
Abstract
Most mitochondrial messenger RNAs in trypanosomatid pathogens undergo a unique type of posttranscriptional modification involving insertion and/or deletion of uridylates. This process, RNA editing, is catalyzed by a multiprotein complex (~1.6 MDa), the editosome. Knockdown of core editosome proteins compromises mitochondrial function and, ultimately, parasite viability. Hence, because the editosome is restricted to trypanosomatids, it serves as a unique drug target in these pathogens. Currently, there is a lack of editosome inhibitors for antitrypanosomatid drug development or that could serve as unique tools for perturbing and characterizing editosome interactions or RNA editing reaction stages. Here, we screened a library of pharmacologically active compounds (LOPAC1280) using high-throughput screening to identify RNA editing inhibitors. We report that aurintricarboxylic acid, mitoxantrone, PPNDS, and NF449 are potent inhibitors of deletion RNA editing (IC50 range, 1–5 µM). However, none of these compounds could specifically inhibit the catalytic steps of RNA editing. Mitoxantrone blocked editing by inducing RNA-protein aggregates, whereas the other three compounds interfered with editosome-RNA interactions to varying extents. Furthermore, NF449, a suramin analogue, was effective at killing Trypanosoma brucei in vitro. Thus, new tools for editosome characterization and downstream RNA editing inhibitor have been identified.
Introduction
Three related trypanosomatids, Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major cause life-threatening tropical diseases—namely, African trypanosomiasis, Chagas disease, and leishmaniasis, respectively. 1 Millions of people are currently affected by these diseases. The chemotherapy available to treat trypanosomes has a limited impact and harmful side effects. In addition, the emergence of drug-resistant parasites is a major driver for drug discovery.2,3
RNA editing, an essential process in trypanosomatids, is a unique and very attractive drug target in these parasites. 4 RNA editing is responsible for the maturation of precursor mitochondrial messenger RNAs (mRNAs). This process, involving only insertion and/or deletion of uridylates (Us), is catalyzed by a 1.6-MDa multiprotein complex called the editosome. The number of Us that require insertion or deletion is dictated by antisense RNA templates known as guide RNAs (gRNAs). 5 The editing reaction commences with endonucleolytic cleavage of the editing site by an endoribonuclease, followed by U insertion by a terminal uridylyltransferase (TUTase), or U deletion by a 3′ U-exoribonuclease (ExoUase); the reaction ends with ligation of the edited site by an RNA ligase.6,7 The core editosome contains over 20 proteins, which include the aforementioned core catalytic enzymes. Various compositions of the multiprotein complex have been observed, with at least three different complexes sedimenting at a density of ~20S on glycerol gradients. 8 These three complexes have different specificities for editing preference (insertion/deletion) or mRNA substrate, suggesting the existence of a complex and dynamic mechanism for substrate selection and editing in vivo, supported by the proposed editosome structure.9,10 Although the compositions of the editosomes have been determined, and the basic biochemistry of the editing reactions has been elucidated, the way in which the proteins in these complexes work together is not clear.
Besides the core editosome, a number of transiently associated accessory proteins indirectly regulate the efficiency of RNA editing and editosome stability. For example, mitochondrial RNA binding complex 1 (MRB1), also known as gRNA binding complex (GRBC), plays a role in diverse aspects of mitochondrial RNA processing and metabolism, such as gRNA utilization and expression. 11 Among the MRB1 proteins, a number of proteins interact with one another or with other complexes (i.e., TbRGG2) in an RNase-sensitive manner. These data have suggested that protein-protein interaction and RNA-dependent interactions are critical for MRB1 integrity with important implications to the core editosome function.
Edited and mature mitochondrial mRNAs code for components of the oxidative phosphorylation system. 12 These mitochondrial genes are essential in trypanosomatids, because the components of the oxidative phosphorylation system are fully functional in the procyclic stage and partially functional in the mammalian stage of the parasite life cycle.13,14 RNA interference (RNAi)–mediated knockdown of the core editosome proteins in both of these life cycle stages has a negative effect on T. brucei growth rate; this in most cases compromises parasite viability. 4 This finding implies that the editosome harbors a large number of drug targets. Hence, identification of novel RNA editing inhibitors should offer unique opportunities for drug discovery. In addition, identification of inhibitors that block editosome assembly at specific steps would provide chemical tools for dissecting the structure and function of the editosome.
Recently, virtual screening of the crystal structure of the T. brucei RNA editing ligase 1 (TbREL1) catalytic domain identified several inhibitors of the ligase in vitro.15–18 However, because of the high editosome concentrations required for completion of an RNA editing cycle in vitro, two of the potent inhibitors were found to affect RNA-protein interactions instead of inhibiting the ligase itself. 18 Another study identified five inhibitors of insertion RNA editing, with two specifically acting at or prior to the endonuclease step. 19 In the present study, we used our high-throughput screening (HTS) assay 17 that targets RNA editing to screen a library of pharmacologically active compounds from Sigma (LOPAC1280; Sigma, St. Louis, MO). The screen identified five inhibitors of deletion RNA editing. However, none of the compounds exhibited catalytic-step inhibition specificity as determined by secondary assays. Instead, these compounds interfered with RNA-protein interaction, making them useful tools for exploring the RNA editing mechanisms mediated by accessory RNA-binding factors. In addition, we show for the first time that a compound identified in this screen, NF449, an analogue of suramin, can effectively kill T. brucei in vitro.
