM HEPES (pH 7.5), glycerol 10%, EDTA 1 mM, 100 mM K-glutamate, 2 mM DTT, 0.002% Brij-35, and 0.1 mg/mL bovine serum albumin (BSA).
Preparation of DNA
Salmon testes DNA (100 mg, D-1626; Sigma-Aldrich) were dissolved in 12 mL of autoclaved distilled water by stirring at room temperature for 2 h. NaCl was added to 1 M and the DNA was extracted with phenol followed by chloroform-isoamyl alcohol. The aqueous phase was diluted 10-fold in 10 mM Tris-HCl (pH 8), 1 mM EDTA and then sonicated to get an average sheared DNA size of 1000 base pairs.
DNA Gyrase ATPase Assays and Determination of IC50 and
Msm GyrB ATPase
Assays were performed at 25 °C in a 50-µL volume in 96-well half-area plates (Costar 3695; Corning, Corning, NY) for 130 min. The reactions contained Msm GyrB (15 nM), 50 mM HEPES (pH 7.7), 250 mM potassium glutamate, 200 mM KCl, 1 mM DTT, 0.001% Brij-35, 2% glycerol, 4% DMSO, and 0.65 mM ATP. The reaction was started by addition of 2.5 mM MgCl2. At the end of the reaction, 12.5 µL of Pi Colorlock Assay reagent (cat. 303-0125; Innova Biosciences) was added, followed 5 min later by addition of 5 µL of stabilizer. Absorbance was measured at 630 nm after 30 min in a Spectramax-250 from Molecular Devices (Sunnyvale, CA). This complete reaction is called the positive control or “Max.” The A630 in the presence of 1 µM novobiocin (Sigma N-1628) was taken as the “background” or “Min” value, and to avoid interference by any novobiocin-resistant ATPase, this value was subtracted from all reactions as a measure of enzyme activity. Compounds were dissolved in DMSO and serially diluted in DMSO, and 2 µL of this stock was used in a 50-µL enzyme reaction to give final compound concentrations in the desired range.
Pi Detection for an HTS-Compatible Assay
For an HTS-compatible format, a published method 15 was modified to enable addition of a single reagent. Reagents A, B, and C were prepared and stored separately (reagent A: 0.12% w/v malachite green in 6 N sulfuric acid; reagent B: 7.5% ammonium molybdate; reagent C: 11% Tween 20). Just before use, the “detection reagent” was prepared by mixing the reagents in a ratio of 10:2.5:0.2 (A:B:C), and this was then diluted 3-fold with water (1 [A-B-C mix]:3 water). An equal volume of detection reagent was added to the reaction in the well of the microplate to stop the enzyme assay, the contents were mixed, and the A630 was read 30 min later.
ATPase reaction:
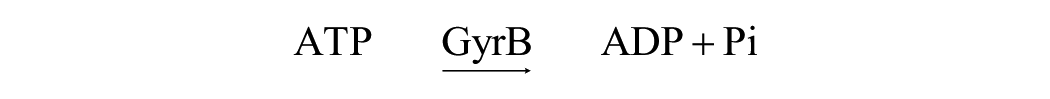
IC50 Determination
The Min value was subtracted from all readings and % activity of test samples was calculated as (Sample – Min)*100/(Max – Min). Percent inhibition (100 – % activity) was fitted to the “four-parameter logistic model” in XLfit for determination of IC50 values.
Determination by the Morrison Equation
The concentration- response data were fitted to a transformed, logarithmic form of the Morrison equation.
Morrison’s equation:
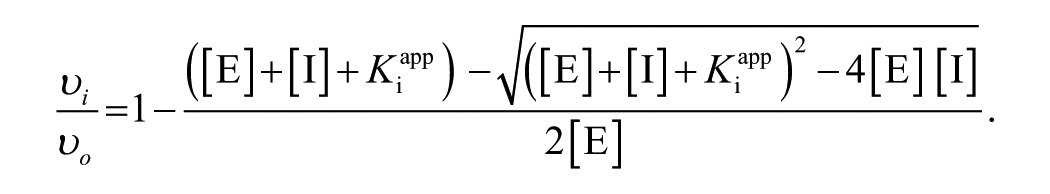
Transformed, the logarithmic form of the Morrison equation:

where [I] is the inhibitor concentration, υ i is ΔA630 in the presence of inhibitor, υo is ΔA630 in the absence of inhibitor, L
The logarithmic form of Morrison’s equation was fitted to υi versus [I] data to estimate the parameters υo, L
Thermofluor Assay
Measurements of shifts in the melting temperature (Tm) of Msm GyrB protein were carried out in 50 mM HEPES (pH 7.7), 250 mM potassium glutamate, 200 mM KCl, 1 mM DTT, 0.001% Brij-35, 2% glycerol and 2.5 mM MgCl2, 4% DMSO, and 0.1 mg/mL of enzyme ± test compounds (50 µM). In addition, 5× Sypro Orange (S-6650; Invitrogen, Carlsbad, CA) was used as a fluorophore. Assays (20 µL) were set up in 96-well PCR plates (Axygen-PCR-96-FLT-C; Corning LifeSciences, Tewksbury, MA, USA), sealed with Microseal B (Bio-Rad, Hercules, CA). The temperature was varied from 25 °C to 95 °C in steps of 0.5 °C, and fluorescence measurements (ex: 470 nm, em: 570 nm) were carried out in an iCycler from Bio-Rad. Fluorescence was plotted versus the temperature. In addition, the rate of change of fluorescence with temperature (–ΔRFU/dT) was plotted versus temperature, and the minimum point of the derivative curve was taken as the melting point (Tm).