Materials and Methods
Editosome Purification
Editosomes were purified from a crude mitochondrial extract by glycerol gradient ultracentrifugation and fractionation as described previously.18,20 The purified editosome was then analyzed and quantified via Western blotting. The most active editosome fractions were identified using the fluorescence resonance energy transfer (FRET)–based in vitro editing assay. 18 Then, 5 µL of this purified editosome extract was used in all in vitro editing assays, including the primary FRET-based and the secondary radiolabeled assays.
Chemical Compounds and LOPAC1280 Screen
RNA molecules, preA6Rbz, gA6Rbz, and gA6Rbz_comp, used in the FRET-based in vitro editing assay (high-throughput screening assay) were prepared by T7 polymerase (Promega, Madison, WI) transcription of synthetic DNA templates as described before. 17 The FRET-based RNA substrate, 5′ FAM (6-carboxyfluorescein)–GAUCUAUUGUCUCACA–Iowa black 3′, was synthesized and high-performance liquid chromatography purified by Integrated DNA Techno-logies (IDT, Coralville, IA). The screening of the Library of Pharmacologically Active Compounds (LOPAC; Sigma) was conducted in 384-well quantitative reverse transcription PCR (RT-qPCR) plates (Bio-Rad, Hercules, CA). The compounds were in a 100-µM stock concentration in the mother plates, and 3 µL of each was transferred to the RT-qPCR plates using a multichannel pipette. For positive and negative control wells, 3 µL of DMSO was pipetted into each well. An assay reaction contained 1 pmol preA6Rbz (preedited ribozyme), 2.5 pmol gA6Rbz (guide ribozyme), and 5 µL editosome extract in a final volume 20 µL of solution containing 1× HHE (25 mM HEPES [pH 7.9], 10 mM Mg (OAc)2, 50 mM KCl, and 1 mM EDTA), 1 mM adenosine triphosphate (ATP), 5 mM CaCl2, 0.5 µg Torula Yeast RNA, and 0.1% Triton X-100. The preedited ribozyme reporter RNA (preA6Rbz) was initially annealed to gA6Rbz by denaturation at 65 °C for 3 min and cooling at room temperature (RT) for 10 min. An enzyme mix was prepared that contained every component of the reaction except for the RNA molecules, and 17 µL of this was then dispensed into each well immediately after mixing the RNA duplex solution with the enzyme mix. A separate enzyme mix was prepared and dispensed in the plates as negative controls; these contained all components of the reaction except for the editosome. The plates were sealed and incubated at 28 °C for 4 h. To stop the editing reaction, 25 pmol of gA6Rbz_comp (guide ribozyme competitor) was added into each well, and the plates were incubated at 85 °C for 5 min. After letting the plates cool at RT for 10 min, 15 pmol of the FRET-based RNA substrate was added to each well. The fluorescence signal was then read kinetically at 430 nm every min at 37 °C in an RT-qPCR machine.
Interference Assay
To eliminate compounds that affect the RNA reporter instead of the editosome, an interference assay was performed. This assay is similar to the primary screening assay except that the reaction contained 1 pmol A6Rbz (active ribozyme) in a 20-µL volume of solution containing 1× HHE (25 mM HEPES [pH 7.9], 10 mM Mg (OAc)2, 50 mM KCl, and 1 mM EDTA), 1 mM ATP, 5 mM CaCl2, 0.5 µg Torula Yeast RNA, and 0.1% Triton X-100. Then, 15 pmol of the FRET-based RNA substrate was added to each well and read kinetically every min at 37 °C in an RT-qPCR machine as described above. A6Rbz used in this assay was prepared by T7 polymerase transcription (Promega) of synthetic DNA templates as previously described. 17 The selected compounds were then purchased from Sigma: PPNDS tetrasodium, aurintricarboxylic acid, mitoxantrone, and NF449.
Dose-Response Studies
For performing a dose-response study on the hits, a serial dilution of the compounds was prepared starting with a 2-mM stock in a 1:2 dilution factor. For the dose-response study using the FRET-based assay, 5 µL of the editosome was incubated with 2 µL of the diluted compounds for 10 min on ice. A master mix containing preA6Rbz, gA6Rbz, and the buffer was prepared as described above and dispensed appropriately to a final volume of 20 µL per reaction. The remaining procedure was followed identically as done for the LOPAC screen, and the IC50 values were calculated using data combined from three replicate sets.
The dose-response study of the hits on the growth of bloodstream form (BF) T. brucei was evaluated via the Alamar blue assay. 21 The assay was carried out in 96-well plates containing growth medium, with the serially diluted compounds. All wells, including the no-compound control, contained 1% DMSO. BF T. brucei were then seeded at a density of 1 × 104 cells/mL to a final volume of 200 µL in each well. The plates were then incubated for 48 h at 37 °C. Subsequently, 20 µL of Alamar blue (Life Technologies, Carlsbad, CA) was added to each well and incubated for 24 h at 37 °C. Fluorescence was measured at 590 nm using a microplate reader, and the EC50 values were calculated using data obtained from three technical replicate sets.
In Vitro Editing Assays
RNA required in the precleaved deletion assay, U5-5′ CL, U5-3′ CLpp, and gA6 [14] PC-del, were prepared in vitro by T7 polymerase (Promega) transcription of PCR-generated DNA templates as described before.22,23 U5-5′ CL was labeled at the 5′ terminus with [γ-32P] ATP using T4 polynucleotide kinase (NEB, Ipswich, MA). The precleaved deletion editing assay was performed using U5-5′ CL, U5-3′ CLpp, and gA6 [14] PC-del, as described previously.22,23 The exoUase assay was performed identically to the precleaved deletion editing assay, except U5-3′ CLpp was eliminated. A6short/TAG.1 pre-mRNA and D34′ gRNA for the endonuclease assay were also prepared as described before.24,25 A6short/TAG.1 pre-mRNA was labeled at the 5′ terminus with [γ-32P] ATP using T4 polynucleotide kinase (NEB, Ipswich, MA). The assay was performed as previously described. 25 All four compounds were screened at a 10-µM concentration in the above-mentioned assays. Equal concentration of DMSO (1%) was included in control reactions that did not contain any compounds. The RNA from each reaction in the assay(s) were purified using phenol/chloroform and resuspended in equal volumes of 7 M urea dye before running on 15% (w/v) denaturing polyacrylamide-urea gels. The gels were visualized using a phosphorimager.
Electrophoretic Mobility Shift Assay
The gA6 [14] guide RNA was prepared in vitro by T7 polymerase (Promega) transcription of a PCR-generated DNA template as described before 18 and labeled internally with [α-32P] UTP. An electrophoretic mobility shift assay (EMSA) was then conducted as previously described. 18 Then, 5 µL of the editosome extract was first incubated with 10 µM (considering final volume of 20 µL) of the selected compounds on ice for 5 min. An equal concentration of DMSO (1%) was used in the control reaction that did not contain a compound. The editosome-compound mixture was then added to a reaction buffer at a final volume of 20 µL containing the 3′ end labeled gA6 [14] guide RNA and RBB50 buffer (20 mM Tris-HCl [pH 7.6], 50 mM KCl, 5 mM MgCl2, 100 µg/mL bovine serum albumin [BSA], 10% (w/v) glycerol, and 1 mM dithiothreitol [DTT]) supplemented with 100 mM KCl and 20 units of RNasin (Promega). The reactions were incubated at RT for 30 min and were then run on 10% (w/v) TBE gels (Bio-Rad) and visualized using a phosphorimager.
Results and Discussion
High-Throughput Assay Identifies RNA Editing Inhibitors
We used a convenient high-throughput, RNA editing-dependent hammerhead ribozyme (HHR) assay 17 to identify RNA editing inhibitors. In this assay, in the presence of editosome and a guide RNA that specifies the removal of three Us (deletion editing), an inactive HHR molecule (preedited A6Rbz) is converted into an active enzyme (edited A6Rbz) ( Fig. 1 ).
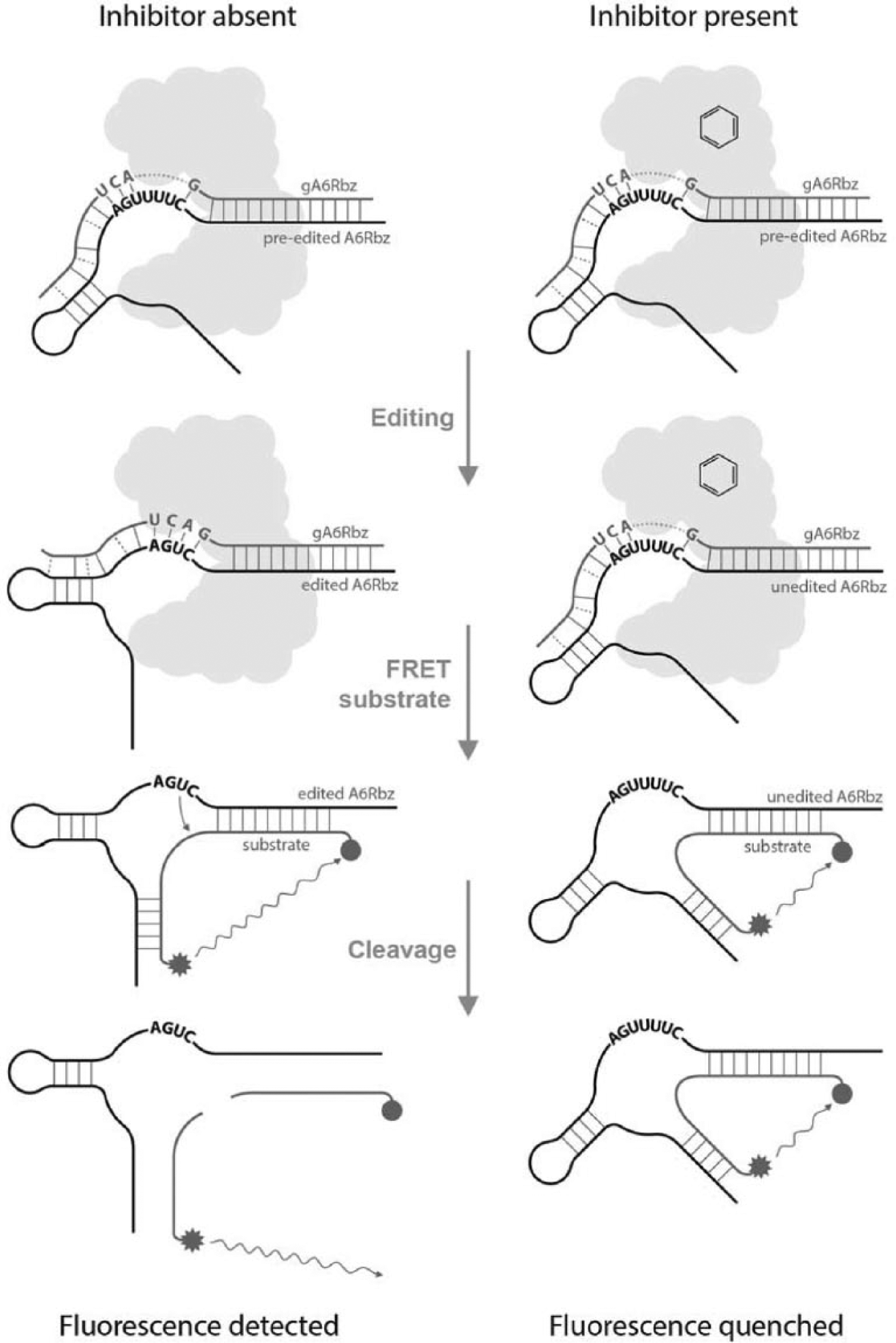
Fluorescence resonance energy transfer (FRET)–based high-throughput in vitro editing assay using hammerhead ribozyme. Editing reaction mixtures containing inactive (preedited) hammerhead ribozyme (preedited A6Rbz), editosome (cloud shape), and gRNA specifying removal of three Us (gA6Rbz) are assembled in the absence (left side) or presence (right side) of each compound (schematic ring structure). Following addition of FAM/Iowa black–labeled substrate, upon cleavage of the substrate by the edited ribozyme (edited A6Rbz), a fluorescence signal is generated that can be quantified. The schematic structural difference between the edited A6Rbz and the preedited/unedited A6Rbz is for representation of the structural and base pairing changes that result from the insertion of three additional Us into the catalytic core.