Mtb DNA Gyrase Supercoiling Assay
The assay was performed in 30 µL for 130 min at 37 °C as described. 14 Supercoiled and relaxed forms of DNA were separated by gel electrophoresis on 0.8% agarose in a buffer containing 45 mM Tris-borate and 1 mM EDTA for 16 h at 1 V/cm. Gels were stained with ethidium bromide (0.7 µg/mL), and the density of relaxed and supercoiled forms was analyzed using the Bio-Rad gel documentation system to compare % conversion of relaxed to supercoiled forms. A unit was defined as the conversion of 100 ng of relaxed plasmid to the supercoiled state in 1 h at 37 °C.
Surface Plasmon Resonance (BIAcore) Studies
Experiments were performed with a BIAcore 3000 system (BIAcore AB, GE Healthcare, Piscataway, NJ, USA) at 25 °C. The N-terminal (26-kDa) fragment of Mtb GyrB was immobilized on a CM5 chip in flow cell 2 (FC2) by covalent amine coupling as recommended by the manufacturer (Biacore Sensor Surface Handbook BR-1005-71 Edition AB). Flow cell 1 (FC1), which was activated and blocked without injecting the protein, was used as a reference cell. Compounds were dissolved in running buffer HBS-P (cat. BR100368; GE Healthcare, Piscataway, NJ) and injected at a flow rate of 60 µL/min. After an association phase of 4 min, the system was switched back to running buffer, and the dissociation phase was followed for at least 10 min; for compounds with longer residence time (>5 min), the dissociation phase was monitored for a longer period (~60 min). BIAevaluation software was used to analyze the data using the 1:1 interaction model with mass transfer correction. The association rate constant (kon), dissociation rate constant (koff), and the equilibrium dissociation constant (Kd) calculated using the kinetic parameters (Kd = koff/kon) are reported. The residence time of the ligand on the protein (1/koff) or the dissociation half-life (t1/2), 0.693/koff, of the enzyme-inhibitor [EI] complex was calculated.
Isothermal Titration Calorimetry
The energetics of binding of novobiocin to full-length Mtb GyrB protein and the 26-kDa fragment was studied using VP-ITC (MicroCal LLC, Northampton, MA, USA). The protein was dialyzed against 50 mM Tris-HCl (pH 7.5), 6% glycerol, 2 mM DTT, 50 mM KCl, and 5 mM MgCl2. The proteins and novobiocin were diluted to a final concentration of 3 µM and ~20 µM, respectively, in degassed dialysis buffer. DMSO and Brij-35 were added to both protein and novobiocin stock to achieve a final concentration of 2% and 0.001%, respectively. The solution of enzyme was titrated with successive additions of 10-µL aliquots of novobiocin (every 3.5 min) at 30 °C. The binding data were corrected for heat of dilution of the compound. The corrected data were analyzed by nonlinear regression using a single-site binding model in Origin software (MicroCal), which yielded the values of equilibrium dissociation constant (Kd) and change in enthalpy (ΔH).
Results
Development of DNA Gyrase Enzyme Assays; Msm as a Surrogate for Mtb
Mtb DNA Gyrase
An assay to measure the ATPase activity of Mtb DNA gyrase was explored. No ATPase activity was detected with Mtb GyrB alone (500 nM) or if only GyrA or DNA was added to it. For Mtb GyrB ATPase activity, addition of both GyrA and DNA was required (
Fig. 1
). The ATPase activity of the Mtb holoenzyme plus DNA was enhanced by the addition of glutamate in the assay buffer; however, despite all attempts to enhance the ATPase activity, by varying assay components or increasing the length of the assay, a concentration of 150 to 300 nM GyrB was needed to give a window that was reproducible and robust (Z′ > 0.5).
16
Having this concentration of enzyme would theoretically limit the minimum measurable IC50 value to 75 nM, since IC50 =
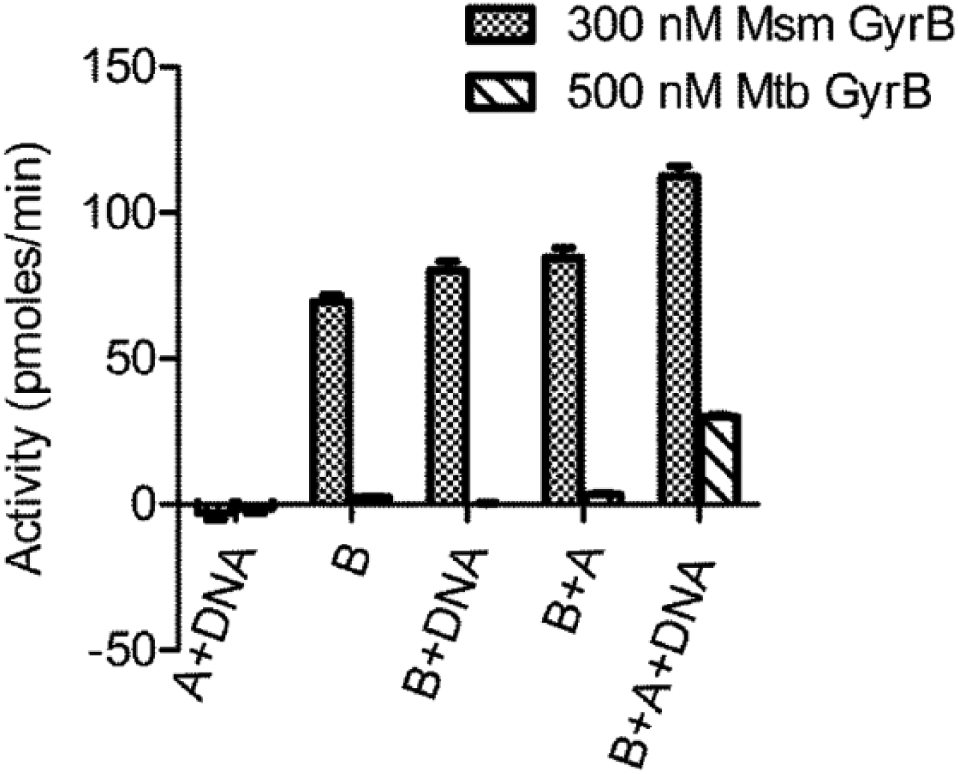
The effect of GyrA and DNA on the ATPase activity of Mtb and Msm GyrB. The bar graph represents ATPase activity of GyrA+DNA, GyrB alone, GyrB+DNA, GyrB+GyrA, and GyrB + GyrA+DNA. Quantities used were 500 nM Mtb GyrB, 1500 nM Mtb GyrA, 300 nM Msm GyrB, 900 nM Msm GyrA, and 1.2 mg sheared salmon testes DNA.
The inability to use IC50s to estimate the affinity of inhibitors with
Msm DNA Gyrase
The ATPase activity of Msm GyrB, unlike that of Mtb GyrB, was only slightly enhanced by the addition of GyrA subunit or DNA and varied from 30% to 100% enhancement of activity across protein batches ( Fig. 1 ). The major ATPase activity was associated with Msm GyrB alone, in contrast to an earlier report 19 in which the protein was exposed to low pH during purification and no ATPase was detectable in a radioactive assay with 450 nM GyrB. The specific activity of GyrB alone was 350 to 900 nmol ATP hydrolyzed/min/mg GyrB protein, and an assay was optimized using 15 nM GyrB. This is 10 to 20 times lower than the concentration of Mtb GyrB needed for a similar window, giving the advantage of being able to theoretically measure IC50s of inhibitors in the ~15 nM range, rather than the ~300 nM range for the Mtb enzyme.
The conditions for a microplate assay for Msm GyrB ATPase were optimized by testing the effect of buffer composition (e.g., salts and pH) on the assay. Both KCl and K-glutamate stimulated the activity of GyrB ATPase, but a combination of the two showed better activity than either KCl or K-glutamate alone. For example, addition of glutamate caused a ~50% to 100% increase in activity even in the presence of 300 mM KCl. The combination of 250 mM K-glutamate and 200 mM KCl was found to give highest activity and was chosen for further assays ( Fig. 2A ).