The substrate for the edited A6Rbz is a FRET-based RNA substrate: a 16-base-long oligoribonucleotide, labeled on the 5′ end with a fluorophore (6-FAM) and on the 3′ end with a dark quencher (Iowa black). Upon cleavage of this substrate by the edited A6Rbz molecule, the fluorescence emitted by FAM can be read as it moves out of the quenching range of Iowa black. The amount of fluorescence measured is proportional to the amount of edited A6Rbz present, which is proportional to the amount of editing that has occurred.
We used this assay to screen a collection of 1280 compounds from LOPAC (Sigma). The compounds were dissolved in DMSO and assayed in duplicate at a final concentration of 15 µM. The editosome used in this screen was prepared from mitochondrial extracts that were partially purified on glycerol gradients at ~20 Svedberg (20S), also known as the “20S editosome.” The editosome from this preparation contains the four key enzyme activities and a large number of mitochondrial RNA-binding proteins, some of which affect editing. 11 The screen was conducted in 384-well RT-qPCR plates, and a screening window coefficient (Z′ factor) value of each plate consistently averaged 0.6. A Z′ factor is a statistical measure of the reliability of an assay; any value above 0.5 is considered suitable for HTS. 26 In this primary screen, NF023, a previously identified inhibitor of insertion editing, was included. 19 Hits were selected at a cutoff stringency of 60%, meaning that any compound that reduced the fluorescence signal by 60% or more was chosen for confirmation assays ( Fig. 2A ). NF023 inhibited the reaction by only ~35%. However, six hits were obtained at the cutoff stringency of 60%—namely, aurintricarboxylic acid (ATA), mitoxantrone, PPNDS, NF449, dequalinium analogue C-14 linker, and ruthenium red ( Fig. 2B ). Dequalinium analogue C-14 linker was unavailable to purchase and was therefore excluded from further analysis.