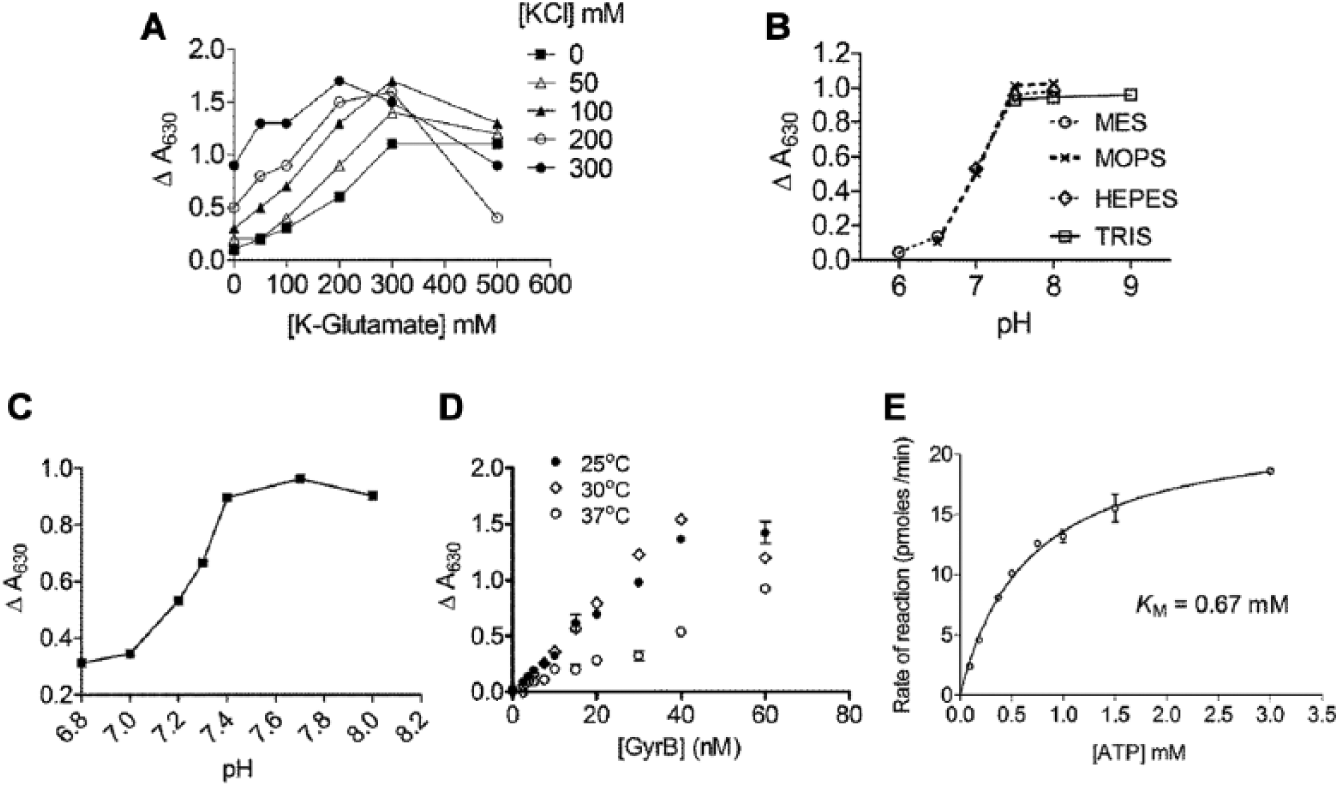
Optimization of a high-throughput screening–compatible assay for the ATPase activity of Msm GyrB. The effect on the ATPase of varying reaction conditions was studied: (
The effect of Brij-35, BSA, and glycerol on the ATPase activity was tested using a “factorial design of experiment”
20
(
A steep drop in activity was observed when the pH reduced below pH 7.4 ( Fig. 2B ). Activity between pH 7 and 8, in smaller increments, was tested in HEPES buffer, and a pH of 7.7 was chosen for further experiments ( Fig. 2C ). Interestingly, the Msm ATPase activity at 37 °C was lower than at 25 ° or 30 °C and could be due to instability of the protein at this temperature. For convenience, 25 °C was chosen for routine screening ( Fig. 2D ). The KM of ATP for Msm GyrB, averaged over four experiments, was 0.65 mM ( Fig. 2E ). This resulted in the final assay screening conditions described in the Materials and Methods section, with ATP concentration close to the KM.
Adaptation to HTS
ATP hydrolysis during assay optimization was measured by detection of inorganic phosphate (Pi) using a commercial kit that gave a signal that was very stable with time, but it needed addition of two separate reagents. Other protocols for malachite green based detection of Pi were explored to make the assay HTS compatible,3,15,22 and one of these 15 was modified to enable a single addition of the detection reagent, which was dispensed using a multidrop. Unlike Pi Colorlock, this reagent caused a gradual increase in A630 over time, but since the rate of increase of the background (Min) and the complete (Max) enzyme reactions was similar, the ΔA630 remained constant over time, and readings could be taken up to 30 min after termination of the enzyme reaction.
The Z′ 16 of the Msm ATPase assay was 0.8, and an HTS of a million compounds from the AstraZeneca library resulted in the identification of the aminopyrazinamide series. 5 The initial hit had an IC50 of 2 µM and cellular activity on Mtb (MIC) of 16 µg/mL. Artifacts due to assay readout and false positives were eliminated by a parallel dose-response plate without enzyme, followed by confirmation of inhibition in the Mtb DNA gyrase gel-based plasmid supercoiling assay. 5
The same microplate assay was also used to progress two other ATPase inhibitor chemical series (pyrrolamides
14
and thiazolopyridine ureas
7
) in a drug discovery program to improve the MIC, pharmacokinetics, and other properties (
Fig. 3A
). The thiazolopyridine ureas were identified from a pharmacophore-based search of the AstraZeneca library,
7
whereas the pyrrolamides
14
were a follow-on of a broad-spectrum antibacterial program within the company. Initial compounds in the thiazolopyridine ureas had an IC50 of ~0.16 µM and an MIC of ~8 µg/mL,
7
whereas the pyrrolamides had similar IC50s but lower MICs (~1 µg/mL).
14
Lead identification efforts resulting in compounds efficacious in mouse models of tuberculosis have been described recently.7,14 Here is described the biochemical and biophysical assays that were necessary and that enabled progression of the three series to compounds with greatly increased potency of enzyme inhibition (≤2 nM IC50s) and Mtb MICs as low as 0.03 µg/ml (
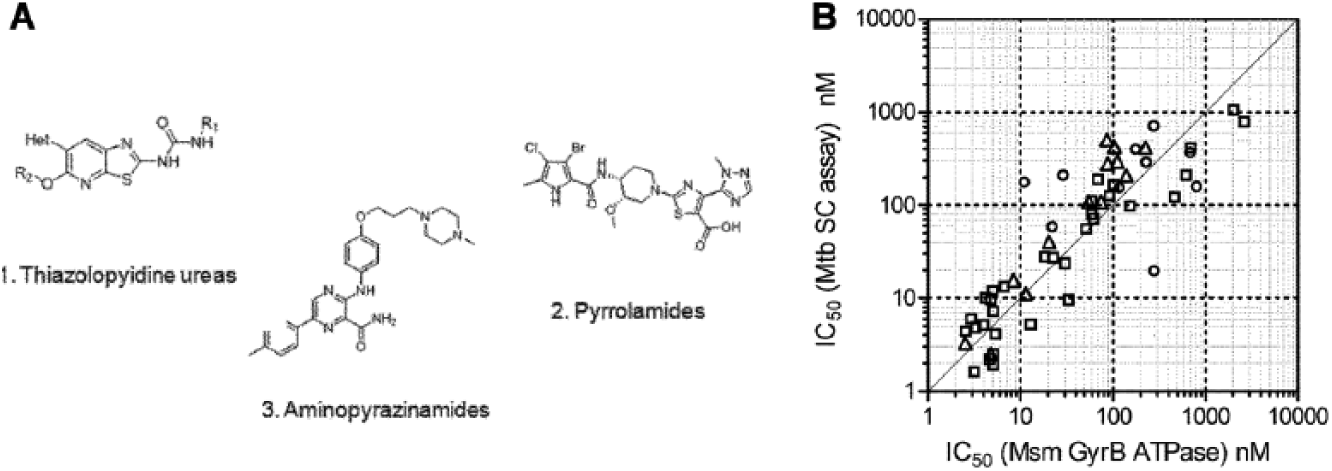
Validation of Msm GyrB ATPase as a surrogate for discovery of inhibitors of Mtb DNA gyrase supercoiling. (
Validation of Msm GyrB as a Surrogate for the Mtb Enzyme
A selection of compounds covering all three series was tested on Msm GyrB ATPase and for inhibition of Mtb DNA gyrase supercoiling activity, with ATP concentration at the respective KMs for the two enzymes. The IC50 in the two assays were similar ( Fig. 3B ), confirming that Msm is a good surrogate for Mtb and that the Msm ATPase assay was able to select ATPase inhibitors that inhibit the supercoiling activity of Mtb DNA gyrase. The Msm ATPase assay was used for compound progression with occasional testing on the Mtb supercoiling assay.
Determining the Relative Potency of Compounds Using Thermal Denaturation
An assay with 15 nM GyrB enzyme was adequate for the primary screen. However, as the SAR was established and potent compounds were synthesized (IC50s approaching 15 nM;
Tm Shift
Msm GyrB enzyme had an average melting temperature (Tm) of 49.6 °C with a standard deviation of 0.25 °C across five experiments. On addition of the ATPase inhibitors, two distinct Tms were observed: one marginally lower than the Tm of the enzyme and the other higher than the Tm of the enzyme; a similar pattern was seen on addition of novobiocin ( Fig. 4A,B ).