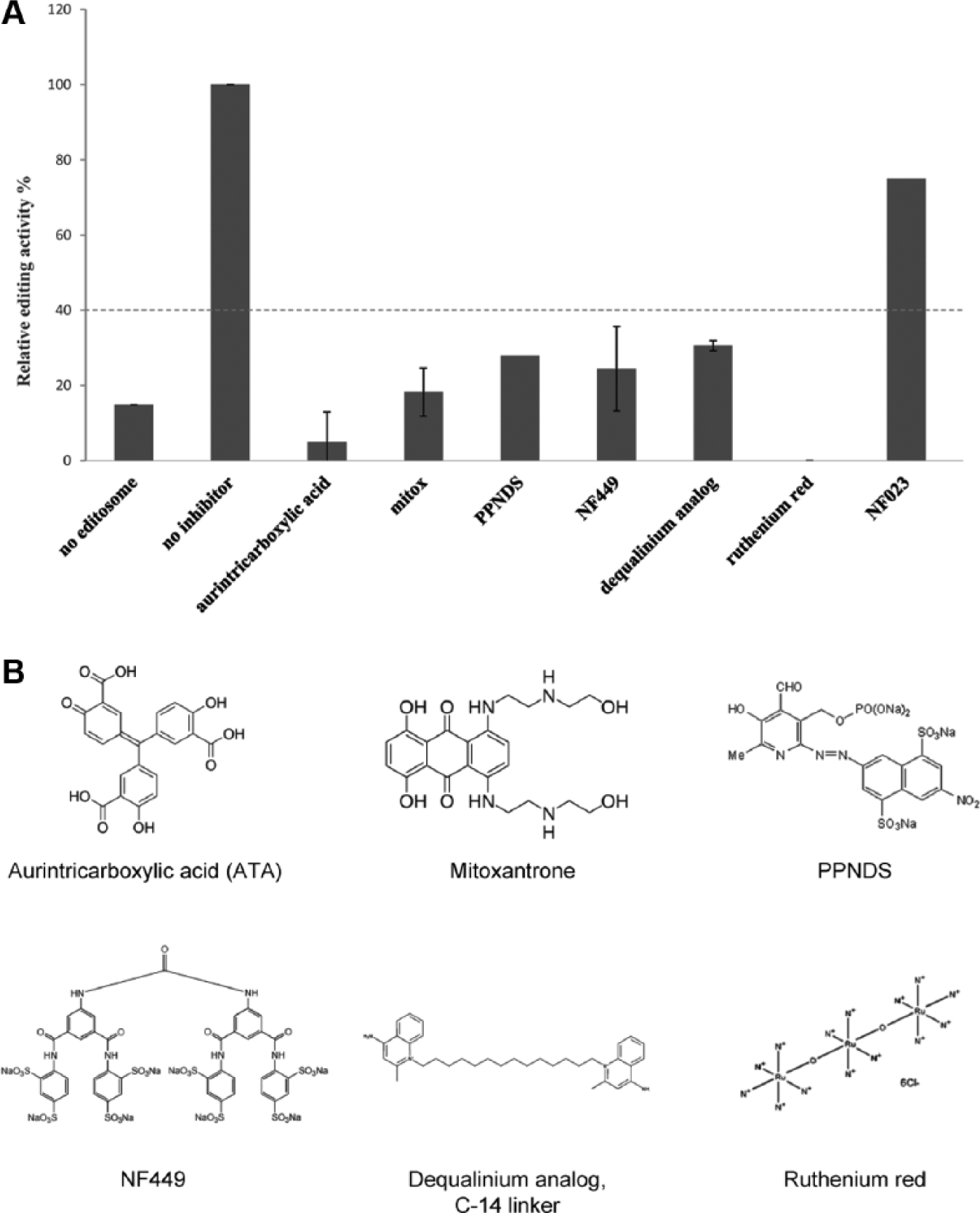
Screening of LOPAC for RNA editing inhibition activity. (
Secondary Assays Identify False Positives
To validate the specificity of the inhibition observed in the primary screen, an interference assay was performed to eliminate compounds that interfere with edited A6Rbz activity instead of the editosome. In this assay, the compounds were screened against the edited A6Rbz in the absence of the editosome. We included ethidium bromide, a known intercalator of nucleic acid interactions, as a positive control. The cleavage efficiency of the edited A6Rbz was significantly reduced in the presence of ethidium bromide ( Fig. 3 ). This assay also eliminates compounds that affect the fluorescence emitted by FAM by nonspecific quenching mechanisms. In the absence of the editosome, ruthenium red was able to reduce the cleavage efficiency of the edited A6Rbz by ~100% at 10 µM or greater and was nearly the same as with no edited A6Rbz (background) control. Therefore, ruthenium red was identified as a false positive and excluded from further characterization ( Fig. 3 ). The compound possibly binds to RNA, because of its dense positive charges, thereby inhibiting edited A6Rbz activity.
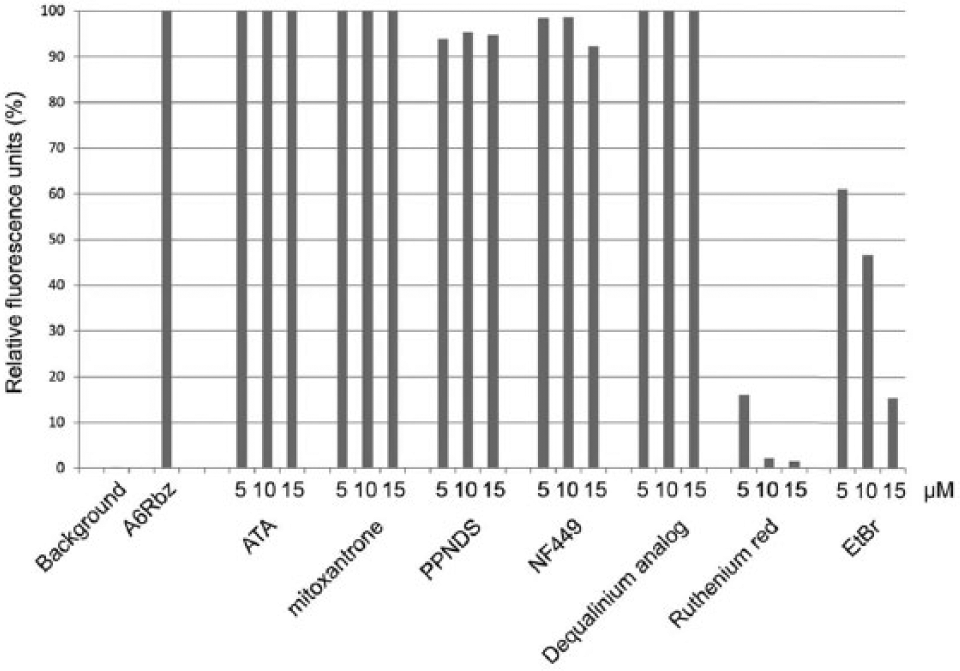
Validation of the hits from the LOPAC high-throughput screening using the interference assay. Edited A6Rbz RNA cleavage activity was monitored in the absence of edited A6Rbz (background) or in the presence of DMSO (A6Rbz); 5, 10, or 15 µM of each compound; and an inhibitory ethidium bromide (EtBr) control. The y-axis represents the fluorescence units relative to the active A6Rbz control (100%).