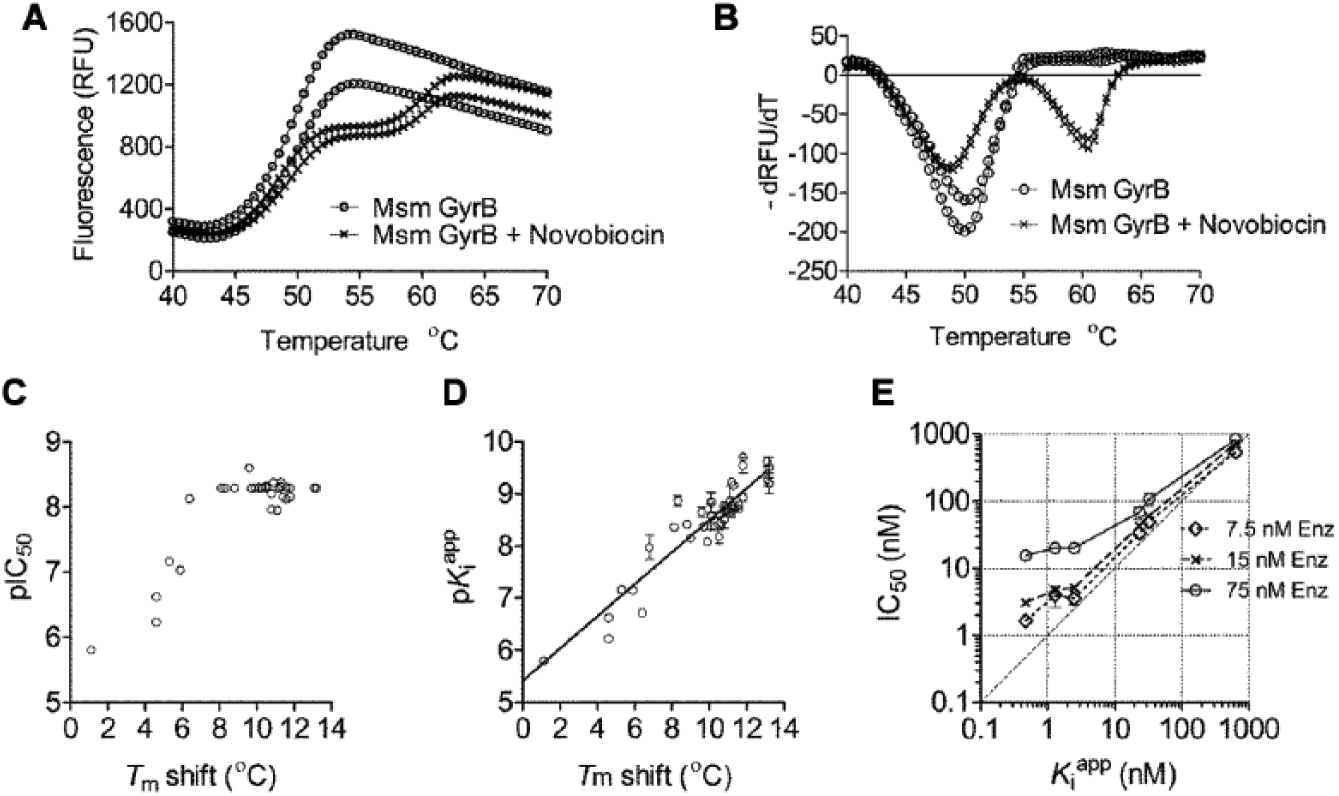
The effect of compounds on the thermal melt of Msm GyrB and comparison of potency determination by different technologies. (
A shift of 11 °C was observed for novobiocin, which had an IC50 of 10 nM. The Tm shift was measured for a selection of compounds from the pyrrolamide series. The shift in Tm correlated well with the IC50 of the compounds on Msm GyrB ATPase (
Fig. 4C
and
The Morrison Equation to Measure
s of Potent Compounds
While the Tm appeared to rank-order compounds, an accurate estimation of binding affinity of the potent compounds was needed to guide the medicinal chemistry. Options that were explored to enable this included (a) lowering the enzyme concentration [E] while simultaneously increasing the reaction time to lower the influence of [E]T in the IC50 determination, but a significant reduction in the enzyme concentration resulted in a loss of signal. (b) The ATP concentration in the assay was increased; this should have the effect of raising the IC50 of a competitive inhibitor and thus enable differentiation of the affinity of compounds in a lower range of
For IC50 measurements in general, it is assumed that the concentration of the EI complex, [EI], is very small compared with [I], and hence the free inhibitor concentration is well modeled by the total inhibitor concentration [I]. In the case of tight binders, [EI] = [I], and IC50 values underestimate the true affinity of the inhibitor for the enzyme.
18
The Morrison equation accounts for tight binding, so it does not assume that the free concentration of inhibitor equals the total concentration and can be used in such cases. A set of compounds with varying potencies from compound series 1 (thiazolopyridine ureas) and 2 (pyrrolamides) was tested for ATPase inhibition, and the same % inhibition data were analyzed by both the standard IC50 method and by the Morrison equation to determine
To investigate the discrepancy between IC50 and
SPR to Measure Kds and Residence Time of Potent Inhibitors
To check if the compounds were more potent than apparent from
Mtb full-length GyrB was covalently bound to the chip using a variety of conditions, and the effectiveness of the immobilization was tested by studying the binding of novobiocin to the protein. During this optimization, it was discovered that the binding response (RU) did not saturate with increasing novobiocin concentration, even up to 100 µM novobiocin (
Fig. 5A
). Considering that the IC50 of novobiocin on the Msm GyrB ATPase is ~20 nM, it suggested that this was due to nonspecific binding. Increasing the salt or adding detergent to the running buffer did not eliminate the nonspecific binding, and no saturation of the signal was observed with either protein. Subsequently, a 45-kDa fragment of Mtb GyrB was tested with essentially the same result. However, a smaller, 26-kDa Mtb GyrB fragment did not have this problem, and novobiocin showed specific binding to this protein, with typical kinetics and a signal that saturated at ~50 nM novobiocin (
Fig. 5B
). The energetics of binding were similar to those obtained by isothermal titration calorimetry measurements of the novobiocin interaction with full-length Mtb GyrB (
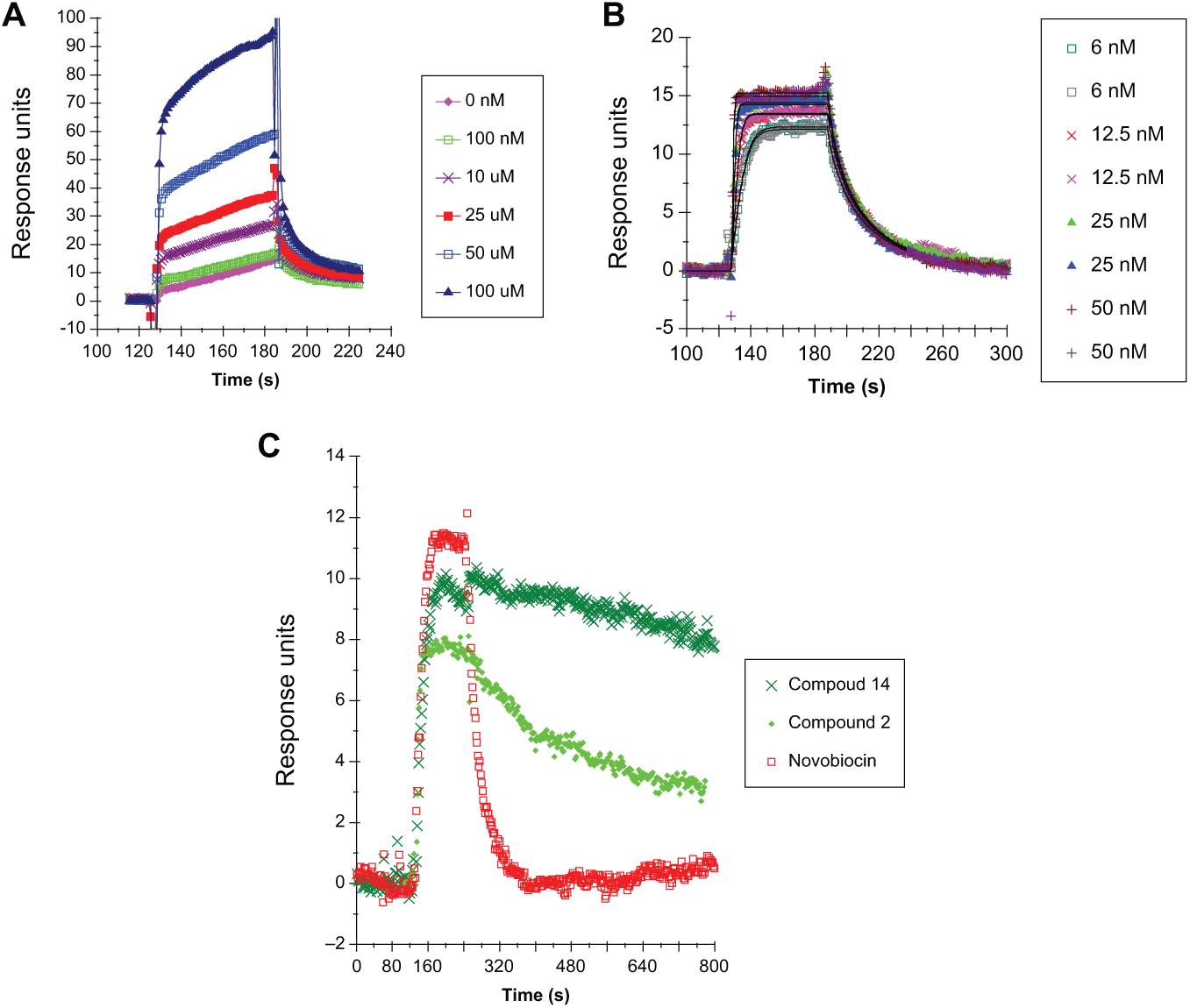
Sensograms of binding of compounds to Mtb GyrB by surface plasmon resonance. Sensograms for a range of concentrations of novobiocin binding to (
Kd values and the residence time were estimated for compounds across all three classes in the drug discovery program, plus three published inhibitors of DNA gyrase ATPase (compounds 12–14;
Discussion
The ATPase active site of DNA gyrase is highly conserved between Msm and Mtb.