Dose-Response Study Identifies Potent Inhibitors of RNA Editing in T. brucei
A dose-response study was conducted on the compounds obtained using the FRET-based primary screening assay. All four compounds showed significant potency, with IC50 values ranging from 1 to 5 µM ( Fig. 4A ). Next, in vitro dose-response studies were conducted on the BF of T. brucei, because BF T. brucei is a favorable target for future drug development. While ATA and PPNDS were not able to kill the parasite (data not shown), mitoxantrone was efficacious in killing the parasite, with an EC50 value of 13 ± 1 nM ( Fig. 4B ). The higher EC50 value compared with the IC50 value of mitoxantrone suggests involvement of noneditosome targets of this compound. Mitoxantrone is known to intercalate DNA duplexes and inhibit various DNA-binding enzymes such as the DNA topoisomerase II, 27 and hence it is likely that its effect is due to additional targets in the cell besides the editosome. However, the EC50 value of NF449, 5.1 ± 0.8 µM ( Fig. 4B ), is close to the IC50 value of 3.5 ± 0.7 µM obtained in the FRET-based in vitro editing assay. Therefore, we suggest that NF449 may target editosomes in cells.
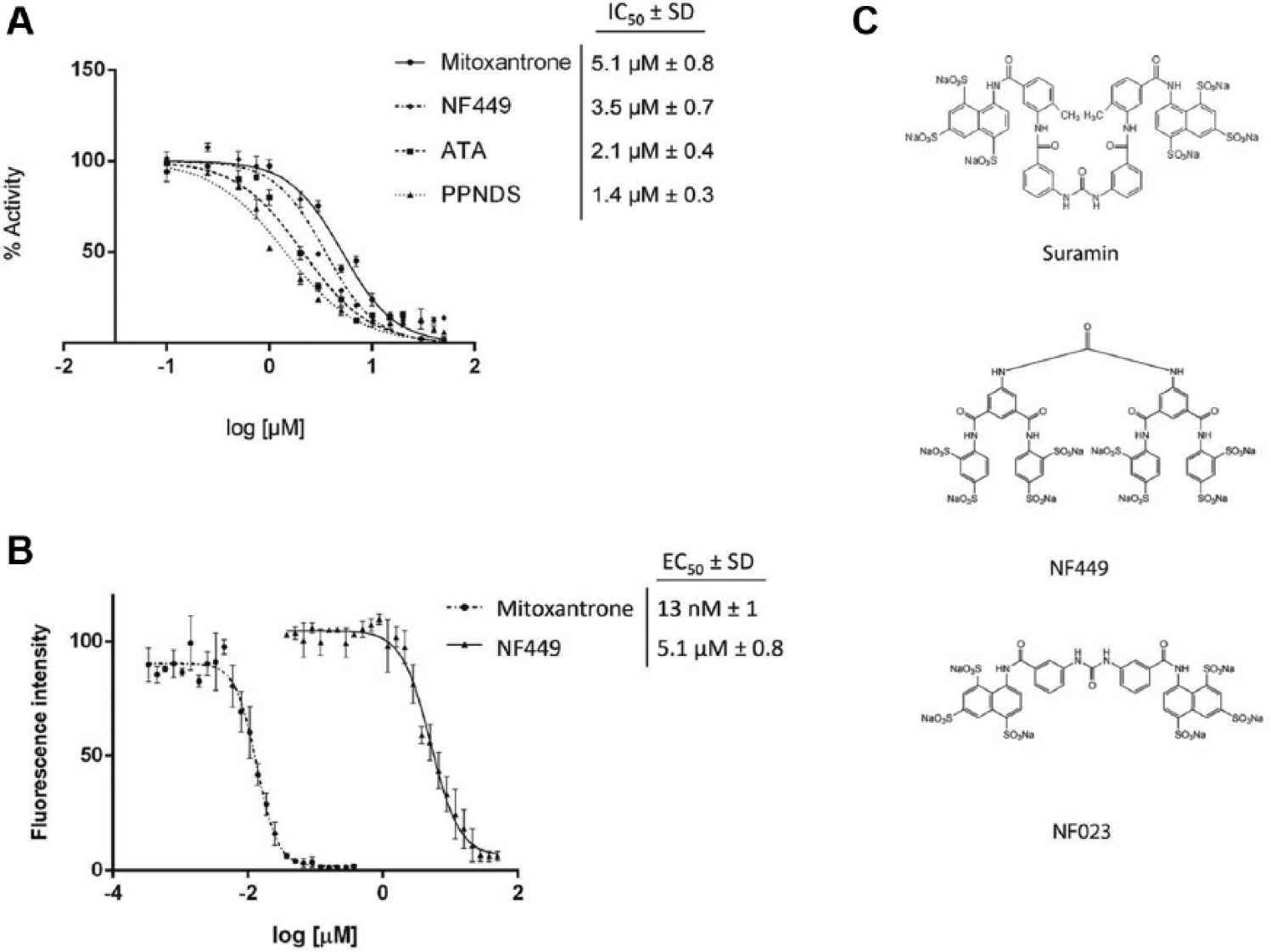
In vitro dose-response studies of compounds determined by the fluorescence resonance energy transfer (FRET)–based assay. (
NF449 is a known purinergic receptor antagonist with high selectivity for P2X1 receptors; it is also an analogue of suramin. 28 Suramin is a drug that has been used to treat trypanosomiasis, and it has been shown to affect metabolic pathways such as glycolysis, mainly due to its high negative charge density. NF023, also a known purinergic antagonist and a suramin analogue, 29 was identified in a similar LOPAC screen against insertion RNA editing. 19 In this previous screen, however, NF449 was not identified as an inhibitor of insertion RNA editing. These results suggest that NF023 may be a specific inhibitor of insertion RNA editing, while NF449 may specifically inhibit deletion RNA editing. Because both NF023 and NF449 are densely anionic like suramin ( Fig. 4C ), it is reasonable to suggest that the dense negative charge does not play a major role in inhibition. While we cannot rule out the possibility that NF449 also affects glycolysis and other metabolic processes as potential noneditosome targets, these data further support NF449 as being a selective inhibitor of deletion RNA editing and that it may target the editosomes in cells.
Secondary Assays Reveal That Hits Interfere with RNA-Protein Interaction
To determine which step of editosome function was affected by the selected compounds, in vitro RNA editing assays were performed. The inhibitors, ATA, mitoxantrone, PPNDS, and NF449, were assayed for their ability to affect the endonuclease, exoUase, or ligase in the editosome. As a control in these experiments, a naphthalene-based compound called Mordant Black 25 (MrB) was used. MrB inhibits REL1 self-adenylation when assayed against REL1 alone, 16 but it affects RNA-protein interaction when assayed against the editosome complex (unpublished data). Because of the unavailability of true inhibitors of the aforementioned editosome enzymes, MrB was used as a positive control compound. As a negative control compound, C53 was used, which was identified as an RNA editing ligase 1 inhibitor from a virtual screen but had no effect on the FRET-based in vitro editing assay. 18
ATA, PPNDS, NF449, or a control compound (MrB) could inhibit the exoUase ( Fig. 5A ), ligase ( Fig. 5B ), and the endonuclease ( Fig. 5C ). However, mitoxantrone or C53 (the negative control compound) had no effect on any of these enzymes and was the same as with no inhibitor treatment. The exoUase assay is based on a previously described precleaved deletion assay, 23 except that the 3′ RNA fragment is excluded from the reaction. By doing so, the assay specifically checks for exoUase activity. In conjunction with this exoUase assay, the precleaved deletion assay (which includes the 3′ RNA fragment) then conveniently tests for ligase activity. These two assays are independent of the endonuclease assay, as the precursor RNA used is already cleaved at its editing site. Hence, a compound inhibiting all three assays cannot be considered an inhibitor of a step prior to the endonuclease step. Inhibition of all the three editing assays implies that the compounds ATA, PPNDS, and NF449 affect RNA-protein interaction. To test this possibility, the compounds were tested against the editosome in an EMSA ( Fig. 5D ). In this assay, the editosome is preincubated with radiolabeled gRNA, which allows detection of RNA-bound protein complexes. Our result showed that there was no shift (RNA-protein complexes) in the reactions containing ATA and PPNDS and was the same as with no editosome. These data confirmed the inhibitory effect of ATA and PPNDS on the RNA-protein interaction. It has been shown that ATA interferes with nucleic acid–protein interaction. 30 Therefore, it is unlikely that ATA specifically selects for RNA-binding proteins in the RNA editing process. The reaction containing NF449 showed a shift of decreased intensity compared with the no-inhibitor control, suggesting that the NF449 block on RNA-protein interaction may specifically target a particular class of RNA-binding proteins that aid RNA editing. Conversely, the reaction containing mitoxantrone did not run into the gel, possibly because it induced RNA-protein aggregation.