5
Hence, it is not surprising that compound affinities for the target measured across the two species and across technologies are very similar (e.g., Msm ATPase vs. binding by SPR to an Mtb GyrB fragment). The use of the Msm protein did not hinder development of a robust SAR or lower the MIC against Mtb. However, despite the similarity of the ATPase site, the specific activity of the Mtb DNA gyrase was much lower than that of Msm. The requirement for both subunits of the Mtb homolog to measure the ATPase versus only GyrB for Msm was another difference, and this was an advantage for Msm protein supply. Despite the similarity between Msm and Mtb at the enzyme level, many compounds that inhibited the growth of Mtb had much higher MICs against Msm (200-fold for compound 6;
The ATPase enzyme assay was the backbone of the program driving the chemistry toward increased affinity for the target and was high throughput. The Morrison analysis was very convenient for estimating inhibitor-target affinity when IC50 values approached the enzyme concentration and was high throughput. Since
The SPR is an attractive technology: it uses a very low quantity of protein and gives rich information (i.e., on and off rates as well as Kds). The dissociation half-life of the [EI] complex for compounds of all three series was ≤8 min. Since the doubling time of Mtb is ~24 h, a t1/2 of 8 min is unlikely to have any impact in vivo. Immobilization of the protein did not affect the affinity of GyrB ATPase inhibitors, and Kds of inhibitors for the target were very similar to affinities obtained by methods where the protein was not immobilized (e.g., isothermal titration calorimetry [ITC], IC50). While not high throughput, the chip with immobilized Mtb GyrB could be reused and the BIAcore machine programmed to measure Kds of ~15 compounds in a 24-h cycle.
The thermal shift analysis is high throughput, while using a fairly large quantity of protein, and has the advantage of being able to determine affinities of slow binders when coupled with differential scanning calorimetry. 26 However, the magnitude of the shift in Tm alone is not directly related to the binding affinity. 27 In the case of this drug discovery program, the Tm shift could be used to rank compounds only because, across and within the series, the binding enthalpies were very similar. The binding of all three scaffolds to Mtb GyrB was driven by enthalpy (preliminary ITC data), with ΔH and TΔS values in a very similar range to that of novobiocin, suggesting the interactions these compounds make are somehow favored by the nature of the protein binding pocket. The Kd of binding of novobiocin to Mtb GyrB is similar to values reported for E. coli and Streptococcus pneumonia.28,29 ITC was useful in determining the energetics of binding, but it needs a high amount of protein, is low throughput, and is not able to determine very potent Kds. Both the thermal shift analysis and ITC, while suitable for compound progression in a lead identification program, are not suitable for HTS.
It was unexpected that binding of both the compounds and novobiocin to Msm GyrB would result in two thermal transitions. Interestingly, the published aminopiperidine inhibitors 30 upon binding to the enzyme also appear to cause two thermal transitions. The reason for two Tms could be that there are two populations of the protein, only one of which binds the compound. Or, alternatively, the enzyme has two domains, only one of which binds the compound, hence melting at a higher temperature, while the other domain remains unaffected. Which one of these hypotheses is true is yet to be investigated.
Most published assays for the DNA gyrase ATPase are those used for hit identification. In summary, described here is a toolkit of assays that were necessary beyond the primary assay to drive a lead generation, drug discovery program targeting Mtb DNA gyrase ATPase. The use of surrogates and alternative technologies enabled the progression of diverse scaffolds to increased potency of enzyme inhibition. Starting with an IC50 of 2 µM, the most potent aminopyrazinamide synthesized had an IC50 of ~2 nM and an MIC of 0.5 µM and were highly bactericidal, killing Mtb in vitro and in macrophages.
5
The most potent thiazolopyridine ureas synthesized had IC50s ~2 nM (
Footnotes
Acknowledgements
We acknowledge and thank Rob Shaw for statistical factorial design tool and Walter Ward and Geoff Holdgate for valuable suggestions and the many people at AstraZeneca, without whose support this work would not have been possible. We thank Peter Doig for critical comments on this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.