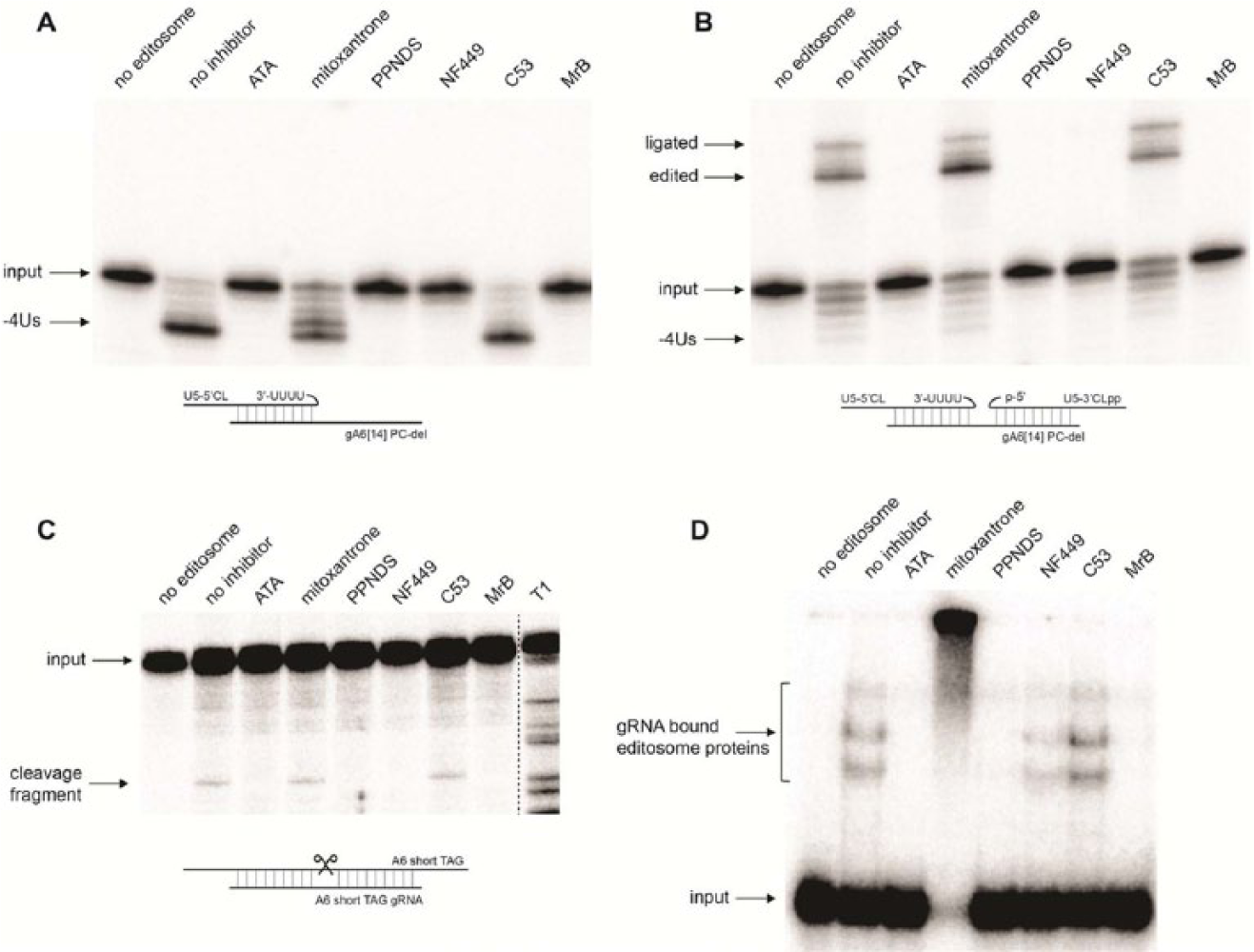
Assessment of in vitro RNA editing inhibition by selected compounds using secondary assays. (
To test whether mitoxantrone induces RNA-protein aggregation, we performed a variation of the interference assay. We screened the four compounds against the edited A6Rbz in presence of the editosome. Reduction in the fluorescence signal would suggest inability of the edited A6Rbz to cleave its substrate because of aggregation; this conclusion can only be made after performing the aforementioned interference assay. Here, mitoxantrone reduced the fluorescence by 50% compared with the positive control (DMSO) ( Fig. 6 ). We conclude that mitoxantrone may not completely inhibit edited A6Rbz molecules in solution (as 50% of edited A6Rbz was still enzymatic), suggesting that the >80% inhibition seen in the primary assay could partly be attributed to aggregation.
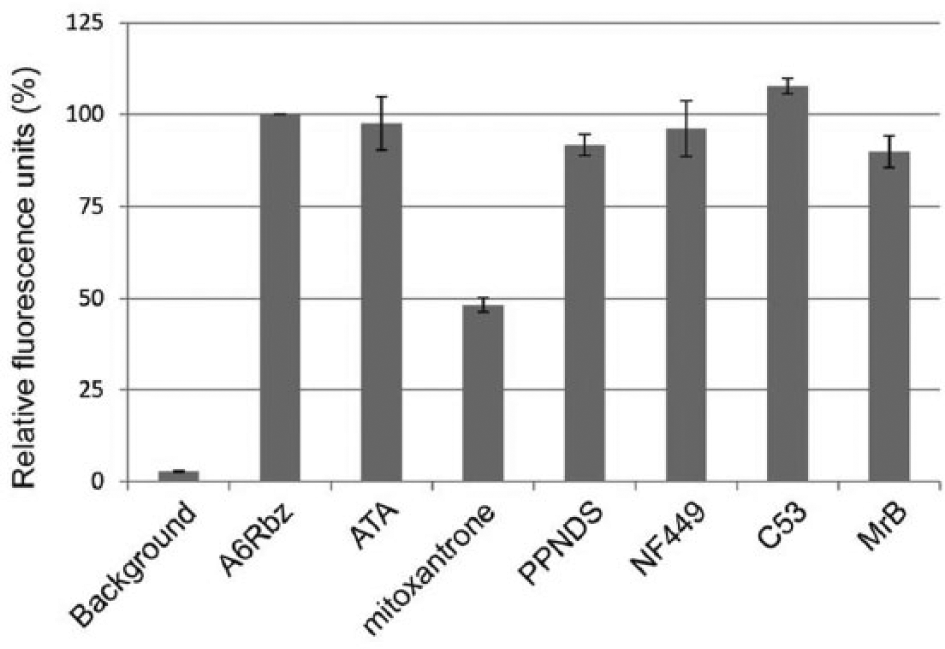
Mitoxantrone induces RNA-protein aggregation. In vitro fluorescence resonance energy transfer–based editing reaction mixtures were monitored in the absence of edited A6Rbz (background), presence of edited A6Rbz/DMSO (A6Rbz), 10 µM aurintricarboxylic acid (ATA), mitoxantrone, PPNDS, NF449, noninhibitory C53, and inhibitory Mordant Black (MrB). The results are expressed as percentages relative to A6Rbz control (100%). The error bars represent standard deviations of three independent experiments.
In conclusion, our screen was successful in identifying inhibitors of RNA editing. While ATA, PPNDS, and NF449 interfere with RNA-protein interaction to different extents, mitoxantrone induces RNA-protein aggregate formation. From our analysis, we have shown that NF449 is selective for deletion RNA editing. Furthermore, because the binding affinity of NF449 is similar to cell growth inhibition activity, we suggest that it may target a specific class of RNA-binding proteins that operate alongside the editosome. Elucidation of the mechanism of inhibition of the leads identified in this study will provide a basis for the selection of drug targets and contribute to the functional studies of an essential gene expression mechanism and to the possibility of future drug development against trypanosomatid pathogens.
Footnotes
Acknowledgements
The authors thank Dr. Zeng and the Conrad Prebys Center for Chemical Genomics of the Sanford-Burnham Medical Reseach Institute for providing the LOPAC plates for this study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Canadian Institutes of Health Research (CIHR) grant 119464 to RS. VM is supported by CIHR Chemical Biology Fellowship.